Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans


Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences and Alignments
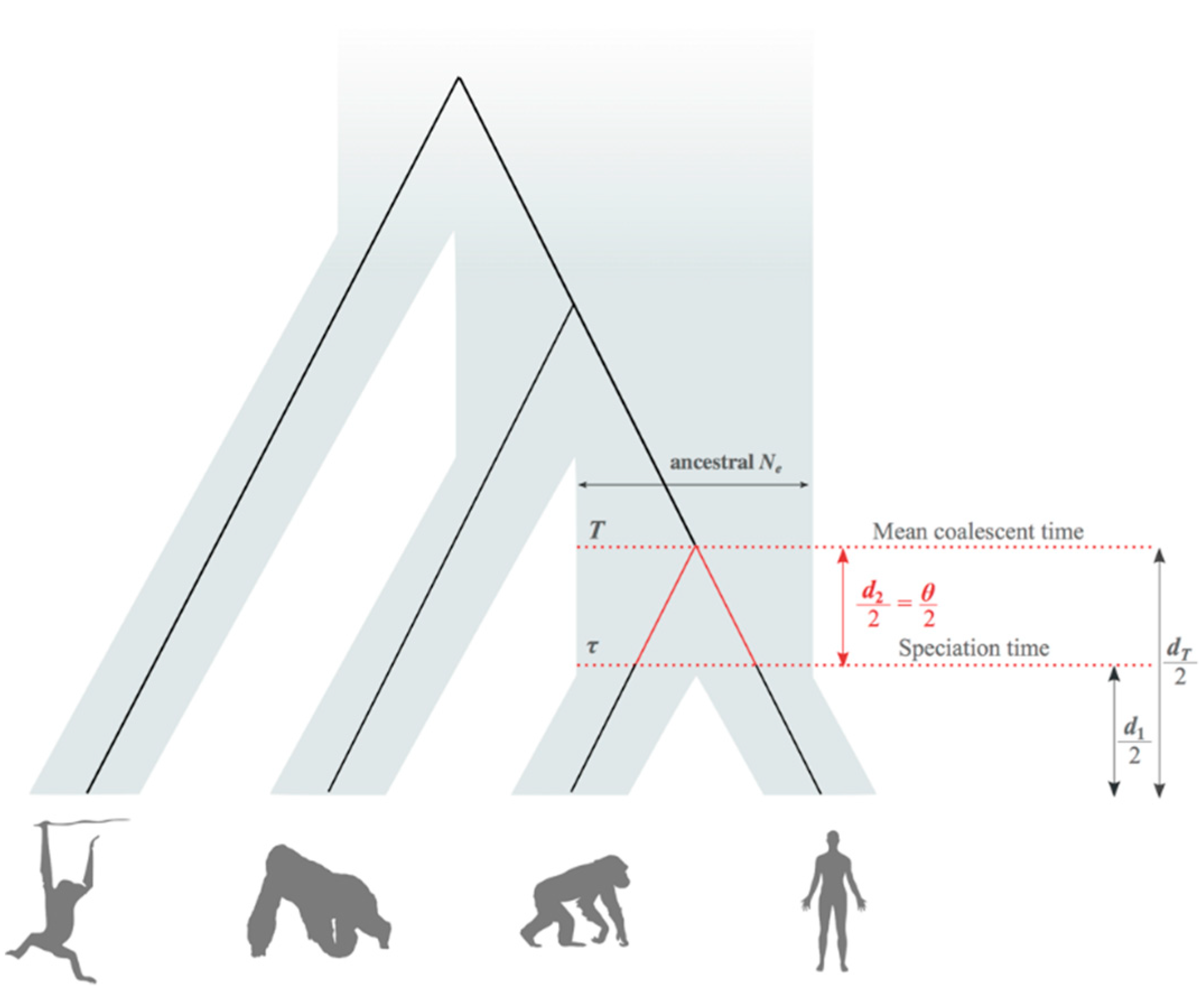
2.2. Average Coalescent Times and Speciation Times
2.3. Estimation of Mean Speciation Times τ
2.4. Fossil African Great Apes and Humans
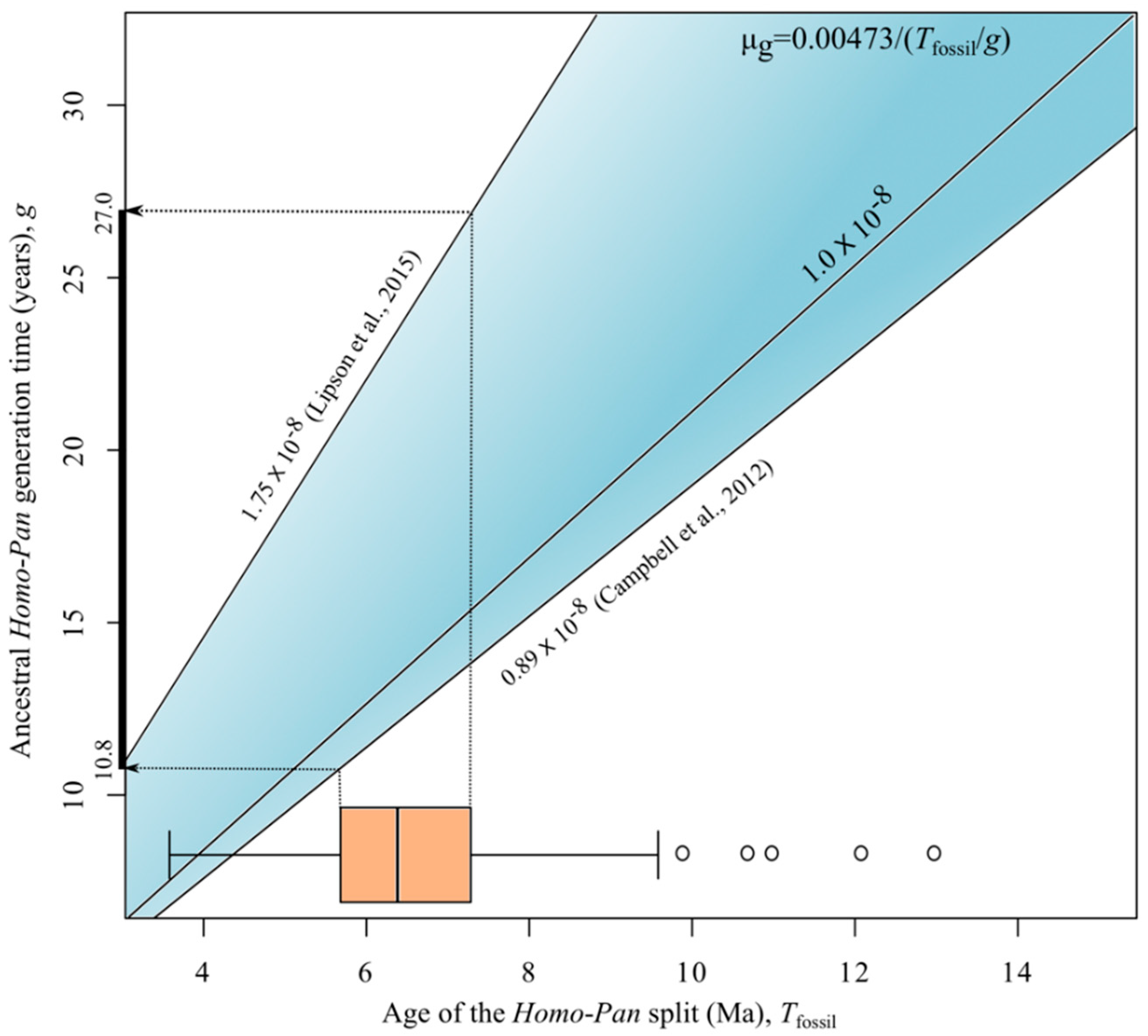
2.5. Generation Times
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: New York, CA, USA, 1983. [Google Scholar]
- Nei, M. Mutation-Driven Evolution; Oxford University Press: New York, CA, USA, 2013. [Google Scholar]
- Haldane, J.B.S. The rate of spontaneous mutation of a human gene. J. Genet. 1935, 31, 317–326. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, CA, USA, 2000. [Google Scholar]
- Benton, M.J.; Donoghue, P.C.J.; Asher, R.J.; Friedman, M.; Near, T.J.; Vinther, J. Constraints on the timescale of animal evolutionary history. Palaeontol. Electron. 2015, 18, 1–107. [Google Scholar] [CrossRef]
- The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.C.; Li, W.H. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 2001, 68, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Ebersberger, I.; Metzler, D.; Schwarz, C.; Paabo, S. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 2002, 70, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.; Guy, F.; Pilbeam, D.; Mackaye, H.T.; Likius, A.; Ahounta, D.; Beauvilain, A.; Blondel, C.; Bocherens, H.; Boisserie, J.R.; et al. A new hominid from the Upper Miocene of Chad, Central Africa. Nature 2002, 418, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Eichler, E.E. Properties and rates of germline mutations in humans. Trends Genet. 2013, 29, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Venn, O.; Turner, I.; Mathieson, I.; de Groot, N.; Bontrop, R.; McVean, G. Strong male bias drives germline mutation in chimpanzees. Science 2014, 344, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Schrago, C.G. The effective population sizes of the anthropoid ancestors of the human-chimpanzee lineage provide insights on the historical biogeography of the great apes. Mol. Biol. Evol. 2014, 31, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 2003, 164, 1645–1656. [Google Scholar] [PubMed]
- Scally, A.; Durbin, R. Revising the human mutation rate: Implications for understanding human evolution. Nat. Rev. Genet. 2012, 13, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M. Rates of Molecular Evolution: The Hominoid Slowdown. Bioessays 1985, 3, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Tanimura, M. The Molecular Clock Runs More Slowly in Man Than in Apes and Monkeys. Nature 1987, 326, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Bailey, W.J.; Fitch, D.H.; Tagle, D.A.; Czelusniak, J.; Slightom, J.L.; Goodman, M. Molecular evolution of the psi eta-globin gene locus: Gibbon phylogeny and the hominoid slowdown. Mol. Biol. Evol. 1991, 8, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Koop, B.F.; Goodman, M.; Xu, P.; Chan, K.; Slightom, J.L. Primate Eta-Globin DNA-Sequences and Mans Place among the Great Apes. Nature 1986, 319, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Elango, N.; Warden, C.; Vigoda, E.; Yi, S.V. Heterogeneous genomic molecular clocks in primates. PLoS Genet. 2006, 2, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Scally, A.; Dutheil, J.Y.; Hillier, L.W.; Jordan, G.E.; Goodhead, I.; Herrero, J.; Hobolth, A.; Lappalainen, T.; Mailund, T.; Marques-Bonet, T.; et al. Insights into hominid evolution from the gorilla genome sequence. Nature 2012, 483, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Phillips, M.J.; Cooper, A.; Drummond, A.J. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 2005, 22, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Emerson, B.C. Alarm bells for the molecular clock? No support for Ho et al.’s model of time-dependent molecular rate estimates. Syst. Biol. 2007, 56, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Beerli, P. Perspective: Gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 2000, 54, 1839–1854. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.I.; Masel, J. Quantitative Prediction of Molecular Clock and K-a/K-s at Short Timescales. Mol. Biol. Evol. 2009, 26, 2595–2603. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-dependent rates of molecular evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Glusman, G.; Smit, A.F.A.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Keebler, J.E.M.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.J.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Awadalla for the 1000 genomes project. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Chong, J.X.; Malig, M.; Ko, A.; Dumont, B.L.; Han, L.; Vives, L.; O’Roak, B.J.; Sudmant, P.H.; Shendure, J.; et al. Estimating the human mutation rate using autozygosity in a founder population. Nat. Genet. 2012, 44, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Wong, W.S.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, J.J.; Shi, Y.J.; Gujral, M.; Zheng, H.C.; Malhotra, D.; Jin, X.; Jian, M.H.; Liu, G.M.; Greer, D.; Bhandari, A.; et al. Whole-Genome Sequencing in Autism Identifies Hot Spots for De Novo Germline Mutation. Cell 2012, 151, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Mittnik, A.; Johnson, P.L.; Bos, K.; Lari, M.; Bollongino, R.; Sun, C.; Giemsch, L.; Schmitz, R.; Burger, J.; et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 2013, 23, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Lipson, M.; Loh, P.R.; Sankararaman, S.; Patterson, N.; Berger, B.; Reich, D. Calibrating the Human Mutation Rate via Ancestral Recombination Density in Diploid Genomes. PLoS Genet. 2015, 11, E1005550. [Google Scholar] [CrossRef] [PubMed]
- Besenbacher, S.; Liu, S.; Izarzugaza, J.M.G.; Grove, J.; Belling, K.; Bork-Jensen, J.; Huang, S.; Als, T.D.; Li, S.; Yadav, R.; et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun. 2015, 6, 5969. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, R.; Wuster, A.; Lindsay, S.J.; Hardwick, R.J.; Alexandrov, L.B.; Al Turki, S.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 2015, 48, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Amster, G.; Sella, G. Life history effects on the molecular clock of autosomes and sex chromosomes. Proc. Natl. Acad. Sci. USA 2016, 113, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.S.W.; Solomon, B.D.; Bodian, D.L.; Kothiyal, P.; Eley, G.; Huddleston, K.C.; Baker, R.; Thach, D.C.; Iyer, R.K.; Vockley, J.G.; et al. New observations on maternal age effect on germline de novo mutations. Nat. Commun. 2016, 7, 10486. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 2010, 107, 9264–9269. [Google Scholar] [CrossRef] [PubMed]
- Takahata, N.; Satta, Y.; Klein, J. Divergence time and population size in the lineage leading to modern humans. Theor. Popul. Biol. 1995, 48, 198–221. [Google Scholar] [CrossRef] [PubMed]
- Hobolth, A.; Dutheil, J.Y.; Hawks, J.; Schierup, M.H.; Mailund, T. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 2011, 21, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Langergraber, K.E.; Prufer, K.; Rowney, C.; Boesch, C.; Crockford, C.; Fawcett, K.; Inoue, E.; Inoue-Muruyama, M.; Mitani, J.C.; Muller, M.N.; et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15716–15721. [Google Scholar] [CrossRef] [PubMed]
- Schrago, C.G. The limiting distribution of the effective population size of the ancestor of humans and chimpanzees. J. Theor. Biol. 2014, 357, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of Life Reveals Clock-Like Speciation and Diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D.; Ellegren, H. Sex biases in the mutation rate. Trends Genet. 1998, 14, 446–452. [Google Scholar] [CrossRef]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdottir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161. [Google Scholar] [CrossRef] [PubMed]
- De Manuel, M.; Kuhlwilm, M.; Frandsen, P.; Sousa, V.C.; Desai, T.; Prado-Martinez, J.; Hernandez-Rodriguez, J.; Dupanloup, I.; Lao, O.; Hallast, P.; et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 2016, 354, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Nater, A.; Mattle-Greminger, M.P.; Nurcahyo, A.; Nowak, M.G.; de Manuel, M.; Desai, T.; Groves, C.; Pybus, M.; Sonay, T.B.; Roos, C.; et al. Morphometric, behavioral, and genomic evidence for a new orangutan species. Curr. Biol. 2017, 27, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Nielsen, R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 2007, 104, 2785–2790. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Yu, Y.; Nakhleh, L. Bayesian inference of reticulate phylogenies under the multispecies network coalescent. PLoS Genet. 2016, 12, e1006006. [Google Scholar] [CrossRef] [PubMed]
- Plouviez, S.; Le Guen, D.; Lecompte, O.; Lallier, F.H.; Jollivet, D. Determining gene flow and the influence of selection across the equatorial barrier of the East Pacific Rise in the tube-dwelling polychaete Alvinella pompejana. BMC Evol. Biol. 2010, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Etter, R.J.; Boyle, E.E.; Glazier, A.; Jennings, R.M.; Dutra, E.; Chase, M.R. Phylogeography of a pan-Atlantic abyssal protobranch bivalve: Implications for evolution in the Deep Atlantic. Mol. Ecol. 2011, 20, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.C. Microevolutionary processes generate phylogenomic discordance at ancient divergences. Evolution 2013, 67, 1823–1830. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Mean Rate (s/s/g) |
|---|---|
| 1000 Genomes Project Consortium [27] | 1.0–1.2 × 10−8 |
| Roach et al. [28] | 1.1 × 10−8 |
| Conrad et al. [29] | 0.97–1.17 × 10−8 |
| Cambpell et al. [30] | 0.89–1.43 × 10−8 |
| Kong et al. [31] | 1.20 × 10−8 |
| Michaelson et al. [32] | 1.0 × 10−8 |
| Fu et al. [33] * | 1.10–1.42 × 10−8 |
| Lipson et al. [34] | 1.55–1.75 × 10−8 |
| Besenbacher et al. [35] | 1.16–1.38 × 10−8 |
| Rahbari et al. [36] | 1.13–1.43 × 10−8 |
| Amster and Seela [37] | 1.2 × 10−8 |
| Wong et al. [38] | 1.05 × 10−8 |
| Mean Coalescent Time (T) | Speciation Time (τ) | ||
|---|---|---|---|
| Genetic Distance (dT/2) 1 | Yearly Evolutionary Rate 2 | Genetic Distance (d1/2) 1 | Yearly Evolutionary Rate 2 |
| 0.00625 * | 0.089 × 10−8 | 0.00473 ± 0.000040 | 0.067–0.068 × 10−8 |
| Ancestral Homo–Pan Generation Time (Years) | Evolutionary Rate |
|---|---|
| 15 | 0.99 × 10−8–1.03 × 10−8 |
| 20 | 1.33 × 10−8–1.37 × 10−8 |
| 26.3 | 1.75 × 10−8–1.81 × 10−8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capellão, R.T.; Costa-Paiva, E.M.; Schrago, C.G. Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans. Life 2018, 8, 49. https://doi.org/10.3390/life8040049
Capellão RT, Costa-Paiva EM, Schrago CG. Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans. Life. 2018; 8(4):49. https://doi.org/10.3390/life8040049
Chicago/Turabian StyleCapellão, Renata T., Elisa M. Costa-Paiva, and Carlos G. Schrago. 2018. "Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans" Life 8, no. 4: 49. https://doi.org/10.3390/life8040049
APA StyleCapellão, R. T., Costa-Paiva, E. M., & Schrago, C. G. (2018). Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans. Life, 8(4), 49. https://doi.org/10.3390/life8040049