Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment



Abstract
1. Introduction
2. History
3. Evaluation of the Causes of ARP and CP
4. Genetic Abnormalities
4.1. CFTR Gene
4.2. PRSS1 Gene
4.3. SPINK1 Gene
4.4. CTRC Gene
4.5. CPA1 Gene
4.6. TRPV6 Gene
4.7. Others
5. Clinical Features of PRSS1 and SPINK1 Gene Mutation-Related Pancreatitis
5.1. Age at Symptom Onset, Pancreatic Exocrine Insufficiency, and Diabetes Mellitus
5.2. Pancreatic Cancer
6. Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitcomb, D.C.; Gorry, M.C.; Preston, R.A.; Furey, W.; Sossenheimer, M.J.; Ulrich, C.D.; Martin, S.P.; Gates, L.K., Jr.; Amann, S.T.; Toskes, P.P.; et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 1996, 14, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Giefer, M.J.; Lowe, M.E.; Werlin, S.L.; Zimmerman, B.; Wilschanski, M.; Troendle, D.; Schwarzenberg, S.J.; Poh, L.J.F.; Palermo, J.; Ooi, C.Y.; et al. Early-Onset Acute Recurrent and Chronic Pancreatitis Is Associated with PRSS1 or CTRC Gene Mutations. J. Pediatr. 2017, 186, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Hamada, S.; Nakano, E.; Kume, K.; Inui, A.; Shimizu, T.; Takeyama, Y.; Nio, M.; Shimosegawa, T. Nationwide survey of hereditary pancreatitis in Japan. J. Gastroenterol. 2018, 53, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Joergensen, M.T.; Brusgaard, K.; Cruger, D.G.; Gerdes, A.M.; Schaffalitzky de Muckadell, O.B. Genetic; epidemiological; and clinical aspects of hereditary pancreatitis, a population-based cohort study in Denmark. Am. J. Gastroenterol. 2010, 105, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Ferec, C.; Le Marechal, C.; Hentic, O.; Maire, F.; Hammel, P.; Ruszniewski, P.; Levy, P. The natural history of hereditary pancreatitis, a national series. Gut 2009, 58, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Toth, M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology 2019, 156, 1951–1968.e1. [Google Scholar] [CrossRef]
- Muller, N.; Sarantitis, I.; Rouanet, M.; de Mestier, L.; Halloran, C.; Greenhalf, W.; Ferec, C.; Masson, E.; Ruszniewski, P.; Levy, P.; et al. Natural history of SPINK1 germline mutation related-pancreatitis. EBioMedicine 2019, 48, 581–591. [Google Scholar] [CrossRef]
- Comfort, M.W.; Steinberg, A.G. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 1952, 21, 54–63. [Google Scholar] [CrossRef]
- Gross, J.B.; Gambill, E.E.; Ulrich, J.A. Hereditary pancreatitis. Description of a fifth kindred and summary of clinical features. Am. J. Med. 1962, 33, 358–364. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene, cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Cohn, J.A.; Friedman, K.J.; Noone, P.G.; Knowles, M.R.; Silverman, L.M.; Jowell, P.S. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med. 1998, 339, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Sharer, N.; Schwarz, M.; Malone, G.; Howarth, A.; Painter, J.; Super, M.; Braganza, J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 1998, 339, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Luck, W.; Hennies, H.C.; Classen, M.; Kage, A.; Lass, U.; Landt, O.; Becker, M. Mutations in the gene encoding the serine protease inhibitor; Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 2000, 25, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Felderbauer, P.; Hoffmann, P.; Einwachter, H.; Bulut, K.; Ansorge, N.; Schmitz, F.; Schmidt, W.E. A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol. 2003, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Felderbauer, P.; Klein, W.; Bulut, K.; Ansorge, N.; Dekomien, G.; Werner, I.; Epplen, J.T.; Schmitz, F.; Schmidt, W.E. Mutations in the calcium-sensing receptor, a new genetic risk factor for chronic pancreatitis? Scand. J. Gastroenterol. 2006, 41, 343–348. [Google Scholar] [CrossRef]
- Muddana, V.; Lamb, J.; Greer, J.B.; Elinoff, B.; Hawes, R.H.; Cotton, P.B.; Anderson, M.A.; Brand, R.E.; Slivka, A.; Whitcomb, D.C. Association between calcium sensing receptor gene polymorphisms and chronic pancreatitis in a US population, role of serine protease inhibitor Kazal 1type and alcohol. World. J. Gastroenterol. 2008, 14, 4486–4491. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Ferec, C. Overrepresentation of Rare CASR Coding Variants in a Sample of Young French Patients with Idiopathic Chronic Pancreatitis. Pancreas 2015, 44, 996–998. [Google Scholar] [CrossRef]
- Rosendahl, J.; Witt, H.; Szmola, R.; Bhatia, E.; Ozsvari, B.; Landt, O.; Schulz, H.U.; Gress, T.M.; Pfutzer, R.; Lohr, M.; et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 2008, 40, 78–82. [Google Scholar] [CrossRef]
- Whitcomb, D.C.; LaRusch, J.; Krasinskas, A.M.; Klei, L.; Smith, J.P.; Brand, R.E.; Neoptolemos, J.P.; Lerch, M.M.; Tector, M.; Sandhu, B.S.; et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat. Genet. 2012, 44, 1349–1354. [Google Scholar] [CrossRef]
- Witt, H.; Beer, S.; Rosendahl, J.; Chen, J.M.; Chandak, G.R.; Masamune, A.; Bence, M.; Szmola, R.; Oracz, G.; Macek, M.; et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat. Genet. 2013, 45, 1216–1220. [Google Scholar] [CrossRef]
- Fjeld, K.; Weiss, F.U.; Lasher, D.; Rosendahl, J.; Chen, J.M.; Johansson, B.B.; Kirsten, H.; Ruffert, C.; Masson, E.; Steine, S.J.; et al. A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat. Genet. 2015, 47, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, J.; Kirsten, H.; Hegyi, E.; Kovacs, P.; Weiss, F.U.; Laumen, H.; Lichtner, P.; Ruffert, C.; Chen, J.M.; Masso, N.E.; et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018, 67, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Lasher, D.; Szabo, A.; Masamune, A.; Chen, J.M.; Xiao, X.; Whitcomb, D.C.; Barmada, M.M.; Ewers, M.; Ruffert, C.; Paliwal, S.; et al. Protease-Sensitive Pancreatic Lipase Variants Are Associated With Early Onset Chronic Pancreatitis. Am. J. Gastroenterol. 2019, 114, 974–983. [Google Scholar] [CrossRef]
- Masamune, A.; Kotani, H.; Sorgel, F.L.; Chen, J.M.; Hamada, S.; Sakaguchi, R.; Masson, E.; Nakano, E.; Kakuta, Y.; Niihori, T.; et al. Variants That Affect Function of Calcium Channel TRPV6 Are Associated With Early-Onset Chronic Pancreatitis. Gastroenterology 2020, 158, 1626–1641.e8. [Google Scholar] [CrossRef] [PubMed]
- Raeder, H.; Johansson, S.; Holm, P.I.; Haldorsen, I.S.; Mas, E.; Sbarra, V.; Nermoen, I.; Eide, S.A.; Grevle, L.; Bjorkhaug, L.; et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat. Genet. 2006, 38, 54–62. [Google Scholar] [CrossRef]
- Kujko, A.A.; Berki, D.M.; Oracz, G.; Wejnarska, K.; Antoniuk, J.; Wertheim-Tysarowska, K.; Kolodziejczyk, E.; Bal, J.; Sahin-Toth, M.; Rygiel, A.M. A novel p.Ser282Pro CPA1 variant is associated with autosomal dominant hereditary pancreatitis. Gut 2017, 66, 1728–1730. [Google Scholar] [CrossRef]
- Witt, H.; Sahin-Toth, M.; Landt, O.; Chen, J.M.; Kahne, T.; Drenth, J.P.; Kukor, Z.; Szepessy, E.; Halangk, W.; Dahm, S.; et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat. Genet. 2006, 38, 668–673. [Google Scholar] [CrossRef]
- Banks, P.A. Epidemiology; natural history; and predictors of disease outcome in acute and chronic pancreatitis. Gastrointest. Endosc. 2002, 56, S226–S230. [Google Scholar] [CrossRef]
- Hegyi, P.; Parniczky, A.; Lerch, M.M.; Sheel, A.R.G.; Rebour, S.V.; Forsmark, C.E.; Del Chiaro, M.; Rosendahl, J.; de-Madaria, E.; Szucs, A.; et al. International Consensus Guidelines for Risk Factors in Chronic Pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with the International Association of Pancreatology; the American Pancreatic Association; the Japan Pancreas Society; and European Pancreatic Club. Pancreatology 2020, 20, 579–585. [Google Scholar]
- DeBanto, J.R.; Goday, P.S.; Pedroso, M.R.; Iftikhar, R.; Fazel, A.; Nayyar, S.; Conwell, D.L.; Demeo, M.T.; Burton, F.R.; Whitcomb, D.C.; et al. Acute pancreatitis in children. Am. J. Gastroenterol. 2002, 97, 1726–1731. [Google Scholar] [CrossRef]
- Nydegger, A.; Couper, R.T.; Oliver, M.R. Childhood pancreatitis. J. Gastroenterol. Hepatol. 2006, 21, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sait, O.N.; Naritaka, N.; Nakano, S.; Minowa, K.; Honda, Y.; Ohtsuka, Y.; Yamataka, A.; Shimizu, T. Scoring system for the prediction of severe acute pancreatitis in children. Pediatr. Int. 2015, 57, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Suzuki, M.; Sakurai, Y.; Nakan, O.S.; Naritaka, N.; Minowa, K.; Sai, J.K.; Shimizu, T. Genetic Analysis of Japanese Children With Acute Recurrent and Chronic Pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Yadav, D.; Garg, P.K. Diagnosis and Management of Chronic Pancreatitis, A Review. JAMA 2019, 322, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Ellis, I.; Lerch, M.M.; Whitcomb, D.C.; Consensus Committees of the European Registry of Hereditary Pancreatic Diseases MM-CPSGIAoP. Genetic testing for hereditary pancreatitis, guidelines for indications; counselling; consent and privacy issues. Pancreatology 2001, 1, 405–415. [Google Scholar] [CrossRef]
- Fink, E.N.; Kant, J.A.; Whitcomb, D.C. Genetic counseling for nonsyndromic pancreatitis. Gastroenterol. Clin. North. Am. 2007, 36, 325–333. [Google Scholar] [CrossRef]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Elli, S.I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- Suzuki, M.; Shimizu, T.; Kudo, T.; Suzuki, R.; Ohtsuka, Y.; Yamashiro, Y.; Shimotakahara, A.; Yamataka, A. Usefulness of nonbreath-hold 1-shot magnetic resonance cholangiopancreatography for the evaluation of choledochal cyst in children. J. Pediatr. Gastroenterol. Nutr. 2006, 42, 539–544. [Google Scholar] [CrossRef]
- Lee, M.G.; Ohana, E.; Park, H.W.; Yang, D.; Muallem, S. Molecular mechanism of pancreatic and salivary gland fluid and HCO3 secretion. Physiol. Rev. 2012, 92, 39–74. [Google Scholar] [CrossRef]
- Freedman, S.D. New concepts in understanding the pathophysiology of chronic pancreatitis. Int. J. Pancreatol. 1998, 24, 1–8. [Google Scholar]
- Fujiki, K.; Ishiguro, H.; Ko, S.B.; Mizuno, N.; Suzuki, Y.; Takemura, T.; Yamamoto, A.; Yoshikawa, T.; Kitagawa, M.; Hayakawa, T.; et al. Genetic evidence for CFTR dysfunction in Japanese, background for chronic pancreatitis. J. Med. Genet. 2004, 41, e55. [Google Scholar] [CrossRef] [PubMed]
- Noone, P.G.; Zhou, Z.; Silverman, L.M.; Jowell, P.S.; Knowles, M.R.; Cohn, J.A. Cystic fibrosis gene mutations and pancreatitis risk, relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology 2001, 121, 1310–1319. [Google Scholar] [CrossRef]
- Rosendahl, J.; Land, T.O.; Bernadova, J.; Kovacs, P.; Teich, N.; Bodeker, H.; Keim, V.; Ruffert, C.; Mossner, J.; Kage, A.; et al. CFTR; SPINK1; CTRC and PRSS1 variants in chronic pancreatitis, is the role of mutated CFTR overestimated? Gut 2013, 62, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Larusch, J.; Sun, X.; Aloe, A.; Lamb, J.; Hawes, R.; Cotton, P.; Brand, R.E.; Anderson, M.A.; Money, M.E.; et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology 2011, 140, 162–171. [Google Scholar] [CrossRef]
- Gorry, M.C.; Gabbaizedeh, D.; Furey, W.; Gates, L.K., Jr.; Preston, R.A.; Aston, C.E.; Zhang, Y.; Ulrich, C.; Ehrlich, G.D.; Whitcom, B.D.C. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology 1997, 113, 1063–1068. [Google Scholar] [CrossRef]
- Szabo, A.; Sahin-Toth, M. Increased activation of hereditary pancreatitis-associated human cationic trypsinogen mutants in presence of chymotrypsin C. J. Biol. Chem. 2012, 287, 20701–20710. [Google Scholar] [CrossRef] [PubMed]
- Nemoda, Z.; Sahin-Toth, M. Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 2006, 281, 11879–11886. [Google Scholar] [CrossRef] [PubMed]
- Kereszturi, E.; Szmola, R.; Kukor, Z.; Simon, P.; Weiss, F.U.; Lerch, M.M.; Sahin-Toth, M. Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen, a novel disease mechanism. Hum. Mutat. 2009, 30, 575–582. [Google Scholar] [CrossRef]
- Schnur, A.; Beer, S.; Witt, H.; Hegyi, P.; Sahin-Toth, M. Functional effects of 13 rare PRSS1 variants presumed to cause chronic pancreatitis. Gut 2014, 63, 337–343. [Google Scholar] [CrossRef]
- Hedstrom, J.; Kemppainen, E.; Andersen, J.; Jokela, H.; Puolakkainen, P.; Stenman, U.H. A comparison of serum trypsinogen-2 and trypsin-2-alpha1-antitrypsin complex with lipase and amylase in the diagnosis and assessment of severity in the early phase of acute pancreatitis. Am. J. Gastroenterol. 2001, 96, 424–430. [Google Scholar] [CrossRef]
- Shimosegawa, T.; Kume, K.; Masamune, A. SPINK1 gene mutations and pancreatitis in Japan. J. Gastroentero. Hepatol. 2006, 21 (Suppl. 3), S47–S51. [Google Scholar] [CrossRef] [PubMed]
- Truninger, K.; Witt, H.; Kock, J.; Kage, A.; Seifert, B.; Ammann, R.W.; Blum, H.E.; Becker, M. Mutations of the serine protease inhibitor, Kazal type 1 gene, in patients with idiopathic chronic pancreatitis. Am. J. Gastroenterol. 2002, 97, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, K.; Hirota, M.; Shimizu, H.; Nakae, M.; Nishihara, S.; Takimoto, A.; Mitsushima, K.; Kikuchi, N.; Endo, K.; Inoue, M.; et al. Functional analysis of recombinant pancreatic secretory trypsin inhibitor protein with amino-acid substitution. J. Gastroenterol. 2002, 37, 928–934. [Google Scholar] [CrossRef]
- Kiraly, O.; Wartmann, T.; Sahin-Toth, M. Missense mutations in pancreatic secretory trypsin inhibitor (SPINK1) cause intracellular retention and degradation. Gut 2007, 56, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, K.M.; Choi, J.H.; Lee, B.H.; Kim, G.H.; Yoo, H.W. High incidence of PRSS1 and SPINK1 mutations in Korean children with acute recurrent and chronic pancreatitis. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Kume, K.; Masamune, A.; Mizutamari, H.; Kaneko, K.; Kikuta, K.; Sato, H.M.; Satoh, K.; Kimura, K.; Suzuki, N.; Nagasaki, Y.; et al. Mutations in the serine protease inhibitor Kazal Type 1 (SPINK1) gene in Japanese patients with pancreatitis. Pancreatology 2005, 5, 354–360. [Google Scholar] [CrossRef]
- Oh, H.C.; Kim, M.H.; Choi, K.S.; Moon, S.H.; Park, D.H.; Lee, S.S.; Seo, D.W.; Lee, S.K.; Yoo, H.W.; Kim, G.H. Analysis of PRSS1 and SPINK1 mutations in Korean patients with idiopathic and familial pancreatitis. Pancreas 2009, 38, 180–183. [Google Scholar] [CrossRef]
- Masamune, A.; Kume, K.; Takagi, Y.; Kikuta, K.; Satoh, K.; Satoh, A.; Shimosegawa, T. N34S mutation in the SPINK1 gene is not associated with alternative splicing. Pancreas 2007, 34, 423–428. [Google Scholar] [CrossRef]
- Beer, S.; Zhou, J.; Szabo, A.; Keiles, S.; Chandak, G.R.; Witt, H.; Sahin-Toth, M. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 2013, 62, 1616–1624. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.M.; Scotet, V.; Le Marechal, C.; Ferec, C. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 2008, 123, 83–91. [Google Scholar] [CrossRef]
- Suzuki, M.; Minowa, K.; Isayama, H.; Shimizu, T. Acute Recurrent and Chronic Pancreatitis in Children. Pediatr Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fecher-Trost, C.; Wissenbach, U.; Weissgerber, P. TRPV6, From identification to function. Cell. Calcium. 2017, 67, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wissenbach, U.; Niemeyer, B.A.; Fixemer, T.; Schneidewind, A.; Trost, C.; Cavalie, A.; Reus, K.; Meese, E.; Bonkhoff, H.; Flockerzi, V. Expression of CaT-like; a novel calcium-selective channel; correlates with the malignancy of prostate cancer. J. Biol. Chem. 2001, 276, 19461–19468. [Google Scholar] [CrossRef]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytto, N.J.; Hebert, S.C. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Racz, G.Z.; Kittel, A.; Riccardi, D.; Case, R.M.; Elliott, A.C.; Varga, G. Extracellular calcium sensing receptor in human pancreatic cells. Gut 2002, 51, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Kukor, Z.; Toth, M.; Sahin-Toth, M. Human anionic trypsinogen, properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur. J. Biochem. 2003, 270, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Jancso, Z.; Hegyi, E.; Sahin-Toth, M. Chymotrypsin Reduces the Severity of Secretagogue-Induced Pancreatitis in Mice. Gastroenterology 2018, 155, 1017–1021. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Abu-El-Haij, A.M.; Husain, S.Z.; Barth, B.; Bellin, M.; Fishman, D.S.; Freedman, S.D.; Gariepy, C.E.; Giefer, M.J.; Gonska, T.; et al. Risk Factors for Rapid Progression From Acute Recurrent to Chronic Pancreatitis in Children, Report from INSPPIRE. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 206–211. [Google Scholar] [CrossRef]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Ferec, C.; Maire, F.; Hammel, P.; Ruszniewski, P.; Levy, P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis, a national exhaustive series. Am. J. Gastroenterol. 2008, 103, 111–119. [Google Scholar] [CrossRef]
- Shelton, C.A.; Umapathy, C.; Stello, K.; Yadav, D.; Whitcomb, D.C. Hereditary Pancreatitis in the United States, Survival and Rates of Pancreatic Cancer. Am. J. Gastroenterol. 2018, 113, 1376. [Google Scholar] [CrossRef]
- Lasson, A.; Ohlsson, K. Consumptive coagulopathy; fibrinolysis and protease-antiprotease interactions during acute human pancreatitis. Thromb. Res. 1986, 41, 167–183. [Google Scholar] [CrossRef]
- Dumnicka, P.; Maduzia, D.; Ceranowicz, P.; Olszanecki, R.; Drozdz, R.; Kusnierz-Cabala, B. The Interplay between Inflammation; Coagulation and Endothelial Injury in the Early Phase of Acute Pancreatitis, Clinical Implications. Int. J. Mol. Sci. 2017, 18, 354. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Hogwood, J.; Mulloy, B. The anticoagulant and antithrombotic mechanisms of heparin. Handb. Exp. Pharmacol. 2012, 207, 43–61. [Google Scholar]
- Finotti, P.; Manente, S. Heparin-induced structural and functional alterations of bovine trypsin. Biochim. Biophys. Acta. 1994, 1207, 80–87. [Google Scholar] [CrossRef]
- Struss, D.; Storck, J.; Zimmermann, R.E. The inhibition of thrombin and chymotrypsin by heparin-cofactor II. Thromb. Res. 1992, 68, 45–56. [Google Scholar] [PubMed]
- Wolosowicz, N.; Prokopowicz, J.; Gabryelewicz, A. The inhibitory effect of heparin on trypsinogen activation with enterokinase. Acta. Hepatogastroenterol. 1977, 24, 368–371. [Google Scholar]
- Bottino, R.; Bertera, S.; Grupillo, M.; Melvin, P.R.; Humar, A.; Mazariegos, G.; Moser, A.J.; Walsh, R.M.; Fung, J.; Gelrud, A.; et al. Isolation of human islets for autologous islet transplantation in children and adolescents with chronic pancreatitis. J. Transplant. 2012, 2012, 642787. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.C.; Sutton, J.M.; Salehi, M.; Schmulewitz, N.; Smith, M.T.; Kucera, S.; Choe, K.A.; Brunner, J.E.; Abbott, D.E.; Sussman, J.J.; et al. Surgical outcomes after total pancreatectomy and islet cell autotransplantation in pediatric patients. Surgery 2013, 154, 777–783. [Google Scholar] [CrossRef]
- Chinnakotla, S.; Radosevic, H.D.M.; Dunn, T.B.; Bellin, M.D.; Freeman, M.L.; Schwarzenberg, S.J.; Balamurugan, A.N.; Wilhelm, J.; Bland, B.; Vickers, S.M.; et al. Long-term outcomes of total pancreatectomy and islet auto transplantation for hereditary/genetic pancreatitis. J. Am. Coll. Surg. 2014, 218, 530–543. [Google Scholar] [CrossRef]
- Agarwal, J.; Reddy, D.N.; Talukdar, R.; Lakhtakia, S.; Ramchandani, M.; Tandan, M.; Gupta, R.; Pratap, N.; Rao, G.V. ERCP in the management of pancreatic diseases in children. Gastrointest. Endosc. 2014, 79, 271–278. [Google Scholar] [CrossRef]
- Nabi, Z.; Reddy, D.N. Advanced Therapeutic Gastrointestinal Endoscopy in Children—Today and Tomorrow. Clin. Endosc. 2018, 51, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Issa, Y.; Bruno, M.J.; Bakker, O.J.; Besselink, M.G.; Schepers, N.J.; van Santvoort, H.C.; Gooszen, H.G.; Boermeester, M.A. Treatment options for chronic pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Chari, S.T. Chronic pancreatitis. Lancet 2016, 387, 1957–1966. [Google Scholar] [CrossRef]
- Kargl, S.; Kienbauer, M.; Duba, H.C.; Schofl, R.; Pumberger, W. Therapeutic step-up strategy for management of hereditary pancreatitis in children. J. Pediatr. Surg. 2015, 50, 511–514. [Google Scholar] [CrossRef]
- Costamagna, G.; Bulajic, M.; Tringali, A.; Pandolfi, M.; Gabbrielli, A.; Spada, C.; Petruzziello, L.; Familiari, P.; Mutignani, M. Multiple stenting of refractory pancreatic duct strictures in severe chronic pancreatitis, long-term results. Endoscopy 2006, 38, 254–259. [Google Scholar] [CrossRef]
- Matsubara, S.; Sasahira, N.; Isayama, H.; Takahara, N.; Mizuno, S.; Kogur, E.H.; Yamamoto, N.; Nakai, Y.; Tada, M.; Koike, K. Prospective pilot study of fully covered self-expandable metal stents for refractory benign pancreatic duct strictures, long-term outcomes. Endosc. Int. Open 2016, 4, E1215–E1222. [Google Scholar]
- Tringali, A.; Bov, E.V.; Vadala di Prampero, S.F.; Boskoski, I.; Familiari, P.; Perri, V.; Costamagna, G. Long-term follow-up after multiple plastic stenting for refractory pancreatic duct strictures in chronic pancreatitis. Endoscopy 2019, 51, 930–935. [Google Scholar] [CrossRef]



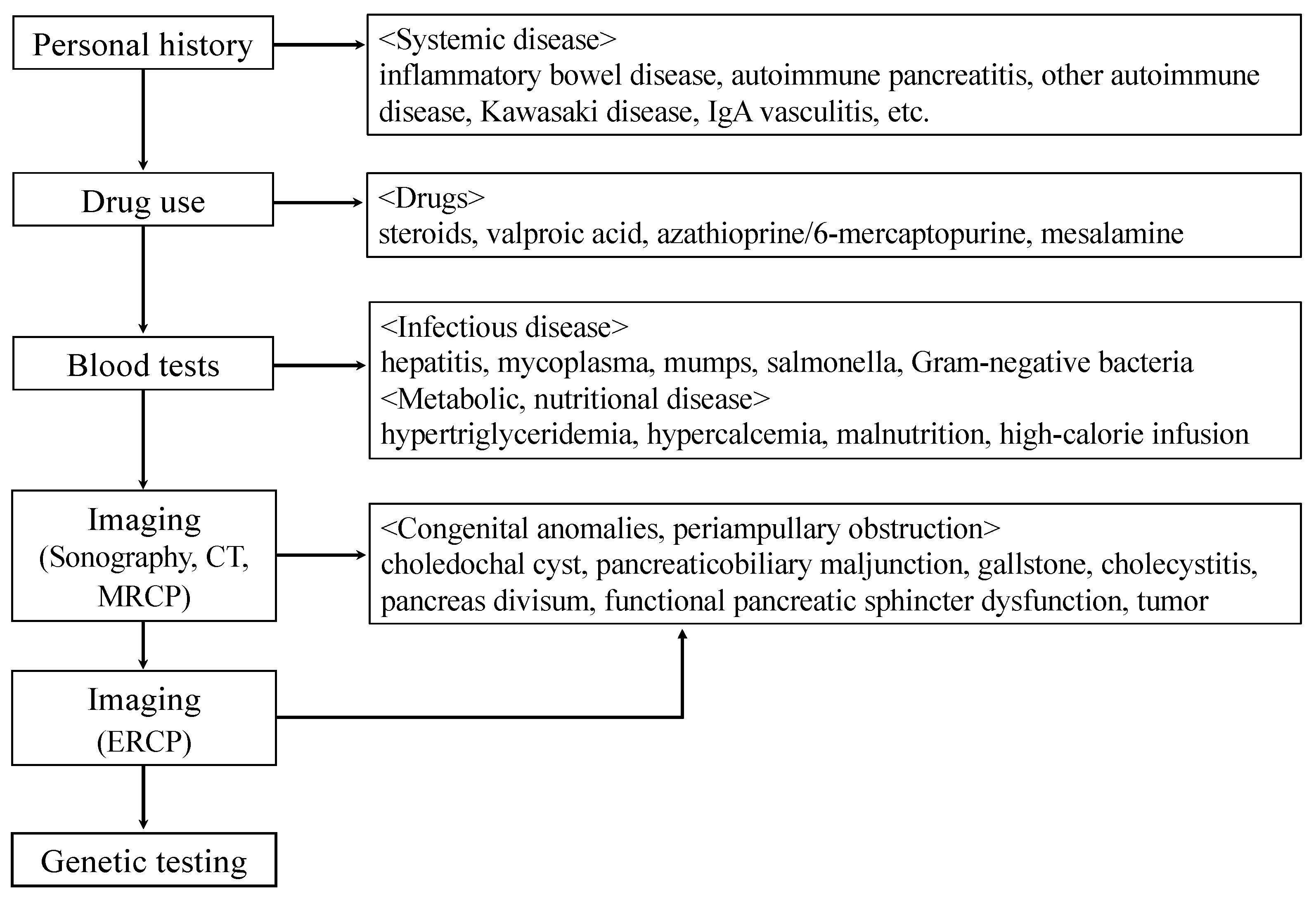{kind=link}
{kind=link}
| Consider When Patients Meet One or More of the Following Criteria: | |
|---|---|
| 1 | A family history of idiopathic CP, ARP, or childhood pancreatitis |
| 2 | Relatives with known mutations associated with HP |
| 3 | Unexpected pancreatitis in a child |
| 4 | Idiopathic CP in patients < 25 years old |
| 5 | ARP of uncertain etiology |
| 6 | Patients who meet criteria for participation in approved research projects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, M.; Minowa, K.; Nakano, S.; Isayama, H.; Shimizu, T. Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment. Diagnostics 2021, 11, 31. https://doi.org/10.3390/diagnostics11010031
Suzuki M, Minowa K, Nakano S, Isayama H, Shimizu T. Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment. Diagnostics. 2021; 11(1):31. https://doi.org/10.3390/diagnostics11010031
Chicago/Turabian StyleSuzuki, Mitsuyoshi, Kei Minowa, Satoshi Nakano, Hiroyuki Isayama, and Toshiaki Shimizu. 2021. "Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment" Diagnostics 11, no. 1: 31. https://doi.org/10.3390/diagnostics11010031
APA StyleSuzuki, M., Minowa, K., Nakano, S., Isayama, H., & Shimizu, T. (2021). Genetic Abnormalities in Pancreatitis: An Update on Diagnosis, Clinical Features, and Treatment. Diagnostics, 11(1), 31. https://doi.org/10.3390/diagnostics11010031