How do Nucleotides Adsorb Onto Clays?

 , , ,
, , ,
Abstract
1. Introduction
2. Study of Adsorption in Aqueous Systems
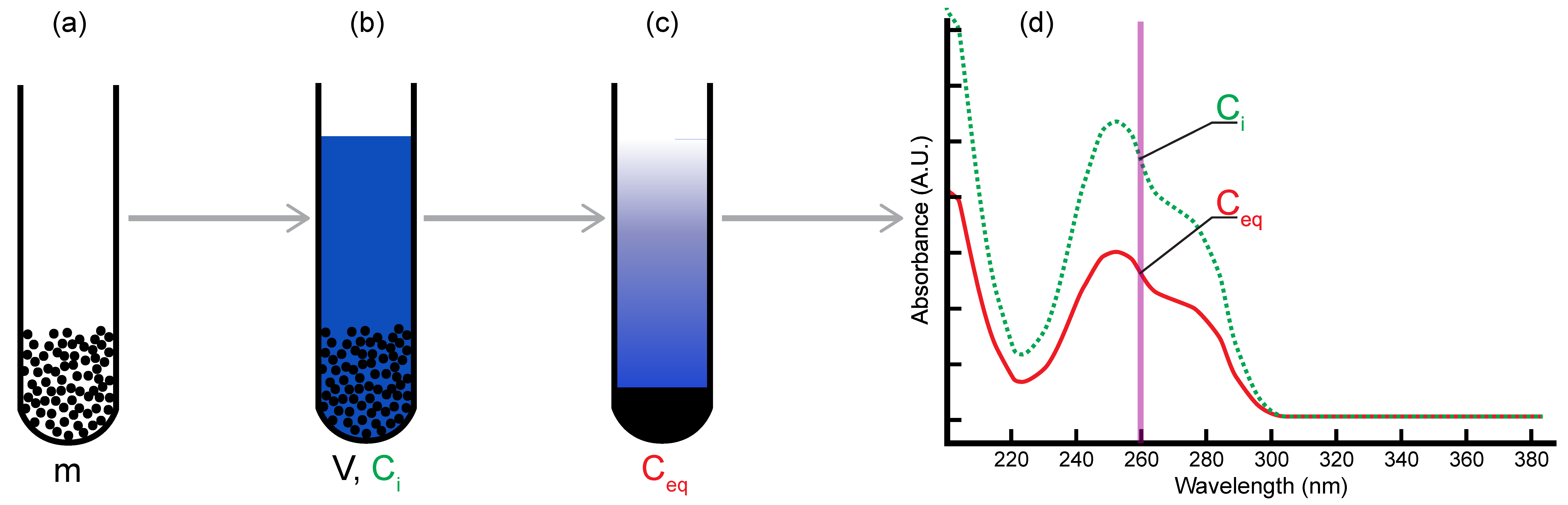
2.1. Batch Adsorption Method
2.2. Limits of Macroscopic Studies
3. Results of Batch Adsorption Studies
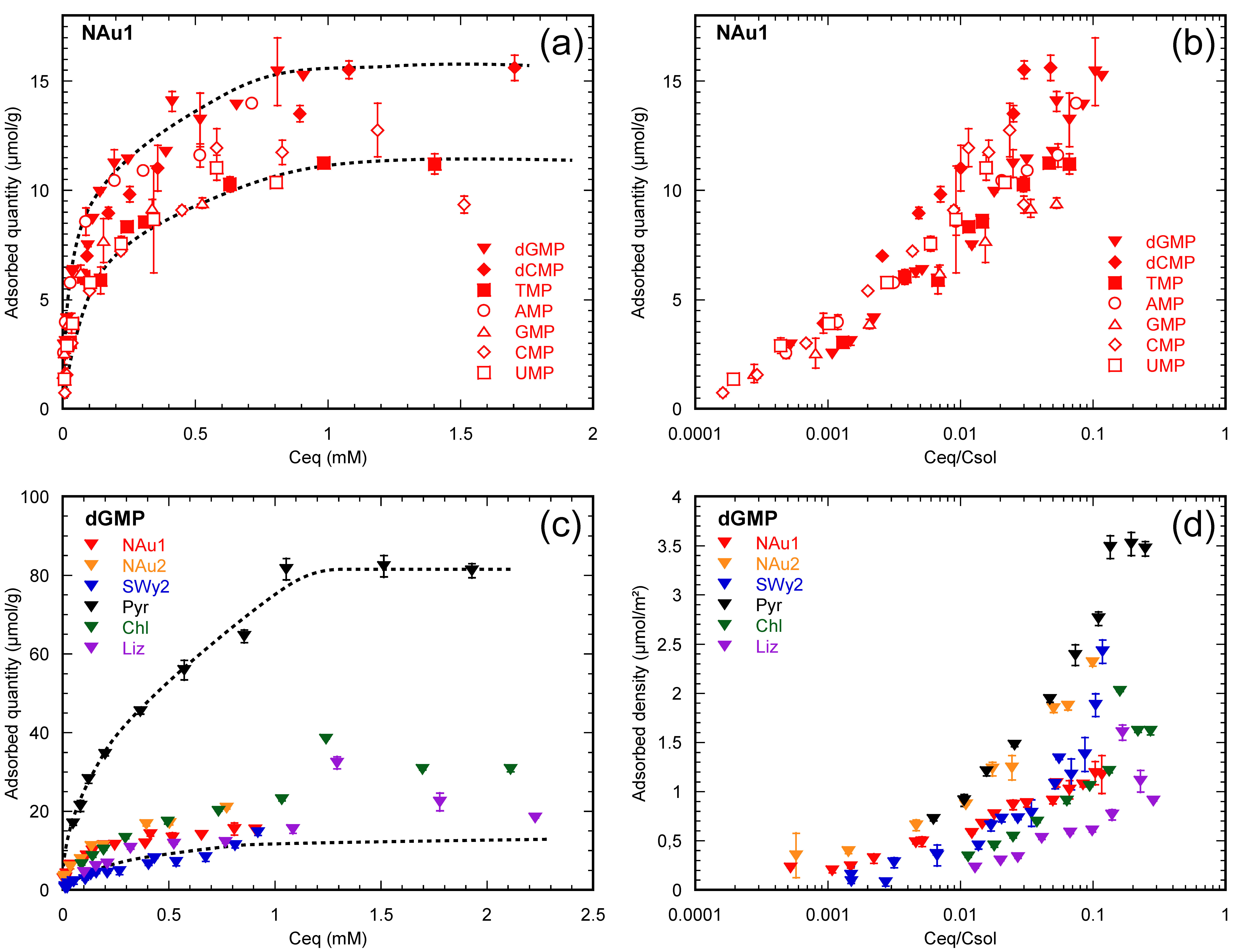
3.1. Half a Century of Data
3.2. How Do Nucleotides Adsorb onto Clay Minerals?
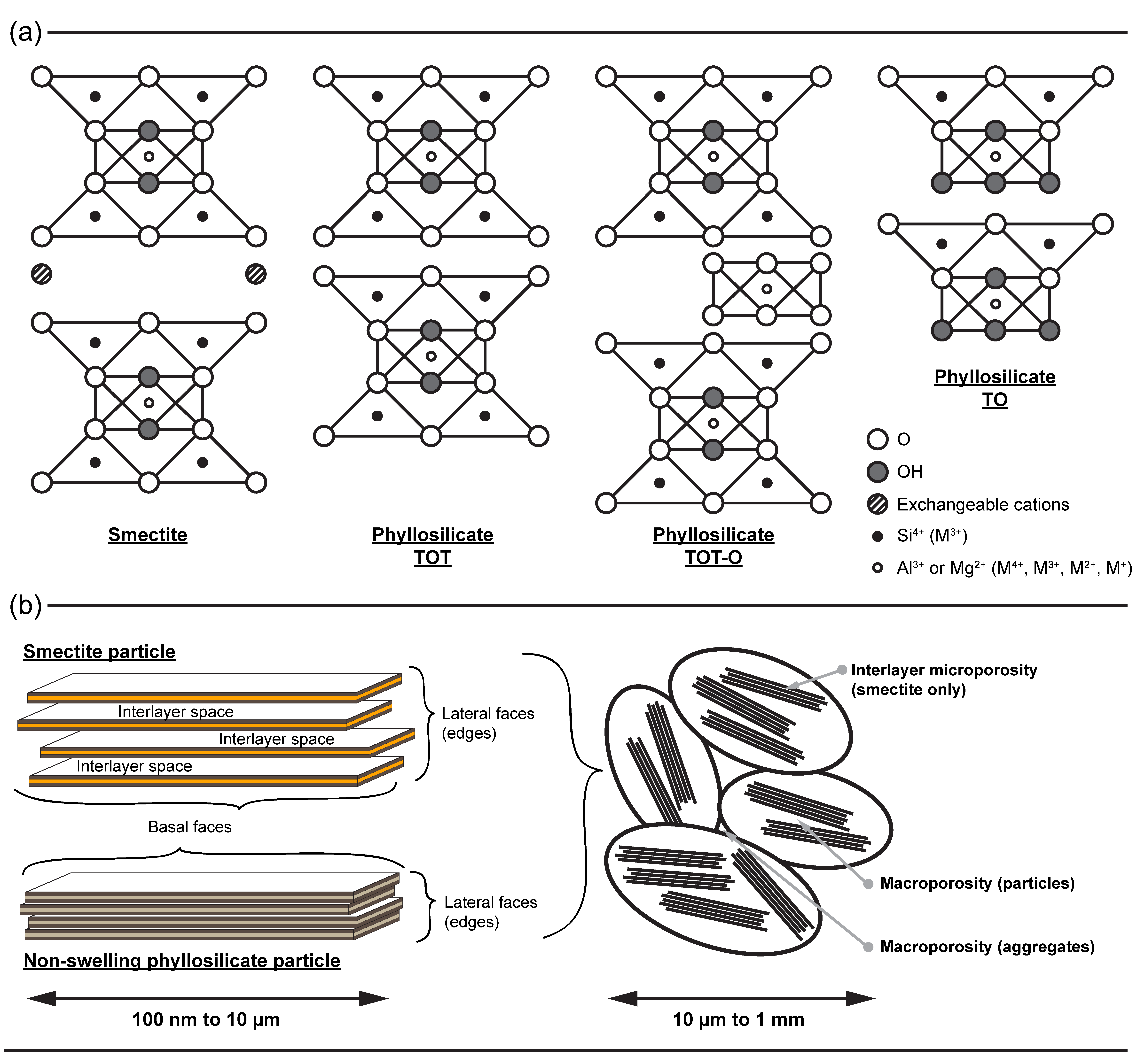
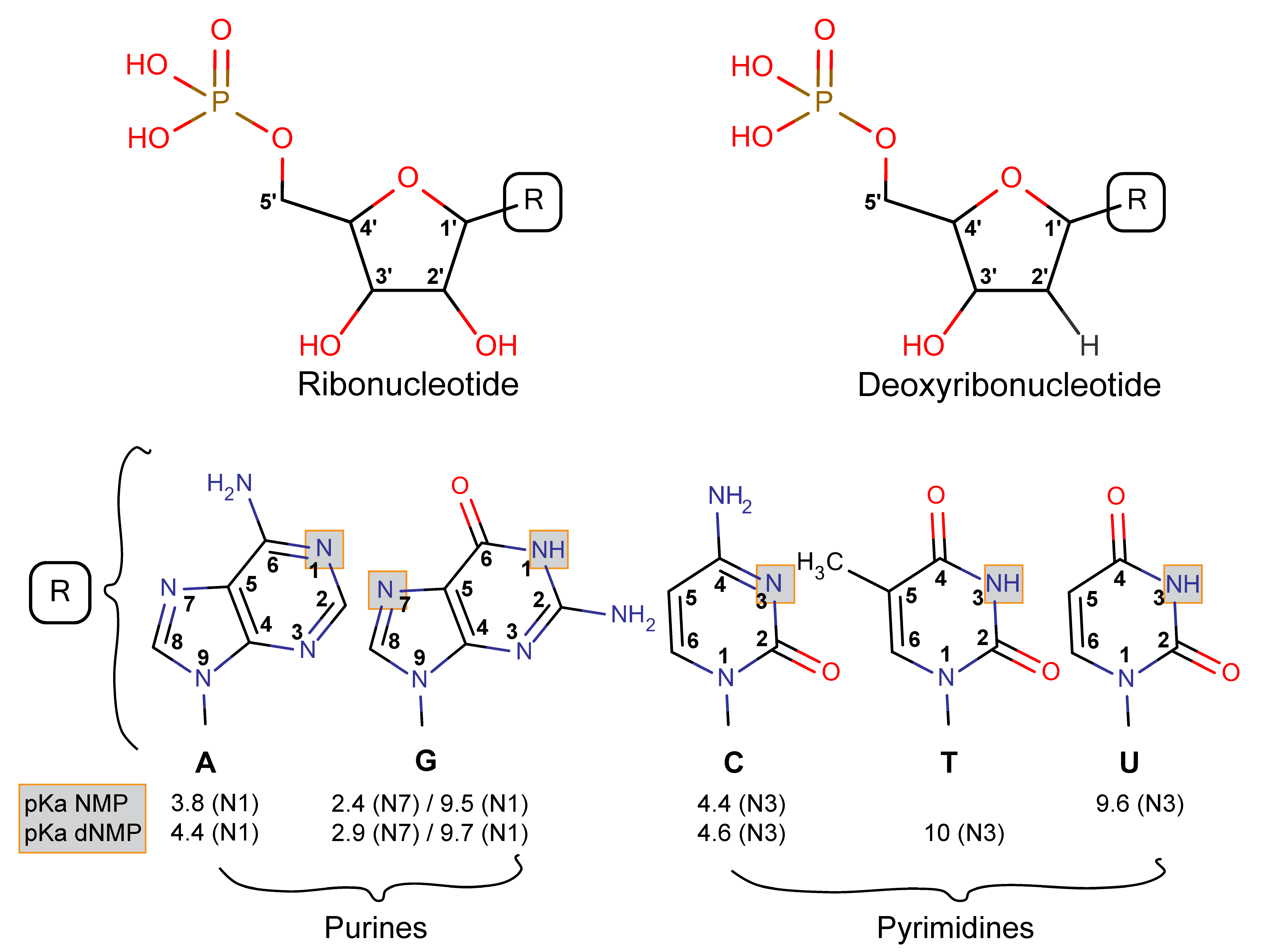
3.2.1. Nucleotides Are Homologous Molecules
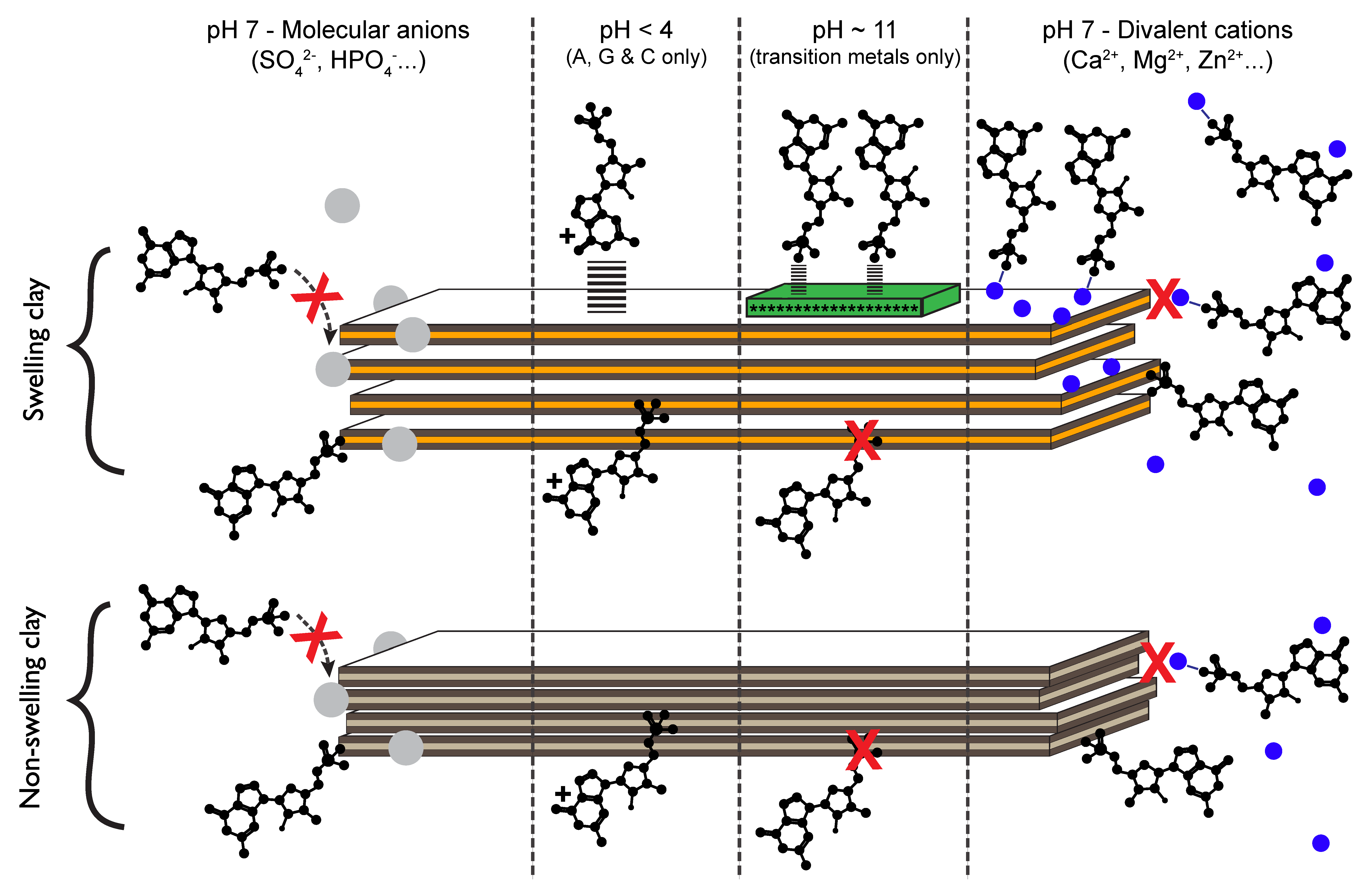
3.2.2. Nucleotides Adsorb on Lateral Faces
3.2.3. The Effect of Extreme Conditions on Adsorption
3.2.4. Ocean Chemistry Controls Adsorption
3.2.5. Implications
4. Recent Developments and New Techniques
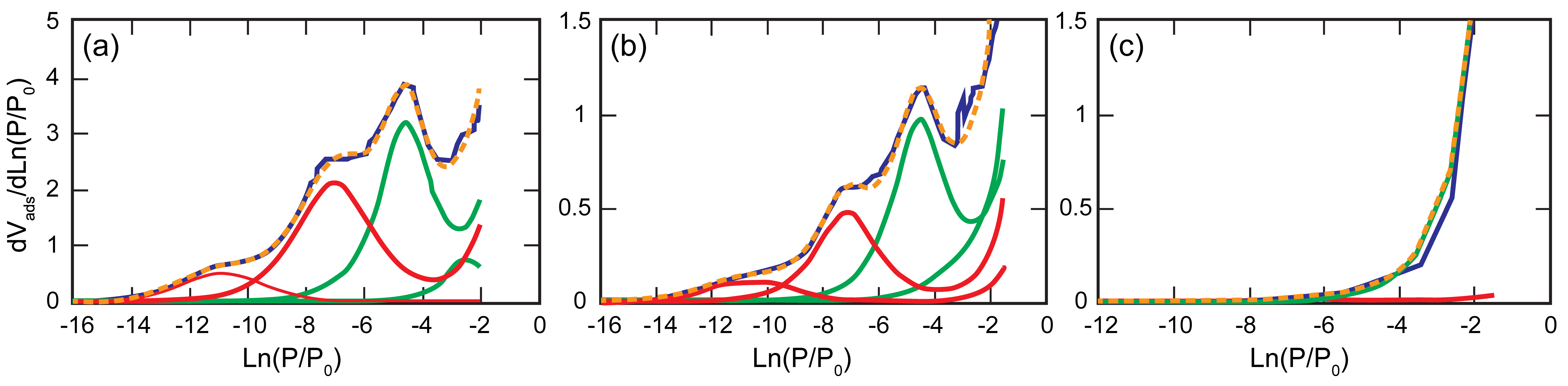
4.1. Low Pressure Gas Adsorption
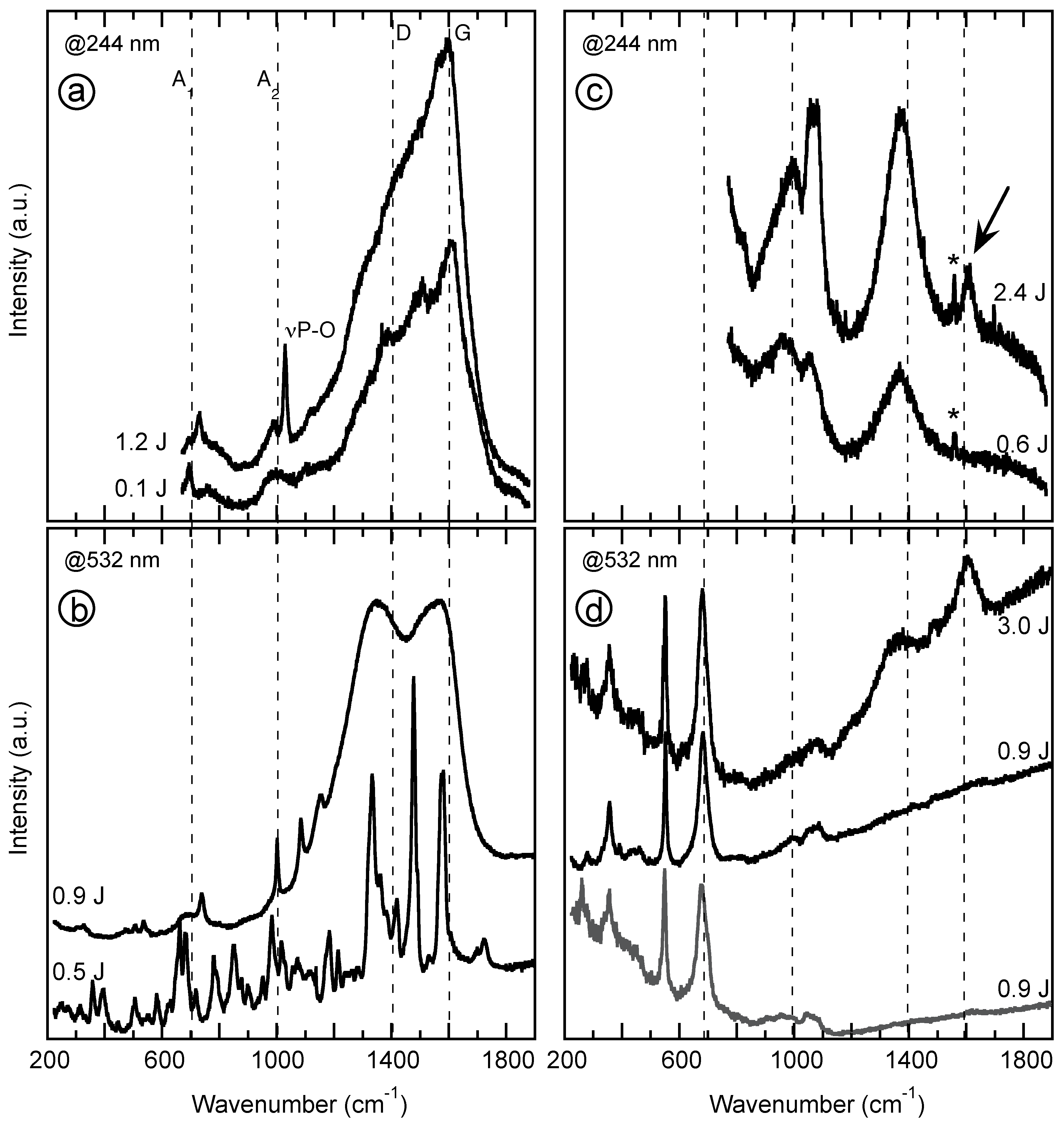
4.2. Vibrational Spectroscopy
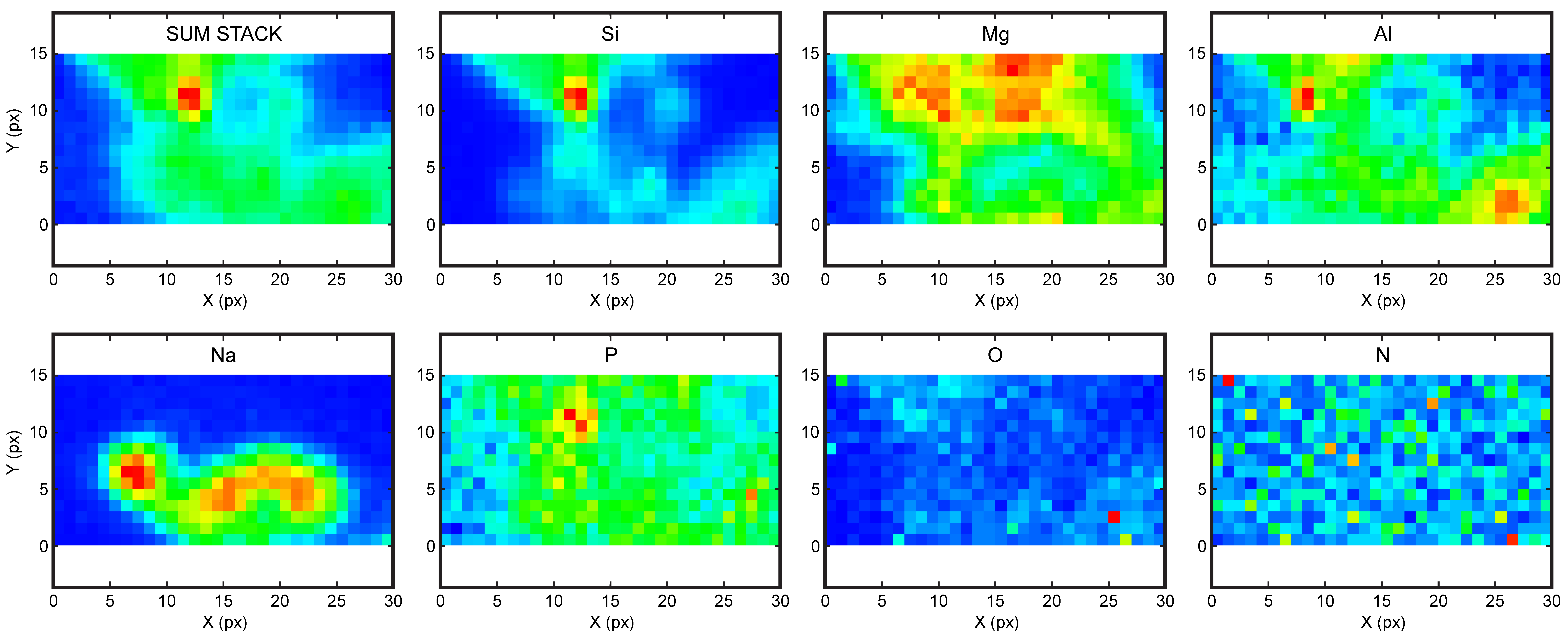
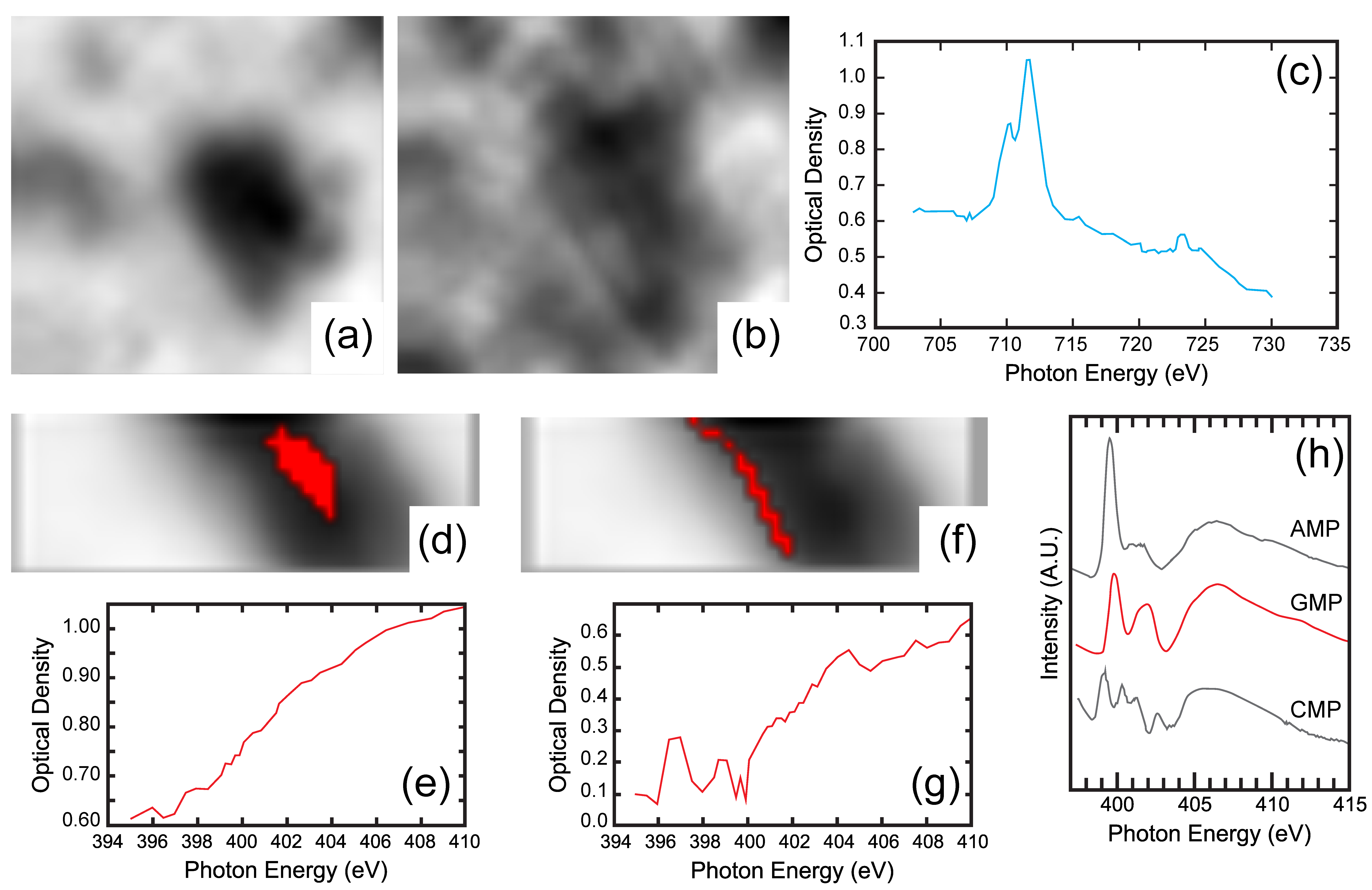
4.3. High Resolution X-ray Absorption and Fluoresence
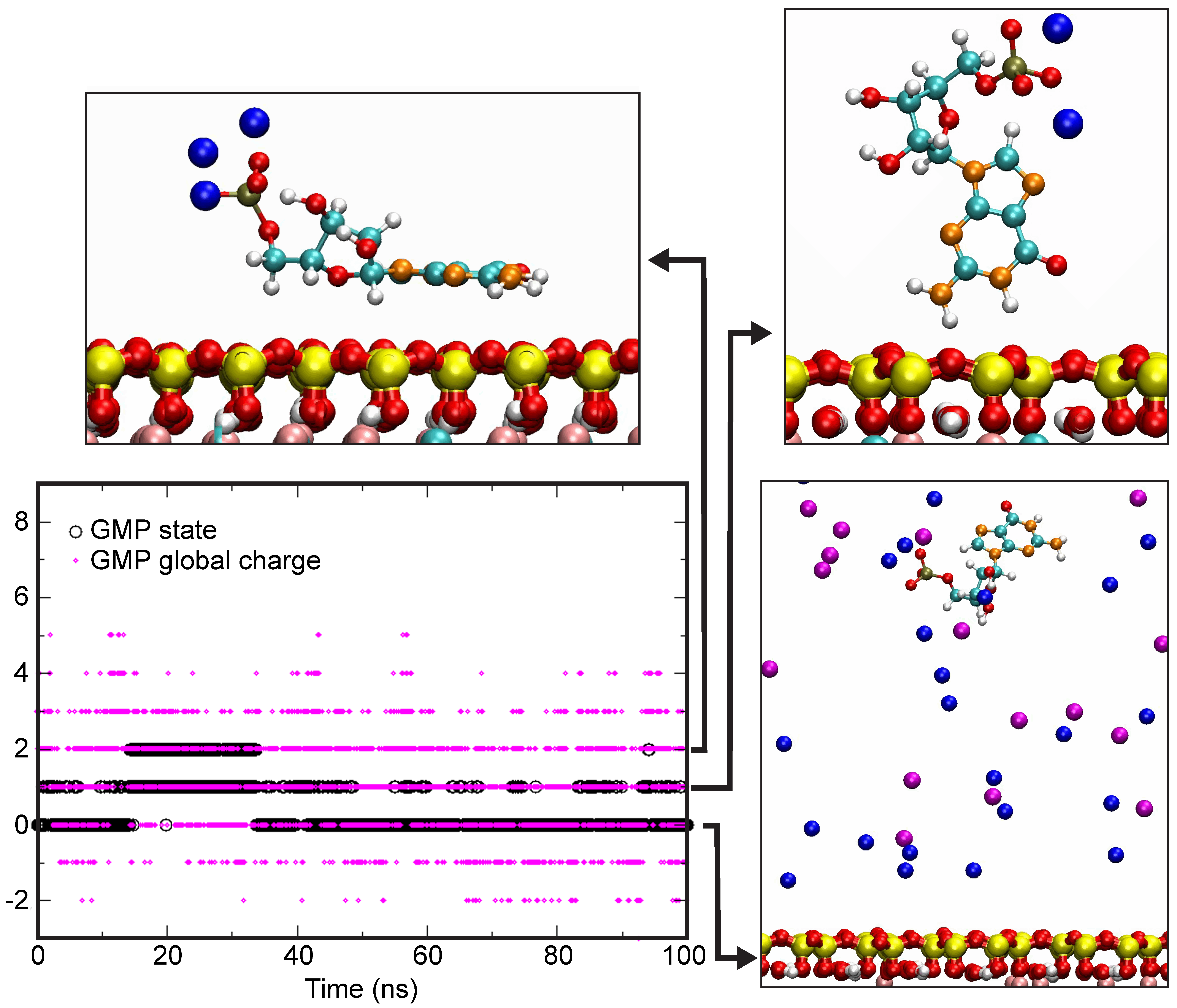
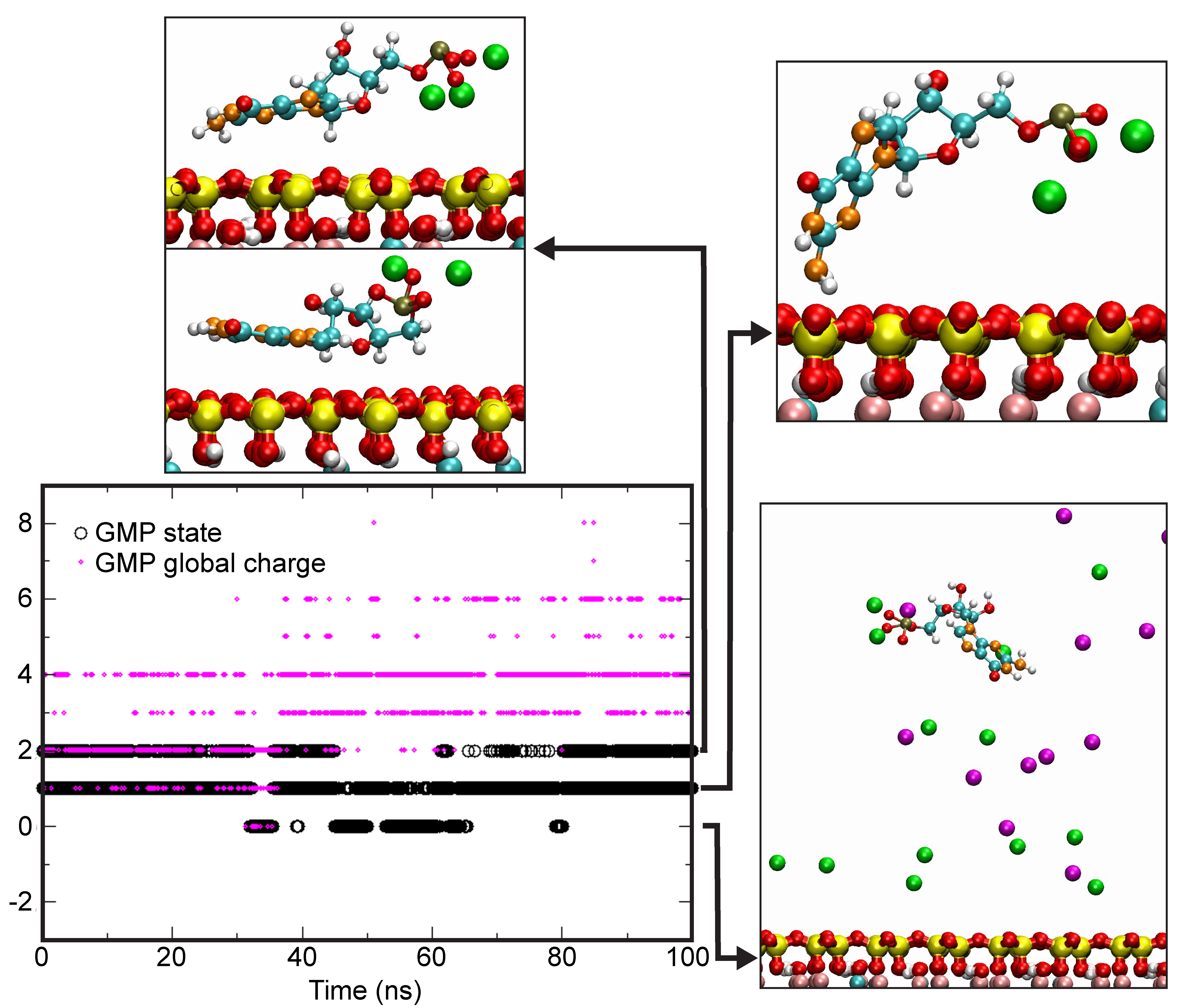
4.4. Theoretical Calculations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poulet, F.; Bibring, J.-P.; Mustard, J.F.; Gendrin, A.; Mangold, N.; Langevin, Y.; Arvidson, R.E.; Gondet, B.; Gomez, C.; Team, T.O. Phyllosilicates on Mars and implications for early Martian climate. Nature 2005, 438, 623. [Google Scholar] [CrossRef] [PubMed]
- Ehlmann, B.L.; Mustard, J.F.; Murchie, S.L.; Bibring, J.P.; Meunier, A.; Fraeman, A.A.; Langevin, Y. Subsurface water and clay mineral formation during the early history of Mars. Nature 2011, 479, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M.; Sverjensky, D.A.; Azzolini, D.; Bish, D.L.; Elmore, S.C.; Hinnov, L.; Milliken, R.E. Clay mineral evolution. Am. Mineral. 2013, 98, 2007–2029. [Google Scholar] [CrossRef]
- Giese, R.F.; Van Oss, C.J.; Yariv, S.; Cross, H. Organophilicity and hydrophobicity of organo-clays. In Organo-Clay Complexes and Interactions; Marcel Dekker: New York, NY, USA, 2002; pp. 175–192. [Google Scholar]
- Bernal, J.D. The Physical Basis of Life. Proc. Phys. Soc. Sect. B 1949, 62, 598–618. [Google Scholar] [CrossRef]
- Corliss, J.B.; Baross, J.A.; Hoffman, S.E. An hypothesis concerning the relationship between submarine hot springs and the origin of life on earth. Oceanol. Acta 1981, 4, 59–69. [Google Scholar]
- Sleep, N.H. Geological and Geochemical Constraints on the Origin and Evolution of Life. Astrobiology 2018, 18, 1199–1219. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.A.; Boehnke, P.; Harrison, T.M.; Mao, W.L. Potentially biogenic carbon preserved in a 4.1 billion-year-old zircon. Proc. Natl. Acad. Sci. USA 2015, 112, 14518–14521. [Google Scholar] [CrossRef] [PubMed]
- Rosing, M.T. 13C-depleted carbon microparticles in >3700-Ma sea-floor sedimentary rocks from West Greenland. Science 1999, 283, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Papineau, D.; De Gregorio, B.T.; Cody, G.D.; Fries, M.D.; Mojzsis, S.J.; Steele, A.; Stroud, R.M.; Fogel, M.L. Ancient graphite in the Eoarchean quartz–pyroxene rocks from Akilia in southern West Greenland I: Petrographic and spectroscopic characterization. Geochim. Cosmochim. Acta 2010, 74, 5862–5883. [Google Scholar] [CrossRef]
- Kasting, J.F. Atmospheric composition of Hadean—Early Archean Earth: The importance of CO. Earth’s Early Atmos. Surf. Environ. Geol. Soc. Am. Spec. Pap. 504 2014, 2504, 19–28. [Google Scholar] [CrossRef]
- Charnay, B.; Le Hir, G.; Fluteau, F.; Forget, F.; Catling, D.C. A warm or a cold early Earth? New insights from a 3-D climate-carbon model. Earth Planet. Sci. Lett. 2017, 474, 97–109. [Google Scholar] [CrossRef]
- Tartèse, R.; Chaussidon, M.; Gurenko, A.; Delarue, F.; Robert, F. Warm Archaean oceans reconstructed from oxygen isotope composition of early-life remnants. Geochem. Perspect. Lett. 2017, 55–65. [Google Scholar] [CrossRef]
- Krissansen-Totton, J.; Arney, G.N.; Catling, D.C. Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl. Acad. Sci. USA 2018, 115, 201721296. [Google Scholar] [CrossRef] [PubMed]
- Albarède, F. Volatile accretion history of the terrestrial planets and dynamic implications. Nature 2009, 461, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Pope, E.C.; Bird, D.K.; Rosing, M.T. Isotope composition and volume of Earth’s early oceans. Proc. Natl. Acad. Sci. USA 2012, 109, 4371–4376. [Google Scholar] [CrossRef] [PubMed]
- Flament, N.; Coltice, N.; Rey, P.F. A case for late-Archaean continental emergence from thermal evolution models and hypsometry. Earth Planet. Sci. Lett. 2008, 275, 326–336. [Google Scholar] [CrossRef]
- Arndt, N. Why was flood volcanism on submerged continental platforms so common in the Precambrian? Precambrian Res. 1999, 97, 155–164. [Google Scholar] [CrossRef]
- Halevy, I.; Bachan, A. The geologic history of seawater pH. Science 2017, 355, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Hall, A.J.; Martin, W. Serpentinization as a source of energy at the origin of life. Geobiology 2010, 8, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Holland, H.D. The Chemical Evolution of the Atmosphere and Oceans; Princeton University Press: Princeton, NJ, USA, 1984; ISBN 0691023816. [Google Scholar]
- Knauth, L.P. Temperature and salinity history of the Precambrian ocean: Implications for the course of microbial evolution. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2005, 219, 53–69. [Google Scholar] [CrossRef]
- Saito, T.; Shibuya, T.; Komiya, T.; Kitajima, K.; Yamamoto, S.; Nishizawa, M.; Ueno, Y.; Kurosawa, M.; Maruyama, S. PIXE and microthermometric analyses of fluid inclusions in hydrothermal quartz from the 2.2 Ga Ongeluk Formation, South Africa: Implications for ancient seawater salinity. Precambrian Res. 2016, 286, 337–351. [Google Scholar] [CrossRef]
- Marty, B.; Avice, G.; Bekaert, D.V.; Broadley, M.W. Salinity of the Archaean oceans from analysis of fluid inclusions in quartz. Comptes Rendus Geosci. 2018, 350, 154–163. [Google Scholar] [CrossRef]
- Kim, H.-J.; Benner, S.A. Prebiotic stereoselective synthesis of purine and noncanonical pyrimidine nucleotide from nucleobases and phosphorylated carbohydrates. Proc. Natl. Acad. Sci. USA 2017, 114, 11315–11320. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Georgiou, C.D. Hydrothermal conditions and the origin of cellular life. Astrobiology 2015, 15, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Coveney, P.V.; Christopher Greenwell, H. Stability of free and mineral-protected nucleic acids: Implications for the RNA world. Geochim. Cosmochim. Acta 2012, 83, 360–378. [Google Scholar] [CrossRef]
- Fornaro, T.; Boosman, A.; Brucato, J.R.; ten Kate, I.L.; Siljeström, S.; Poggiali, G.; Steele, A.; Hazen, R.M. UV irradiation of biomarkers adsorbed on minerals under Martian-like conditions: Hints for life detection on Mars. Icarus 2018, 313, 38–60. [Google Scholar] [CrossRef]
- Fornaro, T.; Brucato, J.R.; Pace, E.; Guidi, M.C.; Branciamore, S.; Pucci, A. Infrared spectral investigations of UV irradiated nucleobases adsorbed on mineral surfaces. Icarus 2013, 226, 1068–1085. [Google Scholar] [CrossRef]
- Meunier, A.; Petit, S.; Cockell, C.S.; El Albani, A.; Beaufort, D. The Fe-rich clay microsystems in basalt-komatiite lavas: Importance of Fe-Smectites for Pre-Biotic molecule catalysis during the Hadean eon. Orig. Life Evol. Biosph. 2010, 40, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Schoonen, M.; Smirnov, A. Staging life in an early warm “seltzer” ocean. Elements 2016, 12, 395–400. [Google Scholar] [CrossRef]
- Ferris, J.P.; Ertem, G.; Agarwal, V.K. The adsorption of nucleotides and polynucleotides on montmorillonite clay. Orig. Life Evol. Biosph. 1989, 19, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Franchi, M.; Bramanti, E.; Bonzi, L.M.; Orioli, P.L.; Vettori, C.; Gallori, E. Clay-nucleic acid complexes: Characteristics and implications for the preservation of genetic material in primeval habitats. Orig. Life Evol. Biosph. 1999, 29, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Fujii, K. Relationship between adsorption and desorption of adenine by montmorillonite. Clay Sci. 2016, 20, 21–25. [Google Scholar]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Maurin, G.; Sing, K.S.W. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Cambridge, MA, USA, 2013; ISBN 0080970362. [Google Scholar]
- Feuillie, C.; Daniel, I.; Michot, L.J.; Pedreira-Segade, U. Adsorption of nucleotides onto Fe-Mg-Al rich swelling clays. Geochim. Cosmochim. Acta 2013, 120, 97–108. [Google Scholar] [CrossRef]
- Pedreira-Segade, U.; Feuillie, C.; Pelletier, M.; Michot, L.J.; Daniel, I. Adsorption of nucleotides onto ferromagnesian phyllosilicates: Significance for the origin of life. Geochim. Cosmochim. Acta 2016, 176, 81–95. [Google Scholar] [CrossRef]
- Pedreira-Segade, U.; Michot, L.J.; Daniel, I. Effects of salinity on the adsorption of nucleotides onto phyllosilicates. Phys. Chem. Chem. Phys. 2018, 20, 1938–1952. [Google Scholar] [CrossRef] [PubMed]
- Pedreira-Segade, U. Nucléotides à l’interface minéral-eau et réactivité des acides aminés en conditions hydrothermales dans le contexte des origines de la vie. Ph.D. Thesis, Université de Lyon, Lyon, France, 2017. [Google Scholar]
- Goring, C.A.I.; Bartholomew, W. V Adsorption of mononucleotides, nucleic acids, and nucleoproteins by clays. Soil Sci. 1952, 74, 149–164. [Google Scholar] [CrossRef]
- Graf, G.; Lagaly, G. Interaction of clay minerals with adenosine-5-phosphates. Clays Clay Miner. 1980, 28, 12. [Google Scholar] [CrossRef]
- Ertem, G. Montmorillonite, oligonucleotides, RNA and origin of life. Orig. Life Evol. Biosph. 2004, 34, 549–570. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Hagan, W.J. The adsorption and reaction of adenine nucleotides on montmorillonite. Orig. Life Evol. Biosph. 1986, 17, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Lawless, J.G.; Banin, A.; Church, F.M.; Mazzurco, J.; Huff, R.; Kao, J.; Cook, A.; Lowe, T.; Orenberg, J.B.; Edelson, E. pH profile of the adsorption of nucleotides onto montmorillonite—I. selected homoionic clays. Orig. Life Evol. Biosph. 1985, 15, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, P.; Loew, G.; Burt, S.; Lawless, J.; MacElroy, R.D. Interaction of Metal Ions and Nucleotides: Possible Mechanisms for the Adsorption of Nucleotides on Homoionic Bentonite Clays. Inorg. Chem. 1982, 21, 1586–1594. [Google Scholar] [CrossRef]
- Rishpon, J.; O’Hara, P.J.; Lahav, N.; Lawless, J.G. Interaction between ATP, metal ions, glycine, and several minerals. J. Mol. Evol. 1982, 18, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.L.; Michalkova, A.; Gorb, L.; Leszczynski, J. Hydrogen bonding of thymine and uracil with surface of dickite: An ab initio study. J. Mol. Struct. 2007, 844–845, 48–58. [Google Scholar] [CrossRef]
- Perezgasga, L.; Serrato-Díaz, A.; Negrón-Mendoza, A.; De Pablo Galán, L.; Mosqueira, F.G. Sites of adsorption of adenine, uracil, and their corresponding derivatives on sodium montmorillonite. Orig. Life Evol. Biosph. 2005, 35, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Villafañe-Barajas, S.A.; Baú, J.P.T.; Colín-García, M.; Negrón-Mendoza, A.; Heredia-Barbero, A.; Pi-Puig, T.; Zaia, D.A.M. Salinity Effects on the Adsorption of Nucleic Acid Compounds on Na-Montmorillonite: A Prebiotic Chemistry Experiment. Orig. Life Evol. Biosph. 2018, 48. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.; Zubay, G. Binding of adenine and adenine-related compounds to the clay montmorillonite and the mineral hydroxylapatite. Orig. Life Evol. Biosph. 1995, 25, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Coveney, P.V.; Greenwell, H.C. Clay minerals mediate folding and regioselective interactions of RNA: A large-scale atomistic simulation study. J. Am. Chem. Soc. 2010, 132, 13750–13764. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Suter, J.L.; Greenwell, H.C.; Coveney, P.V. Influence of surface chemistry and charge on mineral-RNA interactions. Langmuir 2013, 29, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Lailach, G.E.; Thompson, T.D.; Brindley, G.W. Absorption of pyrimidines, purines, and nucleosides by Li-, Na-, Mg-, and Ca-montmorillonite (Clay-organic studies XII). Clays Clay Miner. 1968, 16, 285–293. [Google Scholar] [CrossRef]
- Hao, J.; Mokhtaria, M.; Pedreira-Segade, U.; Michot, L.J.; Daniel, I. Transition metals enhance the adsorption of nucleotides onto clays: Implications for the origin of life. ACS Earth Space Chem. 2018, in press. [Google Scholar] [CrossRef]
- Franchi, M.; Ferris, J.P.; Gallori, E. Cations as mediators of the adsorption of nucleic acids on clay surfaces in prebiotic environments. Orig. Life Evol. Biosph. 2003, 33, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bezanilla, M.; Manne, S.; Laney, D.E.; Lyubchenko, Y.L.; Hansma, H.G.C.N. Adsorption of DNA To Mica, Silylated Mica, And Minerals—Characterization by Atomic-Force Microscopy. Langmuir 1995, 11, 655–659. [Google Scholar] [CrossRef]
- Lorenz, M.G.; Wackernagel, W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl. Environ. Microbiol. 1987, 53, 2948–2952. [Google Scholar] [PubMed]
- Hansma, H.G.; Laney, D.E. DNA binding to mica correlates with cationic radius: Assay by atomic force microscopy. Biophys. J. 1996, 70, 1933–1939. [Google Scholar] [CrossRef]
- Bebié, J.; Schoonen, M.A.A. Pyrite and phosphate in anoxia and an origin-of-life hypothesis. Earth Planet. Sci. Lett. 1999, 171, 1–5. [Google Scholar] [CrossRef]
- Pontes-Buarques, M.; Tessis, A.C.; Bonapace, J.A.P.; Monte, M.B.M.; Cortés-Lopez, G.; De Souza-Barros, F.; Vieyra, A. Modulation of adenosine 5′-monophosphate adsorption onto aqueous resident pyrite: Potential mechanisms for prebiotic reactions. Orig. Life Evol. Biosph. 2001, 31, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, F.; Liu, B.; Kelly, E.Y.; Servos, M.R.; Liu, J. Adsorption of DNA oligonucleotides by titanium dioxide nanoparticles. Langmuir 2014, 30, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Feuillie, C. Détection d’ADN par spectroscopie SERRS et interactions entre nucléotides et surfaces des minéraux phyllosilicatés ferromagnésiens dans le contexte de l’origine de la Vie. Ph.D. Thesis, Ecole Normale Supérieure de Lyon, Lyon, France, 2012. [Google Scholar]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Giles, C.H.; Smith, D.; Huitson, A. A general treatment and classification of the solute adsorption isotherm. I. Theoretical. J. Colloid Interface Sci. 1974, 47, 755–765. [Google Scholar] [CrossRef]
- Hinz, C. Description of sorption data with isotherm equations. Geoderma 2001, 99, 225–243. [Google Scholar] [CrossRef]
- Azizian, S.; Eris, S.; Wilson, L.D. Re-evaluation of the century-old Langmuir isotherm for modeling adsorption phenomena in solution. Chem. Phys. 2018, 513, 99–104. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Anraku, S.; Paineau, E.; Safinya, C.R.; Davidson, P.; Michot, L.J.; Miyamoto, N. Swelling inhibition of liquid crystalline colloidal montmorillonite and beidellite clays by DNA. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Suzzoni, A.; Barre, L.; Kohler, E.; Levitz, P.; Michot, L.J.; M’Hamdi, J. Interactions between kaolinite clay and AOT. Colloids Surf. A Physicochem. Eng. Asp. 2018, 556, 309–315. [Google Scholar] [CrossRef]
- Michot, L.; François, M.; Cases, J.M. Surface heterogeneity studied by a quasi-equilibrium gas adsorption procedure. Langmuir 1990, 6, 677–681. [Google Scholar] [CrossRef]
- Villiéras, F.; Cases, J.M.; François, M.; Michot, L.J.; Thomas, F. Texture and surface energetic heterogeneity of solids from modeling of low pressure gas adsorption isotherms. Langmuir 1992, 8, 1789–1795. [Google Scholar] [CrossRef]
- Villiéras, F.; Michot, L.J.; Cases, J.M.; Berend, I.; Bardot, F.; François, M.; Gérard, G.; Yvon, J. Static and dynamic studies of the energetic surface heterogeneity of clay minerals. Stud. Surf. Sci. Catal. 1997, 104, 573–623. [Google Scholar]
- Villiéras, F.; Michot, L.J.; Bardot, F.; Cases, J.M.; François, M.; Rudzinski, W. An improved derivative isotherm summation method to study surface heterogeneity of clay minerals. Langmuir 1997, 13, 1104–1117. [Google Scholar] [CrossRef]
- Marty, N.C.M.; Cama, J.; Sato, T.; Chino, D.; Villiéras, F.; Razafitianamaharavo, A.; Brendlé, J.; Giffaut, E.; Soler, J.M.; Gaucher, E.C. Dissolution kinetics of synthetic Na-smectite. An integrated experimental approach. Geochim. Cosmochim. Acta 2011, 75, 5849–5864. [Google Scholar] [CrossRef]
- Decarreau, A.; Petit, S.; Andrieux, P.; Villieras, F.; Pelletier, M.; Razafitianamaharavo, A. Study of low-pressure argon adsorption on synthetic nontronite: Implications for smectite crystal growth. Clays Clay Miner. 2014, 62, 102–111. [Google Scholar] [CrossRef]
- Bardot, F.; Villiéras, F.; Michot, L.J.; François, M.; Gérard, G.; Cases, J.M. High Resolution Gas Adsorption Study on Illites Permuted with Various Cations: Assessment of surface energetic properties. J. Dispers. Sci. Technol. 1998, 19, 739–759. [Google Scholar] [CrossRef]
- Michot, L.J.; Villiéras, F. Assessment of surface energetic heterogeneity of synthetic Na-saponites. The role of layer charge. Clay Miner. 2002, 37, 39–57. [Google Scholar] [CrossRef]
- Villiéras, F.; Michot, L.J.; Bardot, F.; Chamerois, M.; Eypert-Blaison, C.; François, M.; Gérard, G.; Cases, J.-M. Surface heterogeneity of minerals. Comptes Rendus Geosci. 2002, 334, 597–609. [Google Scholar] [CrossRef]
- Tournassat, C.; Neaman, A.; Villiéras, F.; Bosbach, D.; Charlet, L. Nanomorphology of montmorillonite particles: Estimation of the clay edge sorption site density by low-pressure gas adsorption and AFM observations. Am. Mineral. 2003, 88, 1989–1995. [Google Scholar] [CrossRef]
- Hassan, M.S.; Villieras, F.; Razafitianamaharavo, A.; Michot, L.J. Role of exchangeable cations on geometrical and energetic surface heterogeneity of kaolinites. Langmuir 2005, 21, 12283–12289. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.S.; Villieras, F.; Gaboriaud, F.; Razafitianamaharavo, A. AFM and low-pressure argon adsorption analysis of geometrical properties of phyllosilicates. J. Colloid Interface Sci. 2006, 296, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Le Forestier, L.; Muller, F.; Villieras, F.; Pelletier, M. Textural and hydration properties of a synthetic montmorillonite compared with a natural Na-exchanged clay analogue. Appl. Clay Sci. 2010, 48, 18–25. [Google Scholar] [CrossRef]
- Fornaro, T.; Brucato, J.R.; Feuillie, C.; Sverjensky, D.A.; Hazen, R.M.; Brunetto, R.; D’Amore, M.; Barone, V. Binding of Nucleic Acid Components to the Serpentinite-Hosted Hydrothermal Mineral Brucite. Astrobiology 2018, 18, 989–1007. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, T.K.; Kumar, M.S.; Arjunan, M.; Pratap, A.; Chandrasekaran, N. Graphene oxide aided structural tailoring of 3-D N-doped amorphous carbon network for enhanced energy storage. RSC Adv. 2015, 5, 93423–93432. [Google Scholar] [CrossRef]
- Bernard, J.M.; Quirico, E.; Brissaud, O.; Montagnac, G.; Reynard, B.; McMillan, P.; Coll, P.; Nguyen, M.J.; Raulin, F.; Schmitt, B. Reflectance spectra and chemical structure of Titan’s tholins: Application to the analysis of Cassini-Huygens observations. Icarus 2006, 185, 301–307. [Google Scholar] [CrossRef]
- Quirico, E.; Montagnac, G.; Lees, V.; McMillan, P.F.; Szopa, C.; Cernogora, G.; Rouzaud, J.N.; Simon, P.; Bernard, J.M.; Coll, P.; et al. New experimental constraints on the composition and structure of tholins. Icarus 2008, 198, 218–231. [Google Scholar] [CrossRef]
- Ukai, M.; Yokoya, A.; Fujii, K.; Saitoh, Y. X-ray absorption spectra of nucleotides (AMP, GMP, and CMP) in liquid water solutions near the nitrogen K-edge. Chem. Phys. Lett. 2010, 495, 90–95. [Google Scholar] [CrossRef]
- Mathew, D.C.; Luthey-Schulten, Z. Influence of montmorillonite on nucleotide oligomerization reactions: A molecular dynamics study. Orig. Life Evol. Biosph. 2010, 40, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Mignon, P.; Sodupe, M. Structural Behaviors of Cytosine into the Hydrated Interlayer of Na+-Montmorillonite Clay. An ab Initio Molecular Dynamics Study. J. Phys. Chem. C 2013, 117, 26179–26189. [Google Scholar] [CrossRef]
- Himbert, S.; Chapman, M.; Deamer, D.W.; Rheinstädter, M.C. Organization of nucleotides in different environments and the formation of pre-polymers. Sci. Rep. 2016, 6, 31285. [Google Scholar] [CrossRef] [PubMed]
- Mignon, P.; Ugliengo, P.; Sodupe, M. Theoretical study of the adsorption of RNA/DNA bases on the external surfaces of Na+-montmorillonite. J. Phys. Chem. C 2009, 113, 13741–13749. [Google Scholar] [CrossRef]
- Mignon, P.; Sodupe, M. Theoretical study of the adsorption of DNA bases on the acidic external surface of montmorillonite. Phys. Chem. Chem. Phys. 2012, 14, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, M.; Greathouse, J.A.; Cygan, R.T.; Kalinichev, A.G. Structure of hydrated gibbsite and brucite edge surfaces: DFT results and further development of the ClayFF classical force field with metal–O–H angle bending terms. J. Phys. Chem. C 2017, 121, 14757–14771. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Jelavić, S.; Tobler, D.J.; Hassenkam, T.; De Yoreo, J.J.; Stipp, S.L.S.; Sand, K.K. Prebiotic RNA polymerisation: Energetics of nucleotide adsorption and polymerisation on clay mineral surfaces. Chem. Commun. 2017, 53, 12700–12703. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GMP State | Na+ | Ca2+ | ||||
|---|---|---|---|---|---|---|
| Sim 1 | Sim 2 | Average | Sim 1 | Sim 2 | Average | |
| 0 | 0.92 | 0.79 | 0.85 | 3.44 | 2.56 | 2.92 |
| 1 | 1.42 | 1.41 | 1.41 | 3.60 | 3.70 | 3.64 |
| 2 | 1.09 | 1.56 | 1.31 | 3.74 | 3.17 | 3.38 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedreira-Segade, U.; Hao, J.; Razafitianamaharavo, A.; Pelletier, M.; Marry, V.; Le Crom, S.; Michot, L.J.; Daniel, I. How do Nucleotides Adsorb Onto Clays? Life 2018, 8, 59. https://doi.org/10.3390/life8040059
Pedreira-Segade U, Hao J, Razafitianamaharavo A, Pelletier M, Marry V, Le Crom S, Michot LJ, Daniel I. How do Nucleotides Adsorb Onto Clays? Life. 2018; 8(4):59. https://doi.org/10.3390/life8040059
Chicago/Turabian StylePedreira-Segade, Ulysse, Jihua Hao, Angelina Razafitianamaharavo, Manuel Pelletier, Virginie Marry, Sébastien Le Crom, Laurent J. Michot, and Isabelle Daniel. 2018. "How do Nucleotides Adsorb Onto Clays?" Life 8, no. 4: 59. https://doi.org/10.3390/life8040059
APA StylePedreira-Segade, U., Hao, J., Razafitianamaharavo, A., Pelletier, M., Marry, V., Le Crom, S., Michot, L. J., & Daniel, I. (2018). How do Nucleotides Adsorb Onto Clays? Life, 8(4), 59. https://doi.org/10.3390/life8040059