Unity Makes Strength: A Review on Mutualistic Symbiosis in Representative Insect Clades


Abstract
1. Introduction
1.1. Brief History of Endosymbiosis and Its Importance in the Evolution of Eukaryotes
1.2. Similar Unbalanced Diets but Different Host-Symbiont Associations
2. Aphids as the First Defined Symbiotic Model: Buchnera and Its Multiple Partners
2.1. Historical View of the Aphid-Buchnera Systems
2.2. From Facultative to Co-Obligate Symbionts: The Establishment of Microbial Consortia
2.3. Replacement of Symbionts
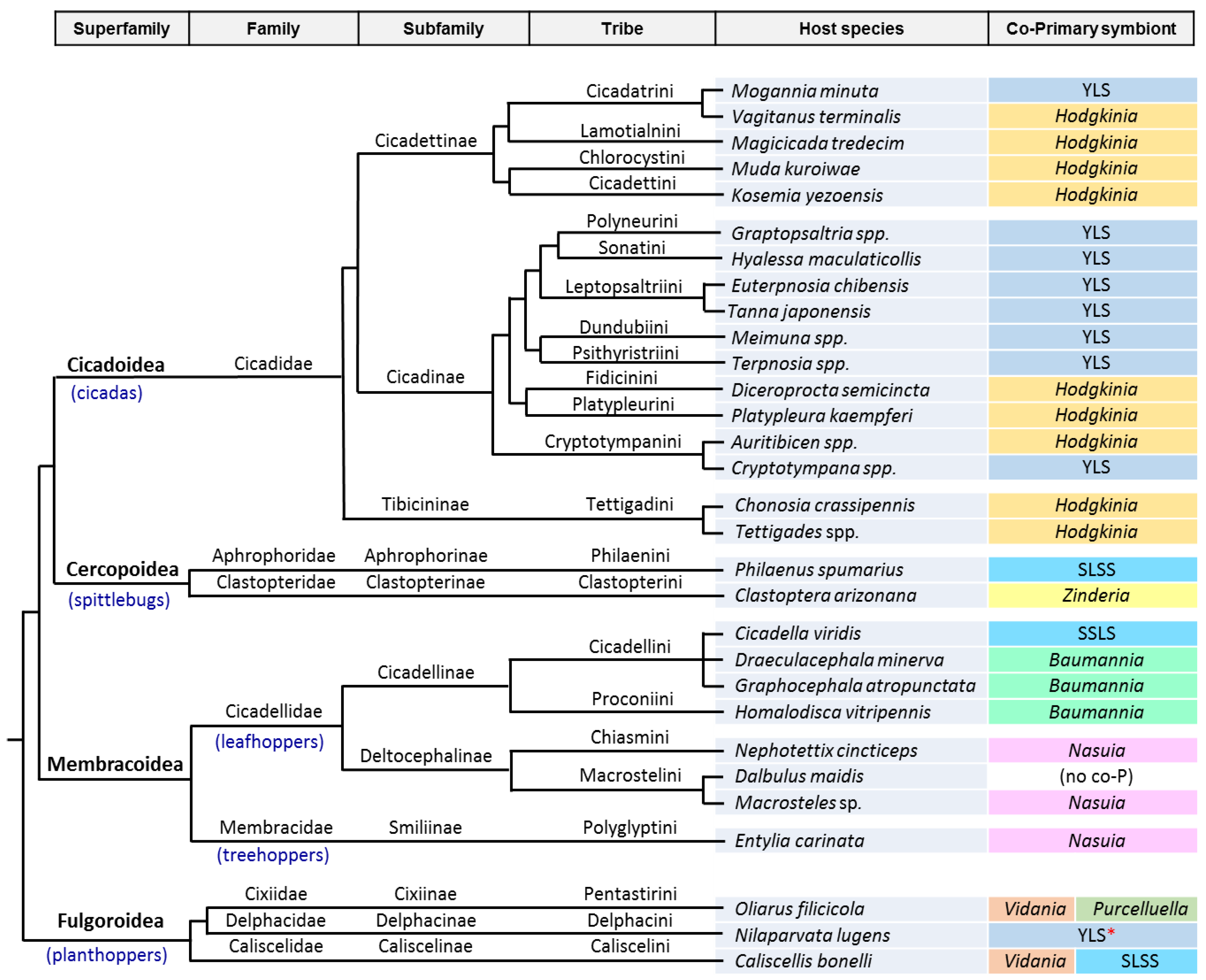
3. The Case of Cicadas and Relatives: Sulcia and Its Multiple Partners
3.1. Consortia and Replacements in the Auchenorrhyncha
3.2. The Peculiarities of the Hodgkinia Genomes and Its Coexisting Interdependent Lineages within a Single Host
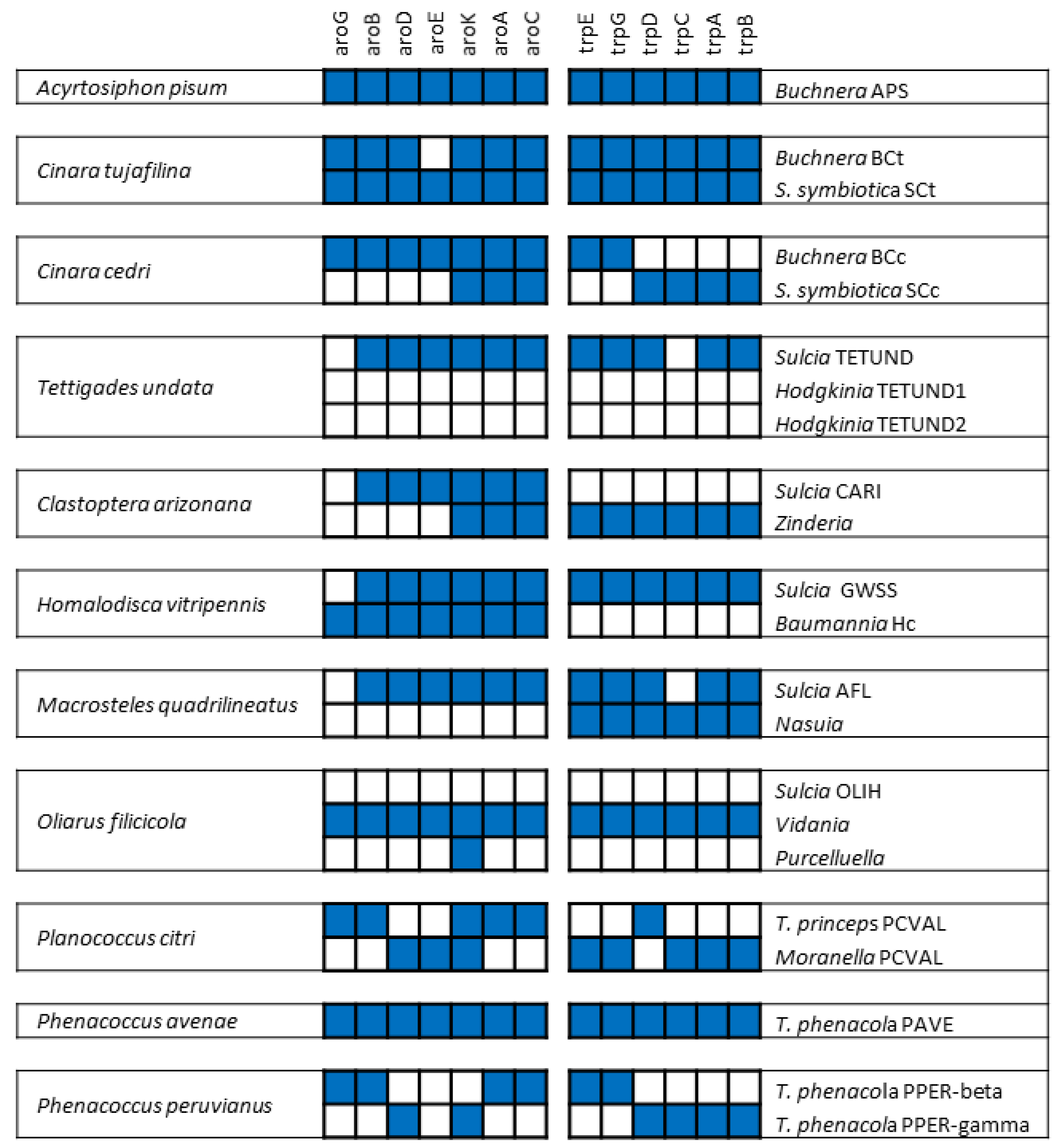
4. The Case of Mealybugs: Not a “Simple” Matryoshka Doll
4.1. A Surprising Nested Endosymbiotic System
4.2. The Fist Chimeric Endosymbiont
5. Concluding Remarks and Future Perspectives in the Field
Funding
Conflicts of Interest
References
- Martin, B.D.; Schwab, E. Current usage of symbiosis and associated terminology. Int. J. Biol. 2012, 5. [Google Scholar] [CrossRef]
- Moya, A.; Pereto, J.; Gil, R.; Latorre, A. Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 2008, 9, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Manzano-Marín, A. Dissecting genome reduction and trait loss in insect endosymbionts. Ann. N. Y. Acad. Sci. 2017, 1389, 52–75. [Google Scholar] [CrossRef] [PubMed]
- Sapp, J. Paul Buchner (1886–1978) and hereditary symbiosis in insects. Int. Microbiol. 2002, 5, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Buchner, P. Endosymbiosis of Animals with Plant Microorganisms; Interscience Publishers: New York, NY, USA, 1965; p. 909. [Google Scholar]
- Douglas, A.E. Mycetocyte symbiosis in insects. Biol. Rev. Camb. Philos. Soc. 1989, 64, 409–434. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef] [PubMed]
- Sudakaran, S.; Kost, C.; Kaltenpoth, M. Symbiont acquisition and replacement as a source of ecological innovation. Trends Microbiol. 2017, 25, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.G.E.; Stackebrandt, E. Taxonomic note: Implementation of the provisional status Candidatus for incompletely described procaryotes. Int. J. Syst. Bacteriol. 1995, 45, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakov, D. The 270 million year history of Auchenorrhyncha (Homoptera). Denisia 2002, 176, 29–36. [Google Scholar]
- Gil, R.; Silva, F.J.; Zientz, E.; Delmotte, F.; Gonzalez-Candelas, F.; Latorre, A.; Rausell, C.; Kamerbeek, J.; Gadau, J.; Holldobler, B.; et al. The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 9388–9393. [Google Scholar] [CrossRef] [PubMed]
- López-Sanchez, M.J.; Neef, A.; Peretó, J.; Patiño-Navarrete, R.; Pignatelli, M.; Latorre, A.; Moya, A. Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 2009, 5, e1000721. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. USA 1996, 93, 2873–2878. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.A.; Baumann, P.; Kinsey, M.G. Buchnera gen. nov. and Buchnera aphidicola sp. nov., a taxon consisting of the mycetocyte-associated, primary endosymbionts of aphids. Int. J. Syst. Bacteriol. 1991, 41, 566–568. [Google Scholar] [CrossRef]
- Dasch, G.A.; Weiss, E.; Chang Sept, K.P.C.N. Endosymbionts of insects. In Bergeys Manual of Systematic Bacteriology; Bergey, D.H., Krieg, N.R., Holt, J.G., Eds.; Williams and Willkins: Baltimore, MD, USA, 1984; Volume 1, pp. 811–833. ISBN 9780683041088. [Google Scholar]
- Douglas, A.E. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu. Rev. Entomol. 1998, 43, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Baumann, L.; Lai, C.Y.; Rouhbakhsh, D.; Moran, N.A.; Clark, M.A. Genetics, physiology, and evolutionary relationships of the genus Buchnera: intracellular symbionts of aphids. Annu. Rev. Microbiol. 1995, 49, 55–94. [Google Scholar] [CrossRef] [PubMed]
- Shigenobu, S.; Watanabe, H.; Hattori, M.; Sakaki, Y.; Ishikawa, H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 2000, 407, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A.; Munson, M.A.; Baumann, P.; Ishikawa, H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc. R. Soc. B Biol. Sci. 1993, 253, 167–171. [Google Scholar] [CrossRef]
- The International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLOS Biol. 2010, 8, e1000313. [Google Scholar] [CrossRef]
- Wilson, A.C.C.; Duncan, R.P. Signatures of host/symbiont genome coevolution in insect nutritional endosymbioses. Proc. Natl. Acad. Sci. USA 2015, 112, 10255–10261. [Google Scholar] [CrossRef] [PubMed]
- Husnik, F.; Nikoh, N.; Koga, R.; Ross, L.; Duncan, R.P.; Fujie, M.; Tanaka, M.; Satoh, N.; Bachtrog, D.; Wilson, A.C.C.; et al. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 2013, 153, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- López-Madrigal, S.; Gil, R. Et tu, brute? Not even intracellular mutualistic symbionts escape horizontal gene transfer. Genes (Basel) 2017, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Nakabachi, A.; Richards, S.; Qu, J.; Murali, S.C.; Gibbs, R.A.; Moran, N.A. Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol. Biol. Evol. 2014, 31, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.B.; Chen, W.; Hasegawa, D.K.; Simmons, A.; Wintermantel, W.M.; Ling, K.S.; Fei, Z.; Liu, S.S.; Douglas, A.E. Metabolic coevolution in the bacterial symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol. Evol. 2015, 7, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Tamas, I.; Klasson, L.; Canback, B.; Naslund, A.K.; Eriksson, A.S.; Wernegreen, J.J.; Sandström, J.P.; Moran, N.A.; Andersson, S.G. 50 million years of genomic stasis in endosymbiotic bacteria. Science 2002, 296, 2376–2379. [Google Scholar] [CrossRef] [PubMed]
- van Ham, R.C.; Kamerbeek, J.; Palacios, C.; Rausell, C.; Abascal, F.; Bastolla, U.; Fernandez, J.M.; Jimenez, L.; Postigo, M.; Silva, F.J.; et al. Reductive genome evolution in Buchnera aphidicola. Proc. Natl. Acad. Sci. USA 2003, 100, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Perez-Brocal, V.; Gil, R.; Ramos, S.; Lamelas, A.; Postigo, M.; Michelena, J.M.; Silva, F.J.; Moya, A.; Latorre, A. A small microbial genome: the end of a long symbiotic relationship? Science 2006, 314, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Lamelas, A.; Gosalbes, M.J.; Moya, A.; Latorre, A. New clues about the evolutionary history of metabolic losses in bacterial endosymbionts, provided by the genome of Buchnera aphidicola from the aphid Cinara tujafilina. Appl. Environ. Microbiol. 2011, 77, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Prieto, M.; Vargas-Chávez, C.; Latorre, A.; Moya, A. SymbioGenomesDB: A database for the integration and access to knowledge on host–symbiont relationships. Database 2015, 2015, bav109. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Hatt, S.; He, K.; Chen, J.; Francis, F.; Wang, Z. Nine facultative endosymbionts in aphids. A review. J. Asia Pac. Entomol. 2017, 20, 794–801. [Google Scholar] [CrossRef]
- Sandström, J.P.; Russell, J.A.; White, J.P.; Moran, N.A. Independent origins and horizontal transfer of bacterial symbionts of aphids. Mol. Ecol. 2001, 10, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Latorre, A.; Sabater-Munoz, B.; Moya, A.; Moran, N.A. Side-stepping secondary symbionts: widespread horizontal transfer across and beyond the Aphidoidea. Mol. Ecol. 2003, 12, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Smith, A.H.; Russell, J.A. Defensive symbiosis in the real world—Advancing ecological studies of heritable, protective bacteria in aphids and beyond. Funct. Ecol. 2014, 28, 341–355. [Google Scholar] [CrossRef]
- Nikoh, N.; Tsuchida, T.; Maeda, T.; Yamaguchi, K.; Shigenobu, S.; Koga, R.; Fukatsu, T. Genomic insight into symbiosis-induced insect color change by a facultative bacterial endosymbiont, “Candidatus Rickettsiella viridis”. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Favret, C. Aphid Species File. Version 5.0/5.0. Available online: http://Aphid.SpeciesFile.org (accessed on 11 December 2018).
- Jousselin, E.; Cœur D’Acier, A.; Vanlerberghe-Masutti, F.; Duron, O. Evolution and diversity of Arsenophonus endosymbionts in aphids. Mol. Ecol. 2013, 22, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Wulff, J.A.; Buckman, K.A.; Wu, K.; Heimpel, G.E.; White, J.A. The endosymbiont Arsenophonus is widespread in soybean aphid, Aphis glycines, but does not provide protection from parasitoids or a fungal pathogen. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Zouari, S.; Ben Halima, M.K.; Reyes-Prieto, M.; Latorre, A.; Gil, R. Natural occurrence of secondary bacterial symbionts in aphids from Tunisia, with a focus on genus Hyalopterus. Environ. Entomol. 2018, 47, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Zytynska, S.E.; Weisser, W.W. The natural occurrence of secondary bacterial symbionts in aphids. Ecol. Entomol. 2016, 41, 13–26. [Google Scholar] [CrossRef]
- Meseguer, A.S.; Manzano-Marín, A.; Coeur d’Acier, A.; Clamens, A.L.; Godefroid, M.; Jousselin, E. Buchnera has changed flatmate but the repeated replacement of co-obligate symbionts is not associated with the ecological expansions of their aphid hosts. Mol. Ecol. 2017, 26, 2363–2378. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.R.; Normark, B.B.; Favret, C.; Moran, N.A. Evolution and diversity of facultative symbionts from the aphid subfamily Lachninae. Appl. Environ. Microbiol. 2009, 75, 5328–5335. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Favret, C.; Jiang, L.; Wang, Z.; Qiao, G. An aphid lineage maintains a bark-feeding niche while switching to and diversifying on conifers. Cladistics 2016, 32, 555–572. [Google Scholar] [CrossRef]
- Lamelas, A.; Perez-Brocal, V.; Gomez-Valero, L.; Gosalbes, M.J.; Moya, A.; Latorre, A. Evolution of the secondary symbiont “Candidatus Serratia symbiotica” in aphid species of the subfamily Lachninae. Appl. Environ. Microbiol. 2008, 74, 4236–4240. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Marín, A.; Szabó, G.; Simon, J.C.; Horn, M.; Latorre, A. Happens in the best of subfamilies: establishment and repeated replacements of co-obligate secondary endosymbionts within Lachninae aphids. Environ. Microbiol. 2017, 19, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.M.; Maiden, M.C.J.; Ferrari, J.; Godfray, H.C.J. Insect life history and the evolution of bacterial mutualism. Ecol. Lett. 2015, 18, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Yu, Y.; Sisneros, N.; Wing, R.A.; Moran, N.A. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc. Natl. Acad. Sci. USA 2009, 106, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.M.; Asplen, M.K.; Desneux, N.; Heimpel, G.E.; Hopper, K.R.; Linnen, C.R.; Oliver, K.M.; Wulff, J.A.; White, J.A. Worldwide populations of the aphid Aphis craccivora are infected with diverse facultative bacterial symbionts. Microb. Ecol. 2014, 67, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Leonardo, T.E.; Cass, B.N.; Hurwitz, B.; Stern, D.; Gibbs, R.A.; Richards, S.; Moran, N.A. Dynamics of genome evolution in facultative symbionts of aphids. Environ. Microbiol. 2010, 12, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Koga, R.; Fujiwara, A.; Fukatsu, T. Phenotypic effect of “Candidatus Rickettsiella viridis,” a Facultative symbiont of the pea aphid (Acyrthosiphon pisum), and Its interaction with a coexisting symbiont. Appl. Environ. Microbiol. 2014, 80, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Koga, R.; Horikawa, M.; Tsunoda, T.; Maoka, T.; Matsumoto, S.; Simon, J.-C.; Fukatsu, T. Symbiotic bacterium modifies aphid body color. Science 2010, 330, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.; Darby, A.C.; Daniell, T.J.; Webster, G.; Van Veen, F.J.; Godfray, H.C.; Prosser, J.I.; Douglas, A.E. Diversity of bacteria associated with natural aphid populations. Appl. Environ. Microbiol. 2003, 69, 7216–7223. [Google Scholar] [CrossRef]
- Burke, G.R.; Moran, N.A. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol. Evol. 2011, 3, 195–208. [Google Scholar] [CrossRef] [PubMed]
- de la Peña, E.; Vandomme, V.; Frago, E. Facultative endosymbionts of aphid populations from coastal dunes of the North Sea. Belgian J. Zool. 2014, 144, 41–50. [Google Scholar]
- Manzano-Marín, A.; Simon, J.C.; Latorre, A. Reinventing the wheel and making it round again: evolutionary convergence in Buchnera-Serratia symbiotic consortia between the distantly related Lachninae aphids Tuberolachnus salignus and Cinara cedri. Genome Biol. Evol. 2016, 8, 1440–1458. [Google Scholar] [CrossRef] [PubMed]
- Lamelas, A.; Gosalbes, M.J.; Manzano-Marín, A.; Peretó, J.; Moya, A.; Latorre, A. Serratia symbiotica from the aphid Cinara cedri: a missing link from facultative to obligate insect endosymbiont. PLoS Genet. 2011, 7, e1002357. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Marín, A.; Latorre, A. Settling down: the genome of Serratia symbiotica from the aphid Cinara tujafilina zooms in on the process of accommodation to a cooperative intracellular life. Genome Biol. Evol. 2014, 6, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xiao, J.H.; Xu, Z.H.; Murphy, R.W.; Huang, D.W. A possibly new Rickettsia-like genus symbiont is found in Chinese wheat pest aphid, Sitobion miscanthi (Hemiptera: Aphididae). J. Invertebr. Pathol. 2011, 106, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xiao, J.H.; Xu, Z.H.; Murphy, R.W.; Huang, D.W. Cellular tropism, population dynamics, host range and taxonomic status of an aphid secondary symbiont, SMLS (Sitobion miscanthi L type symbiont). PLoS ONE 2011, 6, e21944. [Google Scholar] [CrossRef] [PubMed]
- Fukatsu, T.; Tsuchida, T.; Nikoh, N.; Koga, R. Spiroplasma symbiont of the pea aphid, Acyrthosiphon pisum (Insecta: Homoptera). Appl. Environ. Microbiol. 2001, 67, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, Z.R.; Song, Y.; Liu, H.Y.; Li, Z.X. Distribution and diversity of Wolbachia in different populations of the wheat aphid Sitobion miscanthi (Hemiptera: Aphididae) in China. Eur. J. Entomol. 2009, 106, 49–55. [Google Scholar] [CrossRef]
- Augustinos, A.A.; Santos-Garcia, D.; Dionyssopoulou, E.; Moreira, M.; Papapanagiotou, A.; Scarvelakis, M.; Doudoumis, V.; Ramos, S.; Aguiar, A.F.; Borges, P.A.V.; et al. Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS ONE 2011, 6, e28695. [Google Scholar] [CrossRef] [PubMed]
- de Clerck, C.; Tsuchida, T.; Massart, S.; Lepoivre, P.; Francis, F.; Jijakli, M.H. Combination of genomic and proteomic approaches to characterize the symbiotic population of the banana aphid (Hemiptera: Aphididae). Environ. Entomol. 2014, 43, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Jeyaprakash, A.; Hoy, M.A. Long PCR improves Wolbachia DNA amplification: wsp sequences found in 76% of sixty-three arthropod species. Insect Mol. Biol. 2000, 9, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Valero, L.; Soriano-Navarro, M.; Perez-Brocal, V.; Heddi, A.; Moya, A.; Garcia-Verdugo, J.M.; Latorre, A. Coexistence of Wolbachia with Buchnera aphidicola and a secondary symbiont in the aphid Cinara cedri. J. Bacteriol. 2004, 186, 6626–6633. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.-F.; Boudreault, S.; Michaud, D.; Cloutier, C. Impact of environmental stress on aphid clonal resistance to parasitoids: Role of Hamiltonella defensa bacterial symbiosis in association with a new facultative symbiont of the pea aphid. J. Insect Physiol. 2009, 55, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, J.; West, J.A.; Via, S.; Godfray, H.C.J. Population genetic structure and secondary symbionts in host-associated populations of the pea aphid complex. Evolution 2012, 66, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A.; Russell, J.A.; Koga, R.; Fukatsu, T. Evolutionary relationships of three new species of Enterobacteriaceae living as symbionts of aphids and other insects. Appl. Environ. Microbiol. 2005, 71, 3302–3310. [Google Scholar] [CrossRef] [PubMed]
- Gil, R.; Silva, F.J.; Peretó, J.; Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 2004, 68, 518–537. [Google Scholar] [CrossRef] [PubMed]
- Gosalbes, M.J.; Lamelas, A.; Moya, A.; Latorre, A. The striking case of tryptophan provision in the cedar aphid Cinara cedri. J Bacteriol 2008, 190, 6026–6029. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, A.; Nagaya, Y.; Pradel, E.; Ooka, T.; Ogura, Y.; Katsura, K.; Kurokawa, K.; Oshima, K.; Hattori, M.; Parkhill, J.; et al. Genome evolution and plasticity of Serratia marcescens, an important multidrug-resistant nosocomial pathogen. Genome Biol. Evol. 2014, 6, 2096–2110. [Google Scholar] [CrossRef] [PubMed]
- Sabri, A.; Leroy, P.; Haubruge, E.; Hance, T.; Frère, I.; Destain, J.; Thonart, P. Isolation, pure culture and characterization of Serratia symbiotica sp. nov., the R-type of secondary endosymbiont of the black bean aphid Aphis fabae. Int. J. Syst. Evol. Microbiol. 2011, 61, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Foray, V.; Grigorescu, A.S.; Sabri, A.; Haubruge, E.; Lognay, G.; Francis, F.; Fauconnier, M.-L.M.-L.; Hance, T.; Thonart, P. Whole-genome sequence of Serratia symbiotica strain CWBI-2.3T, a free-living symbiont of the black bean aphid Aphis fabae. Genome Announc. 2014, 2, e00767-14. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Marín, A.; Latorre, A. Snapshots of a shrinking partner: genome reduction in Serratia symbiotica. Sci. Rep. 2016, 6, 32590. [Google Scholar] [CrossRef]
- Gil, R.; Latorre, A. Factors behind junk DNA in bacteria. Genes (Basel) 2012, 3, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Fukatsu, T.; Ishikawa, H. Phylogenetic position of yeast-like symbiont of Hamiltonaphis styraci (Homoptera, Aphididae) based on 18S rDNA sequence. Insect Biochem. Mol. Biol. 1996, 26, 383–388. [Google Scholar] [CrossRef]
- Vogel, K.J.; Moran, N.A. Functional and evolutionary analysis of the genome of an obligate fungal symbiont. Genome Biol. Evol. 2013, 5, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.A.; Moran, N.A. Evolutionary loss and replacement of Buchnera, the obligate endosymbiont of aphids. ISME J. 2018, 12, 898–980. [Google Scholar] [CrossRef] [PubMed]
- Fukatsu, T.; Watanabe, K.; Sekiguchi, Y. Specific detection of intracellular symbiotic bacteria of aphids by oligonucleotide-probed in situ hybridization. Appl. Entomol. Zool. 1998, 33, 461–472. [Google Scholar] [CrossRef]
- Pyka-Fosciak, G.; Szklarzewicz, T. Germ cell cluster formation and ovariole structure in viviparous and oviparous generations of the aphid Stomaphis quercus. Int. J. Dev. Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Jousselin, E.; Clamens, A.L.; Galan, M.; Bernard, M.; Maman, S.; Gschloessl, B.; Duport, G.; Meseguer, A.S.; Calevro, F.; Coeur d’acier, A. Assessment of a 16S rRNA amplicon Illumina sequencing procedure for studying the microbiome of a symbiont-rich aphid genus. Mol. Ecol. Resour. 2016, 16, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.M.; Cryan, J.R. Two ancient bacterial endosymbionts have coevolved with the planthoppers (Insecta: Hemiptera: Fulgoroidea). BMC Evol. Biol. 2012, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A.; Tran, P.; Gerardo, N.M. Symbiosis and insect diversification: an ancient symbiont of sap-feeding insects from the bacterial phylum Bacteroidetes. Appl. Environ. Microbiol. 2005, 71, 8802–8810. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Daugherty, S.C.; Van Aken, S.E.; Pai, G.H.; Watkins, K.L.; Khouri, H.; Tallon, L.J.; Zaborsky, J.M.; Dunbar, H.E.; Tran, P.L.; et al. Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol. 2006, 4, e188. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Moriyama, M.; Łukasik, P.; Vanderpool, D.; Tanahashi, M.; Meng, X.-Y.; McCutcheon, J.P.; Fukatsu, T. Recurrent symbiont recruitment from fungal parasites in cicadas. Proc. Natl. Acad. Sci. USA 2018, 115, 5970–5979. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Van Leuven, J.T.; Meister, R.C.; Carey, K.M.; Simon, C.; McCutcheon, J.P. Genome expansion via lineage splitting and genome reduction in the cicada endosymbiont Hodgkinia. Proc. Natl. Acad. Sci. USA 2015, 112, 10192–10199. [Google Scholar] [CrossRef] [PubMed]
- Łukasik, P.; Nazario, K.; Van Leuven, J.T.; Campbell, M.A.; Meyer, M.; Michalik, A.; Pessacq, P.; Simon, C.; Veloso, C.; McCutcheon, J.P. Multiple origins of interdependent endosymbiotic complexes in a genus of cicadas. Proc. Natl. Acad. Sci. USA 2018, 115, 226–235. [Google Scholar] [CrossRef] [PubMed]
- van Leuven, J.T.; Meister, R.C.; Simon, C.; McCutcheon, J.P. Sympatric speciation in a bacterial endosymbiont results in two genomes with the functionality of one. Cell 2014, 158, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Koga, R.; Moran, N.A. Swapping symbionts in spittlebugs: evolutionary replacement of a reduced genome symbiont. ISME J. 2014, 8, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol. Evol. 2010, 2, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Woyke, T.; Tighe, D.; Mavromatis, K.; Clum, A.; Copeland, A.; Schackwitz, W.; Lapidus, A.; Wu, D.; McCutcheon, J.P.; McDonald, B.R.; et al. One bacterial cell, one complete genome. PLoS ONE 2010, 5, e10314. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.M.; McCutcheon, J.P.; MacDonald, B.R.; Romanovicz, D.; Moran, N.A. Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. MBio 2014, 5, e01697-14. [Google Scholar] [CrossRef]
- Chang, H.-H.; Cho, S.-T.; Canale, M.C.; Mugford, S.T.; Lopes, J.R.S.; Hogenhout, S.A.; Kuo, C.-H. Complete genome sequence of “Candidatus Sulcia muelleri” ML, an obligate nutritional symbiont of maize leafhopper ( Dalbulus maidis ). Genome Announc. 2015, 3, e01483. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.M.; Moran, N.A. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 2013, 5, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.M.; Abbà, S.; Kube, M.; Marzachì, C. Complete genome sequences of the obligate symbionts “Candidatus Sulcia muelleri” and “Ca. Nasuia deltocephalinicola” from the pestiferous leafhopper Macrosteles quadripunctulatus (Hemiptera: Cicadellidae). Genome Announc. 2016, 4, e01604-15. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Yang, X.; Poff, K.; Bennett, G. Comparative genomics of the dual-obligate symbionts from the treehopper, entylia carinata (Hemiptera:Membracidae), Provide insight into the origins and evolution of an ancient symbiosis. Genome Biol. Evol. 2017, 9, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.M.; Mao, M. Comparative genomics of a quadripartite symbiosis in a planthopper host reveals the origins and rearranged nutritional responsibilities of anciently diverged bacterial lineages. Environ. Microbiol. 2018, 20, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Watanabe, K.; Kawai, S.; Yukuhiro, F.; Miyoshi, T.; Tomizawa, M.; Koizumi, Y.; Nikoh, N.; Fukatsu, T. Bacteriome-associated endosymbionts of the green rice leafhopper Nephotettix cincticeps (Hemiptera: Cicadellidae). Appl. Entomol. Zool. 2012, 47, 217–225. [Google Scholar] [CrossRef]
- Gonella, E.; Negri, I.; Marzorati, M.; Mandrioli, M.; Sacchi, L.; Pajoro, M.; Crotti, E.; Rizzi, A.; Clementi, E.; Tedeschi, R.; et al. Bacterial endosymbiont localization in Hyalesthes obsoletus, the insect vector of bois noir in Vitis vinifera. Appl. Environ. Microbiol. 2011, 77, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Gil, R. The minimal gene-set machinery. In Encyclopedia of Molecular Cell Biology and Molecular Medicine: Synthetic Biology, 2nd ed.; Meyers, R.A., Ed.; Wiley-VCH Verlag GmbH. & Co.: Weinheim, Germany, 2015; Volume 2, pp. 443–478. ISBN 978-3-527-33482-7. [Google Scholar]
- Takiya, D.M.; Tran, P.L.; Dietrich, C.H.; Moran, N.A. Co-cladogenesis spanning three phyla: leafhoppers (Insecta: Hemiptera: Cicadellidae) and their dual bacterial symbionts. Mol. Ecol. 2006, 15, 4175–4191. [Google Scholar] [CrossRef] [PubMed]
- Brentassi, M.E.; Franco, E.; Balatti, P.; Medina, R.; Bernabei, F.; Marino de Remes Lenicov, A.M. Bacteriomes of the corn leafhopper, Dalbulus maidis (DeLong & Wolcott, 1923) (Insecta, Hemiptera, Cicadellidae: Deltocephalinae) harbor Sulcia symbiont: molecular characterization, ultrastructure, and transovarial transmission. Protoplasma 2017, 254, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Bressan, A.; Arneodo, J.; Simonato, M.; Haines, W.P.; Boudon-Padieu, E. Characterization and evolution of two bacteriome-inhabiting symbionts in cixiid planthoppers (Hemiptera: Fulgoromorpha: Pentastirini). Environ. Microbiol. 2009, 11, 3265–3279. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liang, A.P. A Preliminary Molecular Phylogeny of Planthoppers (Hemiptera: Fulgoroidea) Based on Nuclear and Mitochondrial DNA Sequences. PLoS ONE 2013, 8, e58400. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.C.; Moulds, M.; Hill, K.B.R.; Price, B.W.; Wade, E.J.; Owen, C.L.; Goemans, G.; Marathe, K.; Sarkar, V.; Cooley, J.R.; et al. A molecular phylogeny of the cicadas (Hemiptera: Cicadidae) with a review of tribe and subfamily classification. Zootaxa 2018, 4424, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Zahniser, J.N.; Dietrich, C. A review of the tribes of Deltocephalinae (Hemiptera: Auchenorrhyncha: Cicadellidae). Eur. J. Taxon. 2013, 45, 1–211. [Google Scholar] [CrossRef]
- Tang, M.; Lv, L.; Jing, S.; Zhu, L.; He, G. Bacterial symbionts of the brown planthopper, Nilaparvata lugens (Homoptera: Delphacidae). Appl. Environ. Microbiol. 2010, 76, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Koizumi, Y.; Zhang, Q.; Deng, K. Infection density of Wolbachia and incompatibility level in two planthopper species, Laodelphax striatellus and Sogatella furcifera. Insect Biochem. Mol. Biol. 2001, 31, 727–737. [Google Scholar] [CrossRef]
- Kobiałka, M.; Michalik, A.; Walczak, M.; Szklarzewicz, T. Dual “bacterial-fungal” symbiosis in deltocephalinae leafhoppers (Insecta, Hemiptera, Cicadomorpha: Cicadellidae). Microb. Ecol. 2018, 75, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, C.; Moharramipour, S.; Seyahooei, M.A.; Bagheri, A.; Mehrabadi, M. Identification of yeast and yeast-like symbionts associated with Hishimonus phycitis (Hemiptera: Cicadellidae), the insect vector of lime witches’ broom phytoplasma. J. Crop Prot. 2017, 6, 439–446. [Google Scholar]
- Michalik, A.; Jankowska, W.; Kot, M.; Gołas, A.; Szklarzewicz, T. Symbiosis in the green leafhopper, Cicadella viridis (Hemiptera, Cicadellidae). Association in statu nascendi? Arthropod Struct. Dev. 2014, 43, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Tanahashi, M.; Lin, C.P.; Koga, R.; Fukatsu, T. Fungal and bacterial endosymbionts of eared leafhoppers of the subfamily Ledrinae (Hemiptera: Cicadellidae). Appl. Entomol. Zool. 2016, 51, 465–477. [Google Scholar] [CrossRef]
- Sasaki, T.; Kawamura, M.; Ishikawa, H. Nitrogen recycling in the brown planthopper, Nilaparvata lugens: involvement of yeast-like endosymbionts in uric acid metabolism. J. Insect Physiol. 1996, 42, 125–129. [Google Scholar] [CrossRef]
- Koga, R.; Bennett, G.M.; Cryan, J.R.; Moran, N.A. Evolutionary replacement of obligate symbionts in an ancient and diverse insect lineage. Environ. Microbiol. 2013, 15, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Michalik, A.; Szwedo, J.; Stroiński, A.; Świerczewski, D.; Szklarzewicz, T. Symbiotic cornucopia of the monophagous planthopper Ommatidiotus dissimilis (Fallén, 1806) (Hemiptera: Fulgoromorpha: Caliscelidae). Protoplasma 2018, 255, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Kobiałka, M.; Michalik, A.; Walczak, M.; Junkiert, Ł.; Szklarzewicz, T. Sulcia symbiont of the leafhopper Macrosteles laevis (Ribaut, 1927) (Insecta, Hemiptera, Cicadellidae: Deltocephalinae) harbors Arsenophonus bacteria. Protoplasma 2016, 253, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Matsuura, Y.; Kakizawa, S.; Nikoh, N.; Fukatsua, T. Diversity of bacterial endosymbionts associated with macrosteles leafhoppers vectoring phytopathogenic phytoplasmas. Appl. Environ. Microbiol. 2013, 79, 5013–5022. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, L.; Genchi, M.; Clementi, E.; Bigliardi, E.; Avanzati, A.M.; Pajoro, M.; Negri, I.; Marzorati, M.; Gonella, E.; Alma, A.; et al. Multiple symbiosis in the leafhopper Scaphoideus titanus (Hemiptera: Cicadellidae): details of transovarial transmission of Cardinium sp. and yeast-like endosymbionts. Tissue Cell 2008, 40, 231–242. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; McDonald, B.R.; Moran, N.A. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc. Natl. Acad. Sci. USA 2009, 106, 15394–15399. [Google Scholar] [CrossRef] [PubMed]
- Yamao, F. UGA is read as tryptophan in Mycoplasma capricolum. Proc. Natl. Acad. Sci. USA 1985, 82, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.D.; Landweber, L.F.; Yarus, M. How mitochondria redefine the code. J. Mol. Evol. 2001, 53, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Łukasik, P.; Simon, C.; McCutcheon, J.P. Idiosyncratic genome degradation in a bacterial endosymbiont of periodical cicadas. Curr. Biol. 2017, 27, 3568–3575.e3. [Google Scholar] [CrossRef] [PubMed]
- Thao, M.L.; Gullan, P.J.; Baumann, P. Secondary (gamma-Proteobacteria) endosymbionts infect the primary (beta-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl. Environ. Microbiol. 2002, 68, 3190–3197. [Google Scholar] [CrossRef]
- von Dohlen, C.D.; Kohler, S.; Alsop, S.T.; McManus, W.R. Mealybug beta-proteobacterial endosymbionts contain gamma-proteobacterial symbionts. Nature 2001, 412, 433–436. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Von Dohlen, C.D. An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr. Biol. 2011, 21, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- López-Madrigal, S.; Latorre, A.; Porcar, M.; Moya, A.; Gil, R. Complete genome sequence of “Candidatus Tremblaya princeps” strain PCVAL, an intriguing translational machine below the living-cell status. J. Bacteriol. 2011, 193, 5587–5588. [Google Scholar] [CrossRef] [PubMed]
- López-Madrigal, S.; Latorre, A.; Porcar, M.; Moya, A.; Gil, R. Mealybugs nested endosymbiosis: going into the “matryoshka” system in Planococcus citri in depth. BMC Microbiol. 2013, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- Husnik, F.; McCutcheon, J.P. Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proc. Natl. Acad. Sci. USA 2016, 113, 5416–5424. [Google Scholar] [CrossRef] [PubMed]
- Szabó, G.; Schulz, F.; Toenshoff, E.R.; Volland, J.M.; Finkel, O.M.; Belkin, S.; Horn, M. Convergent patterns in the evolution of mealybug symbioses involving different intrabacterial symbionts. ISME J. 2017, 11, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Baumann, L.; Thao, M.L.; Hess, J.M.; Johnson, M.W.; Baumann, P. The genetic properties of the primary endosymbionts of mealybugs differ from those of other endosymbionts of plant sap-sucking insects. Appl. Environ. Microbiol. 2002, 68, 3198–3205. [Google Scholar] [CrossRef]
- López-Madrigal, S.; Latorre, A.; Moya, A.; Gil, R. The link between independent acquisition of intracellular gamma-endosymbionts and concerted evolution in Tremblaya princeps. Front. Microbiol. 2015, 6, 642. [Google Scholar] [CrossRef] [PubMed]
- López-Madrigal, S.; Beltrà, A.; Resurrección, S.; Soto, A.; Latorre, A.; Moya, A.; Gil, R. Molecular evidence for ongoing complementarity and horizontal gene transfer in endosymbiotic systems of mealybugs. Front. Microbiol. 2014, 5, 449. [Google Scholar] [CrossRef] [PubMed]
- Hardy, N.B.; Gullan, P.J.; Hodgson, C.J. A subfamily-level classification of mealybugs (Hemiptera: Pseudococcidae) based on integrated molecular and morphological data. Syst. Entomol. 2008, 33, 51–71. [Google Scholar] [CrossRef]
- Gruwell, M.E.; Hardy, N.B.; Gullan, P.J.; Dittmar, K. Evolutionary relationships among primary endosymbionts of the mealybug subfamily Phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Appl. Environ. Microbiol. 2010, 76, 7521–7525. [Google Scholar] [CrossRef] [PubMed]
- Gil, R.; Vargas-Chavez, C.; López-Madrigal, S.; Santos-García, D.; Latorre, A.; Moya, A. Tremblaya phenacola PPER: an evolutionary beta-gammaproteobacterium collage. ISME J. 2018, 12, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; DePamphilis, C.W.; et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Ankrah, N.Y.D.; Chouaia, B.; Douglas, A.E. The cost of metabolic interactions in symbioses between insects and bacteria with reduced genomes. MBio 2018, 9, e01433-18. [Google Scholar] [CrossRef] [PubMed]
- Nakabachi, A.; Shigenobu, S.; Sakazume, N.; Shiraki, T.; Hayashizaki, Y.; Carninci, P.; Ishikawa, H.; Kudo, T.; Fukatsu, T. Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc. Natl. Acad. Sci. USA 2005, 102, 5477–5482. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.K.; Moran, N.A. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. USA 2011, 108, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Masson, F.; Copete, S.C.; Schüpfer, F.; Garcia-Arraez, G.; Lemaitre, B. In vitro culture of the insect endosymbiont Spiroplasma poulsonii highlights bacterial genes involved in host- symbiont interaction. MBio 2018, 9, e00024-18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Co-Symbiont: Bacterium (Class) | Aphid Subfamily: Tribe | Host Examples (genus) | Sequenced Genome (Host Strain) | Genome Size (Mb) | GC (%) | CDS | Refs. |
|---|---|---|---|---|---|---|---|
| Arsenophonus (γ-proteobacteria) | Aphidinae: Aphidini | Aphis, Hyalopterus, Melanaphis | [37,38,39,40] | ||||
| Acinetobacter | Lachninae: Eulachnini * | Cinara | [41] | ||||
| (γ-proteobacteria) | Lachninae: Stomaphidini * | Stomaphis | [42,43] | ||||
| Erdwardsiella | Lachninae: Eulachnini * | Cinara | [41] | ||||
| (γ-proteobacteria) | |||||||
| Erwinia-like symbiont | Aphidinae: Aphidini | Hyalopterus | [39] | ||||
| (γ-proteobacteria) | Lachninae: Eulachnini * | Cinara | [41] | ||||
| GLSS (γ-proteobacteria) | Lachninae: Stomaphidini * | Stomaphis | [44,45] | ||||
| Hamiltonella defensa (γ-proteobacteria) | Aphidinae: Macrosiphi | Acyrthosiphon, Myzus Macrosiphon, Sitobion | A. pisum 5AT | 2.17 | 40.5 | 2,158 | [40,46,47] |
| Aphidinae: Aphidini | Aphis, Hyalopterus | [39,40,48] | |||||
| Lachninae: Tuberolachnini * | Nippolachnus | [42] | |||||
| Lachninae: Eulachnini * | Eulachnus, Cinara | [41,42,44] | |||||
| Regiella insecticola (γ-proteobacteria) | Aphidinae: Macrosiphini | Acyrthosiphon, Myzus Macrosiphum, Sitobion | A. pisum LSR1 | 2.07 | 42.5 | 1,769 | [40,46,49] |
| Aphidinae: Aphidini | Aphis | [40,46,48] | |||||
| Lachninae: Eulachnini * | Cinara | [41] | |||||
| Ricketsiella viridis (γ-proteobacteria) | Aphidinae: Macrosiphini | Acyrthosiphon | A. pisum RA04 | 1.6 | 39 | 1,378 | [35,50] |
| Rickettsia (α-proteobacteria) | Aphidinae: Macrosiphini | Acyrthosiphon, Uroleucon | [40,48,51] | ||||
| Aphidinae: Aphidini | Aphis | [40,48,51] | |||||
| Lachninae: Eulachnini * | Cinara | [41] | |||||
| Serratia symbiotica (γ-proteobacteria) | Aphidinae: Macrosiphini | Acyrthosiphon, Myzus, Macrosiphum, Sitobion, Uroleucon | A. pisum TUC | 2.57 | 52.1 | 2,098 | [40,46,52,53] |
| Aphidinae: Aphidini | Aphis, Rhopalosiphum, Hyalopterus | A. fabae CWBI-2.3 | 3.58 | 52.1 | 3,398 | [39,40,46,48,54] | |
| Lachninae: Lachnini * | Pterochloroides, Lachnus | [45] | |||||
| Lachninae: Stomaphidini * | Stomaphis | [42,43] | |||||
| Lachninae: Tramini * | Trama | [45] | |||||
| Lachninae: Tuberolachnini * | Tuberolachnus | T. salignus STs | 0.65 | 20.9 | 495 | [55] | |
| Lachninae: Eulachnini * | Cinara | C. cedri SCc | 1.76 | 29.2 | 677 | [41,56] | |
| C. tujafilina SCt-VCL | 2.49 | 52.2 | 1,601 | [57] | |||
| SLSS (γ-proteobacteria) | Lachninae: Tuberolachnini * | Nippolachnus | [42] | ||||
| Lachninae: Eulachnini * | Eulachnus, Cinara | [33,41,42,45] | |||||
| SMLSS (γ-proteobacteria) | Aphidinae: Macrosiphini | Acyrthosiphon, Sitobion | [58] | ||||
| Aphidinae: Aphidini | Rhopalosiphum | [59] | |||||
| Lachninae: Stomaphidini * | Stomaphis | [42] | |||||
| Spiroplasma (Mollicutes) | Aphidinae: Macrosiphini | Acyrthosiphon | [60] | ||||
| Aphidinae: Aphidini | Aphis | [40,48] | |||||
| Wolbachia (α-proteobacteria) | Aphidinae: Macrosiphini | Sitobion, Macrosiphum, Aulacorthum, Pentalonia | [61,62,63] | ||||
| Aphidinae: Aphidini | Aphis, Aphis (Toxoptera) | [64] | |||||
| Chaitophorinae: Siphini | Sipha | [62] | |||||
| Eriosomatinae: Fordini | Baizongia | [62] | |||||
| Neophyllaphidinae | Neophyllaphis | [62] | |||||
| Lachninae: Stomaphidini | Stomaphis | [43] | |||||
| Lachninae: Eulachnini | Cinara | [62,65] | |||||
| Fukatsia symbiotica (X-type) | Aphidinae: Macrosiphini | Acyrthosiphon | [66,67] | ||||
| (γ-proteobacteria) | Lachninae: Lachnini * | Maculolachnus | [42,44,45] | ||||
| Lachninae: Eulachnini * | Cinara | [41,44,45] |
| Insect host | P-endosymbiont | Genome size (kb) | GC (%) | CDS | Ref. |
|---|---|---|---|---|---|
| Mogannia minuta | Sulcia SMMOGMIN | 243,55 | 22.30 | 220 | [85] |
| Vagitanus terminalis | Sulcia SMVAGTER | 245,30 | 22.70 | 227 | [86] |
| Hodgkinia HCVAGTER | 353 | 30.0 | nd | ||
| Magicicada tredecim | Sulcia SMMAGTRE | 268,54 | 22.70 | 224 | [85] |
| Hodgkinia HCMAGTRE | 1571 | 29.1 | 252 | ||
| Muda kuroiwae | Sulcia SMMUDKUR | 266,95 | 22.60 | 248 | [85] |
| Hodgkinia HCMUDKUR | 909 | 27.1 | nd | ||
| Kosemia yezoensis | Sulcia SMKOSYEZ | 244,20 | 22.80 | 221 | [85] |
| Hodgkinia HCKOSYEZ | 1863 | 30.0 | nd | ||
| Graptopsaltria bimaculata | Sulcia SMGRABIM | 271,62 | 22.60 | 253 | [85] |
| Graptopsaltria nigrofuscata | Sulcia SMGRANIG | 271,57 | 22.60 | 253 | [85] |
| Hyalessa maculaticollis | Sulcia SMHYAMAC | 272,58 | 22.50 | 249 | [85] |
| Euterpnosia chibensis | Sulcia SMEUTCHI | 273,71 | 22.60 | 257 | [85] |
| Tanna japonensis | Sulcia SMTANJAP | 278,30 | 22.50 | 256 | [85] |
| Meimuna iwasakii | Sulcia SMMEIIWA | 272,32 | 22.60 | 253 | [85] |
| Meimuna kuroiwae | Sulcia SMMEIKUR | 271,07 | 22.60 | 253 | [85] |
| Meimuna opalifera | Sulcia SMMEIOPA | 271,56 | 22.60 | 252 | [85] |
| Meimuna oshimensis | Sulcia SMMEIOSH | 270,60 | 22.60 | 253 | [85] |
| Terpnosia nigricosta | Sulcia SMTERNIG | 273,63 | 22.70 | 256 | [85] |
| Terpnosia vacua | Sulcia SMTERVAC | 273,80 | 22.60 | 256 | [85] |
| Diceroprocta semicincta | Sulcia SMDSEM | 276,98 | 22.60 | 242 | [85] |
| Hodgkinia Dsem | 144 | 58.4 | 169 | ||
| Platypleura kaempferi | Sulcia SMPLAKAE | 268,04 | 22.50 | 248 | [85] |
| Hodgkinia HCPLAKAE | 349 | 47.9 | nd | ||
| Auritibicen bihamatus | Sulcia SMAURBIH | 276,77 | 22.80 | 256 | [85] |
| Hodgkinia HCAURBIH | 474 | 45.0 | nd | ||
| Auritibicen japonicus | Sulcia SMAURJAP | 278,18 | 22.80 | 259 | [85] |
| Hodgkinia HCAURJAP | 438 | 45.8 | nd | ||
| Cryptotympana atrata | Sulcia SMCRYATR | 273,23 | 22.70 | 252 | [85] |
| Cryptotympana facialis | Sulcia SMCRYFAC | 270,78 | 22.70 | 238 | [85] |
| Chonosia crassipennis | Hodgkinia CHOCRA | 149 | 38.7 | 170 | [87] |
| Tettigades limbata | Hodgkinia TETLIM1 | 145 | 45.4 | 130 | [87] |
| TETLIM2 | 131 | 45.1 | 73 | ||
| TETLIM3 | 128 | 47.8 | 50 | ||
| TETLIM4 | 126 | 47.2 | 47 | ||
| TETLIM5 | 122 | 45.8 | 39 | ||
| Tettigades auropilosa | Hodgkinia TETAUR | 126 | 46.3 | 117 | [87] |
| Tettigades chilensis | Hodgkinia TETCHI1a | 130 | 44.9 | 163 | [87] |
| TETCHI1b | 129 | 44.8 | 156 | ||
| TETCHI2 | 117 | 45.8 | 115 | ||
| TETCHI4 | 106 | 45.6 | 114 | ||
| Tettigades ulnaria | Hodgkinia TETULN | 150 | 46.4 | 170 | [87] |
| Tettigades undata | Sulcia TETUND | 270,03 | 23.00 | 247 | [88] |
| Hodgkinia TETUND1 | 134 | 46.8 | 121 | ||
| TETUND2 | 141 | 46.2 | 140 | ||
| Tettigades undata | Hodgkinia TETLON1 | 133 | 47.7 | 104 | [87] |
| TETLON2a | 140 | 46.5 | 128 | ||
| TETLON2b | 137 | 46.7 | 109 | ||
| Philaenus spumarius | Sulcia PSPU | 285,35 | 20.90 | 257 | [89] |
| Clastoptera arizonana | Sulcia CARI | 276,51 | 21.10 | 246 | [90] |
| Zinderia | 209 | 13.5 | 206 | ||
| Draeculacephala minerva | Sulcia DMIN | 243,93 | 22.50 | 226 | [91] |
| Baumannia | 636 + 3.5 | 31.6 | 517 + 5 | ||
| Graphocephala atropunctata | Sulcia BGSS | 244,62 | 22.50 | 227 | [92] |
| Baumannia | 759 | 39 | 669 | ||
| Homalodisca vitripennis | Sulcia GWSS | 245,53 | 22.40 | 227 | [84] |
| Baumannia | 686 | 33.2 | 595 | ||
| Nephotettix cincticeps | Sulcia NC | 192,24 | 23.70 | 176 | U |
| Dalbulus maidis | Sulcia ML | 190,41 | 24.10 | 187 | [93] |
| Macrosteles quadrilineatus | Sulcia ALF | 190,73 | 24.00 | 188 | [94] |
| Nasuia | 112 | 17.1 | 138 | ||
| Macrosteles quadripunctulatus | Sulcia PUNC | 190,66 | 24.00 | 181 | [95] |
| Nasuia | 112 | 16,6 | 138 | ||
| Entylia carinata | Sulcia ENCA | 218,03 | 23.00 | 198 | [96] |
| Nasuia | 144.6 | 15.2 | 159 | ||
| Oliarus filicicola | Sulcia OLIH | 156,58 | 24.90 | 152 | [97] |
| Vidania | 136 | 18.2 | 154 | ||
| Purcelluella | 480 | 21.2 | 431 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, R.; Latorre, A. Unity Makes Strength: A Review on Mutualistic Symbiosis in Representative Insect Clades. Life 2019, 9, 21. https://doi.org/10.3390/life9010021
Gil R, Latorre A. Unity Makes Strength: A Review on Mutualistic Symbiosis in Representative Insect Clades. Life. 2019; 9(1):21. https://doi.org/10.3390/life9010021
Chicago/Turabian StyleGil, Rosario, and Amparo Latorre. 2019. "Unity Makes Strength: A Review on Mutualistic Symbiosis in Representative Insect Clades" Life 9, no. 1: 21. https://doi.org/10.3390/life9010021
APA StyleGil, R., & Latorre, A. (2019). Unity Makes Strength: A Review on Mutualistic Symbiosis in Representative Insect Clades. Life, 9(1), 21. https://doi.org/10.3390/life9010021