Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis



Abstract
:1. Introduction
2. Case Report
2.1. Diagnosis
2.2. Hematopoietic Stem Cell Transplantation (HSCT)
2.3. Clinical Course after HSCT
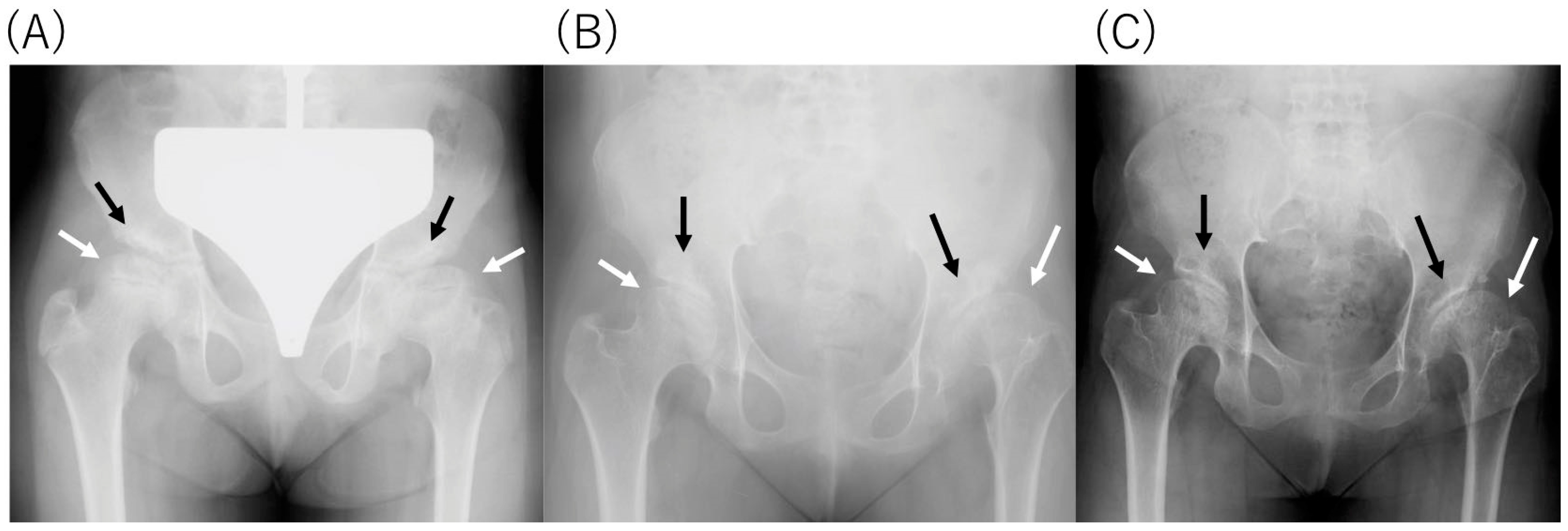
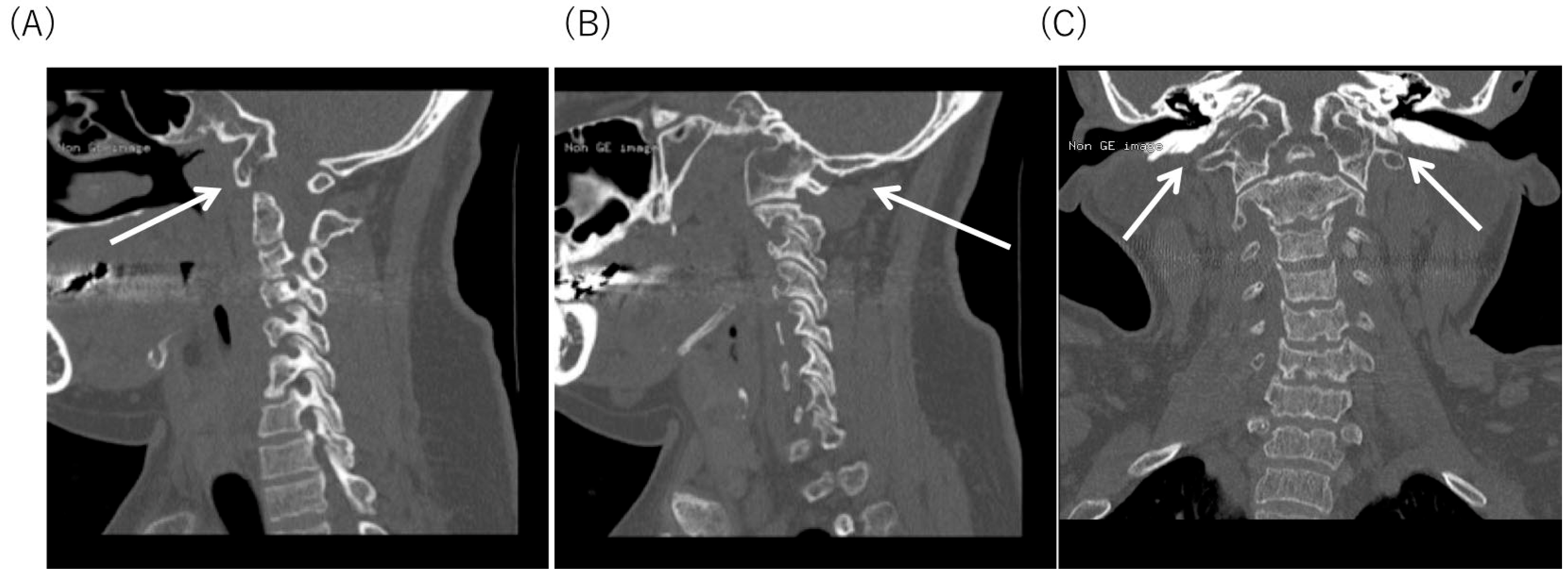
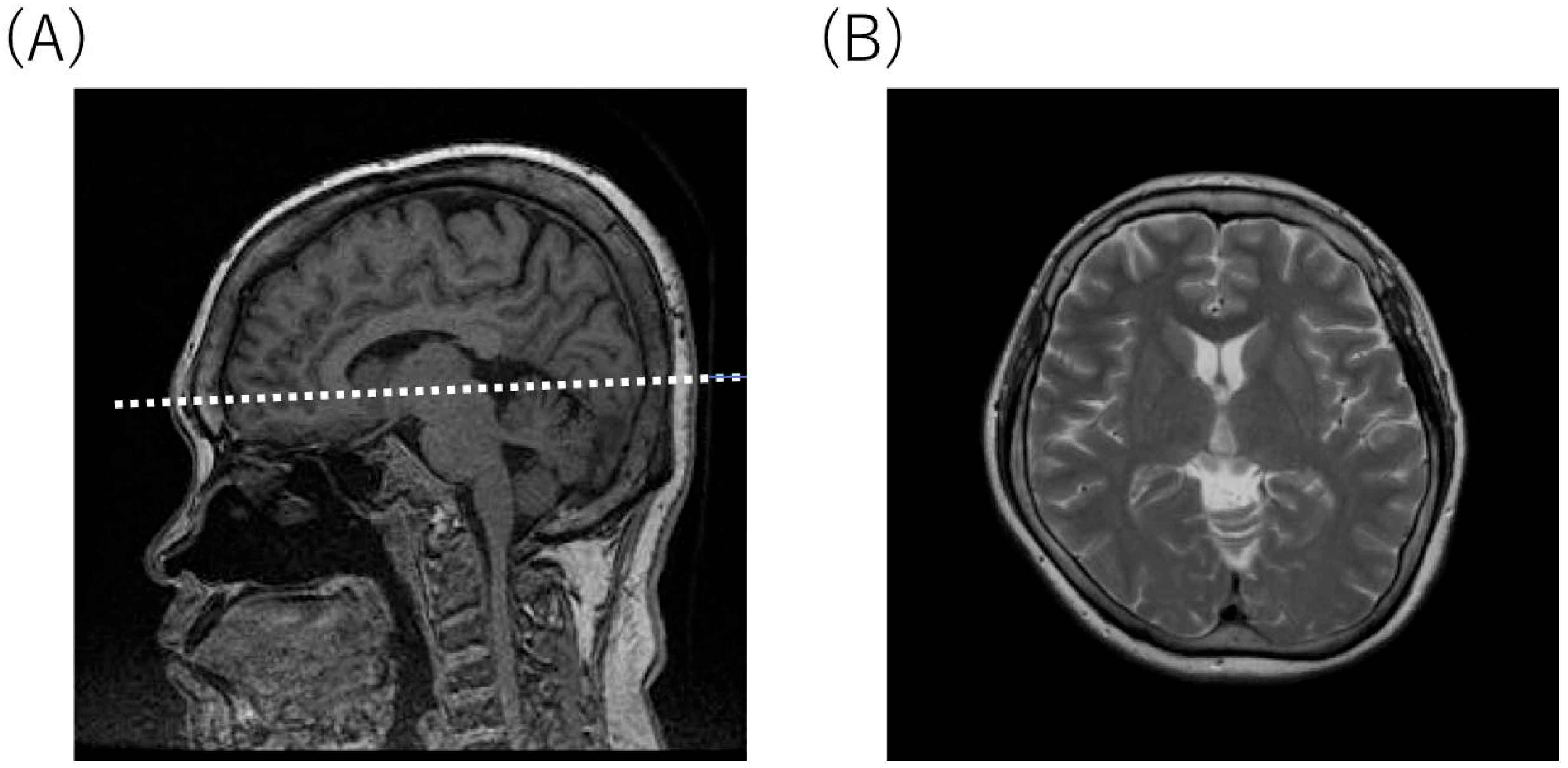
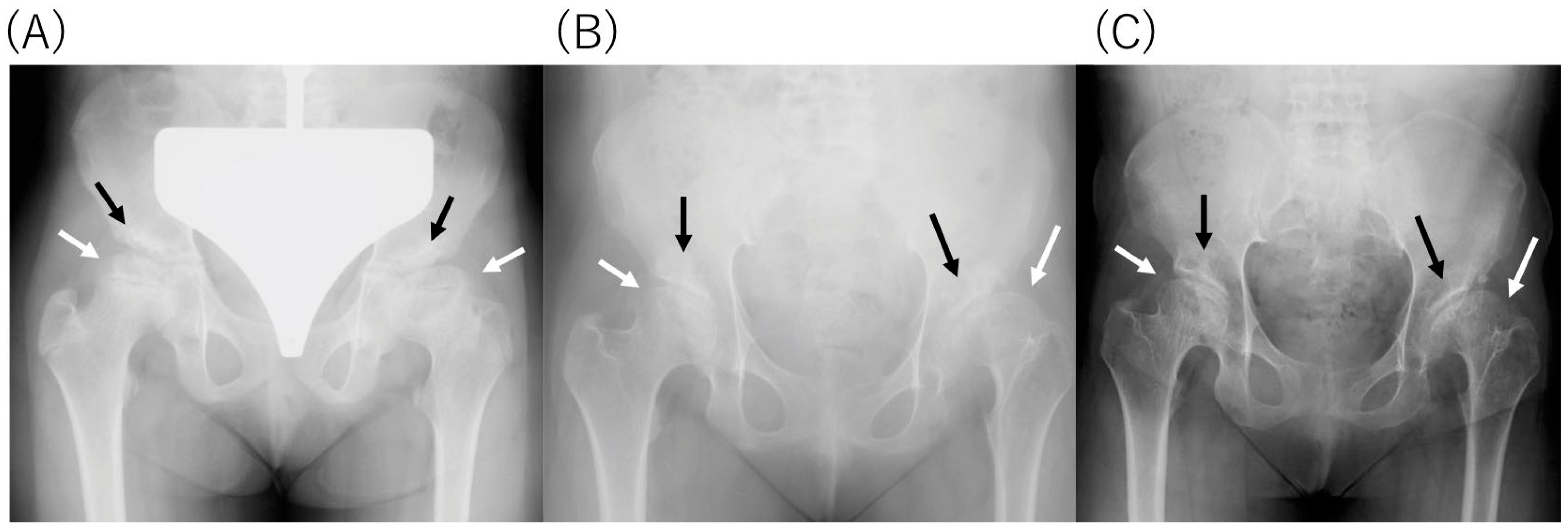
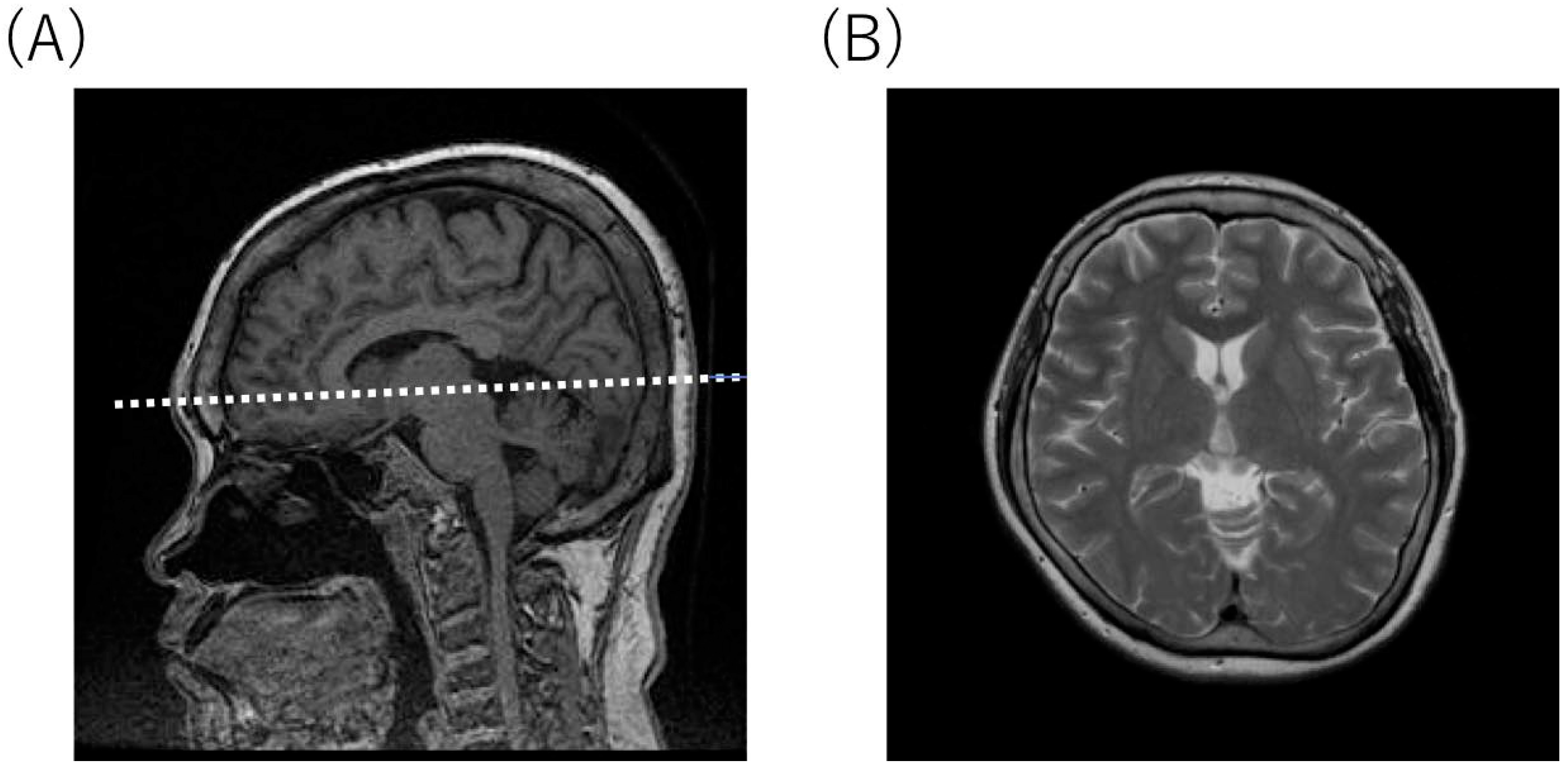
2.4. Radiological Findings
3. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HSCT | Hematopoietic stem cell transplantation |
| MPS VII | Mucopolysaccharidosis type VII (Sly syndrome) |
| ADL | Activity of living |
| GAGs | Glycosaminoglycans |
| DS | Dermatan sulfate |
| HS | Heparan sulfate |
| CS | Chondroitin sulfate |
| LSD | Lysosomal storage disorder |
| CNS | Central nervous system |
| GUS | Glucuronidase |
| rhGUS | Recombinant human glucuronidase |
| ERT | Enzyme replacement therapy |
| MRI | Magnetic resonance imaging |
| IQ | Intelligence quotient |
| SD | Standard deviation |
| MR | Mitral valve regurgitation |
| AR | Aortic valve regurgitation |
References
- Sly, W.S.; Quinton, B.A.; McAlister, W.H.; Rimoin, D.L. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J. Pediatr. 1973, 82, 249–257. [Google Scholar] [CrossRef]
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic & Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.; Graham, B.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Coker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef] [PubMed]
- HGMD. Available online: http://www.hgmd.cf.ac.uk./ac/index.php (accessed on 13 January 2020).
- McCafferty, E.H.; Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs 2019, 33, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.E.; Volpe, L.; Bullaro, J.; Kakkis, E.D.; Sly, W.S. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015, 114, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Boil. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, K.; Sukegawa, K.; Tomatsu, S.; Fukuda, S.; Emura, S.; Kojima, S.; Matsuyama, T.; Sly, W.S.; Kondo, N.; et al. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998, 21, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Fukuda, S.; Sukegawa, K.; Ikedo, Y.; Yamada, S.; Yamada, Y.; Sasaki, T.; Okamoto, H.; Kuwahara, T.; Yamaguchi, S.; et al. Mucopolysaccharidosis type VII: Characterization of mutations and molecular heterogeneity. Am. J. Hum. Genet. 1991, 48, 89–96. [Google Scholar] [PubMed]
- Hess, D.C.; Abe, T.; Hill, W.D.; Studdard, A.M.; Carothers, J.; Masuya, M.; Fleming, P.A.; Drake, C.J.; Ogawa, M. Hematopoietic origin of microglial and perivascular cells in brain. Exp. Neurol. 2004, 186, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Vervoort, R.; Lissens, W.; Hoo, J.J.; Valentino, L.A.; Sly, W.S. β-Glucuronidase P408S, P415L mutations: Evidence that both mutations combine to produce an MPS VII allele in certain Mexican patients. Qual. Life Res. 1996, 98, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Furlan, F.; Rovelli, A.; Rigoldi, M.; Filocamo, M.; Tappino, B.; Friday, D.; Gasperini, S.; Mariani, S.; Izzi, C.; Bondioni, M.P.; et al. A new case report of severe mucopolysaccharidosis type VII: Diagnosis, treatment with haematopoietic cell transplantation and prenatal diagnosis in a second pregnancy. Ital. J. Pediatr. 2018, 44, 128. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Pineda, M.; Coll, M.J.; Gort, L.; Turon, E.; Torrent, M.; Ey, A.; Tobajas, E.; López-Granados, L. Haematopoietic stem cell transplantation for mucopolysaccharidosis type VII: A case report. Pediatr. Transplant. 2018, 22, e13278. [Google Scholar] [CrossRef] [PubMed]
- Dubot, P.; Sabourdy, F.; Plat, G.; Jubert, C.; Cances, C.; Broué, P.; Touati, G.; Levade, T. First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation. Int. J. Mol. Sci. 2019, 20, 5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, M.; Garbade, S.F.; Kolker, S.; Hoffmann, G.F.; Ries, M. Quantitatibe clinical characteristics of 53 patients with MPS VII: A cross-sectional analysis. Genet. Med. 2017, 19, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Harrower, G. Variations in the Region of the Foramen Magnum. J. Anat. 1923, 57, 178–192. [Google Scholar] [PubMed]
- Menezes, A.H. Craniocervical developmental anatomy and its implications. Child’s Nerv. Syst. 2008, 24, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Gholve, H.A.; Hosalkar, H.S.; Ricchetti, E.T.; Pollock, A.N.; Dormans, J.P.; Drummond, D.S. Occipitalzation of the atlas in children. Morphologic classification, associations, and clinical relevance. J. Bone Joint Surg. Am. 2007, 89, 571–578. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| 5 Months before HSCT | 15 Months after HSCT | 17 Years after HSCT | 21 Years after HSCT | |
|---|---|---|---|---|
| age | 12y2m | 13y10m | 29y11m | 34y2m |
| Height (cm) | 141.5 (−1.4 SD) | 141.5 (−2.6 SD) | 144.2 (−2.6 SD) | 143.8 (−2.7 SD) |
| Weight (kg) | 47.0 (+0.5 SD) | 41.3 (−0.9 SD) | 53.6 (+0.1 SD) | 54.3 (+0.2 SD) |
| Body mass index | 23.5 | 20.6 | 25.8 | 26.3 |
| Intelligence quotient | 50 | 47 | 37 | 37 |
| Coarse facies | coarse | coarse | coarse | coarse |
| Corneal clouding | normal | normal | normal | normal |
| Thoracolumbar gibbus | abnormal | abnormal | abnormal | abnormal |
| Bone deformity | abnormal | abnormal | abnormal | abnormal |
| Hepatosplenomegaly | none | none | none | none |
| Snoring | severe | diminished | diminished | diminished |
| Deafness | moderate | moderate | moderate | moderate |
| Heart disease | Moderate MR Moderate AR | Moderate MR Moderate AR | Moderate MR Moderate AR | Moderate MR Moderate AR |
| Walking | Wheelchair | Ambulatory | Ambulatory | Ambulatory |
| before HSCT | 17 Years after HSCT | Control | |
|---|---|---|---|
| β-glucuronidase (nmol/mg protein/h) | ND | 144.5 | 116.4–240.4 |
| Arylsulfatase (nmol/mg protein/h) | NT | 132.4 | 109.0–217.2 |
| β-hexosaminidase (nmol/mg protein/h) | 1427 | NT | 562–882 |
| 5 Months before HSCT | 15 Months after HSCT | 17 Years after HSCT | |
|---|---|---|---|
| Urinary GAGs (mg/g creatinine) | 44.0 | 18.0 | 12.1 |
| Control | 14.2 ± 6.8 | 11.7 ± 5.7 | 10.3 ± 2.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orii, K.; Suzuki, Y.; Tomatsu, S.; Orii, T.; Fukao, T. Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis. Diagnostics 2020, 10, 105. https://doi.org/10.3390/diagnostics10020105
Orii K, Suzuki Y, Tomatsu S, Orii T, Fukao T. Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis. Diagnostics. 2020; 10(2):105. https://doi.org/10.3390/diagnostics10020105
Chicago/Turabian StyleOrii, Kenji, Yasuyuki Suzuki, Shunji Tomatsu, Tadao Orii, and Toshiyuki Fukao. 2020. "Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis" Diagnostics 10, no. 2: 105. https://doi.org/10.3390/diagnostics10020105
APA StyleOrii, K., Suzuki, Y., Tomatsu, S., Orii, T., & Fukao, T. (2020). Long-Term Follow-up Posthematopoietic Stem Cell Transplantation in a Japanese Patient with Type-VII Mucopolysaccharidosis. Diagnostics, 10(2), 105. https://doi.org/10.3390/diagnostics10020105