Formamide Adsorption at the Amorphous Silica Surface: A Combined Experimental and Computational Approach

 ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Experimental Approach
2.2. Computational Approach
3. Results
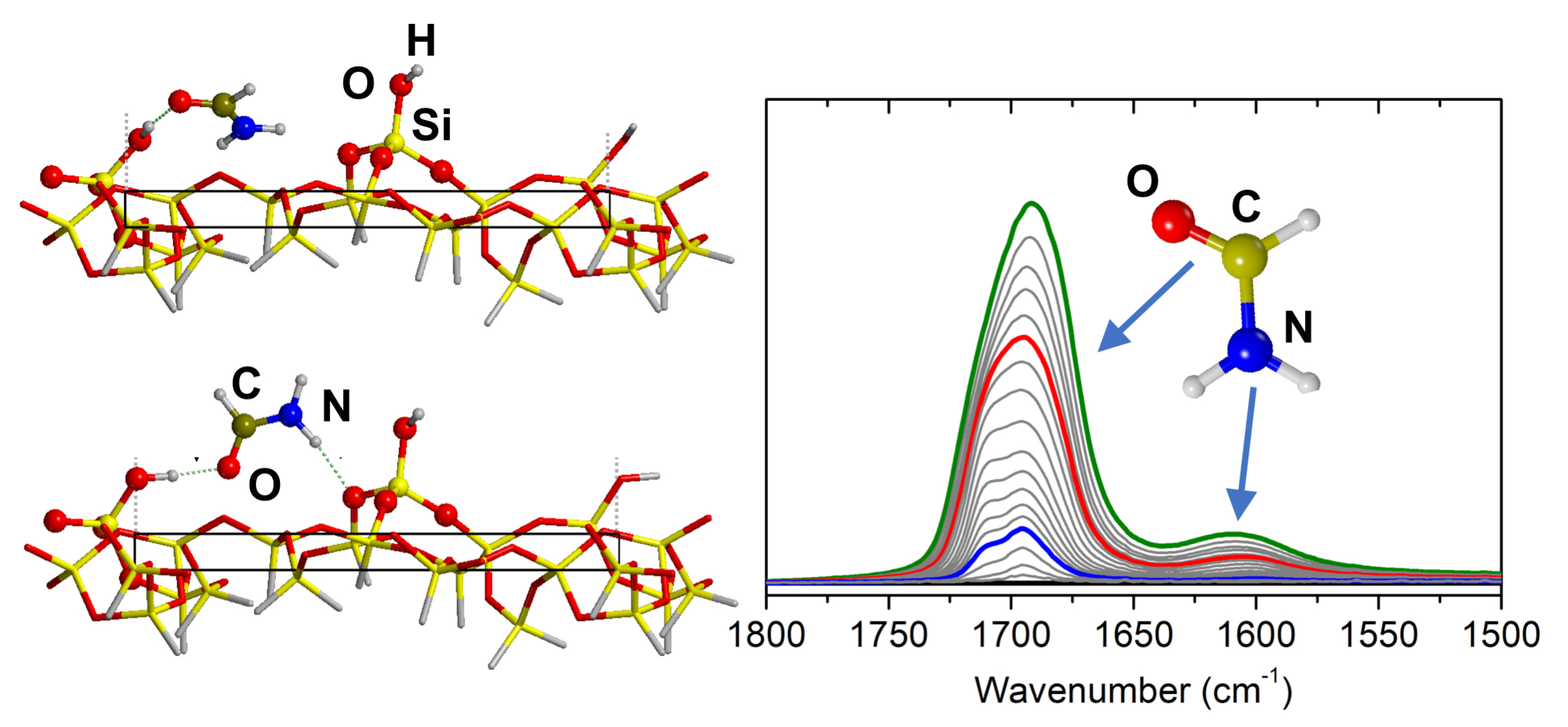
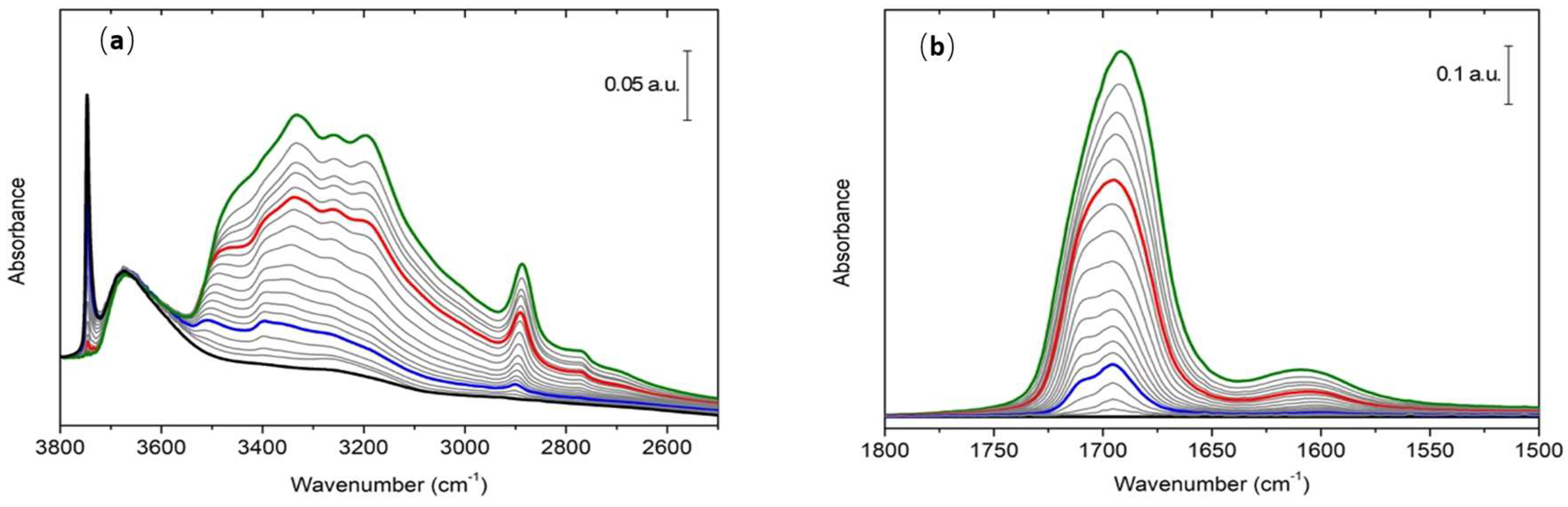
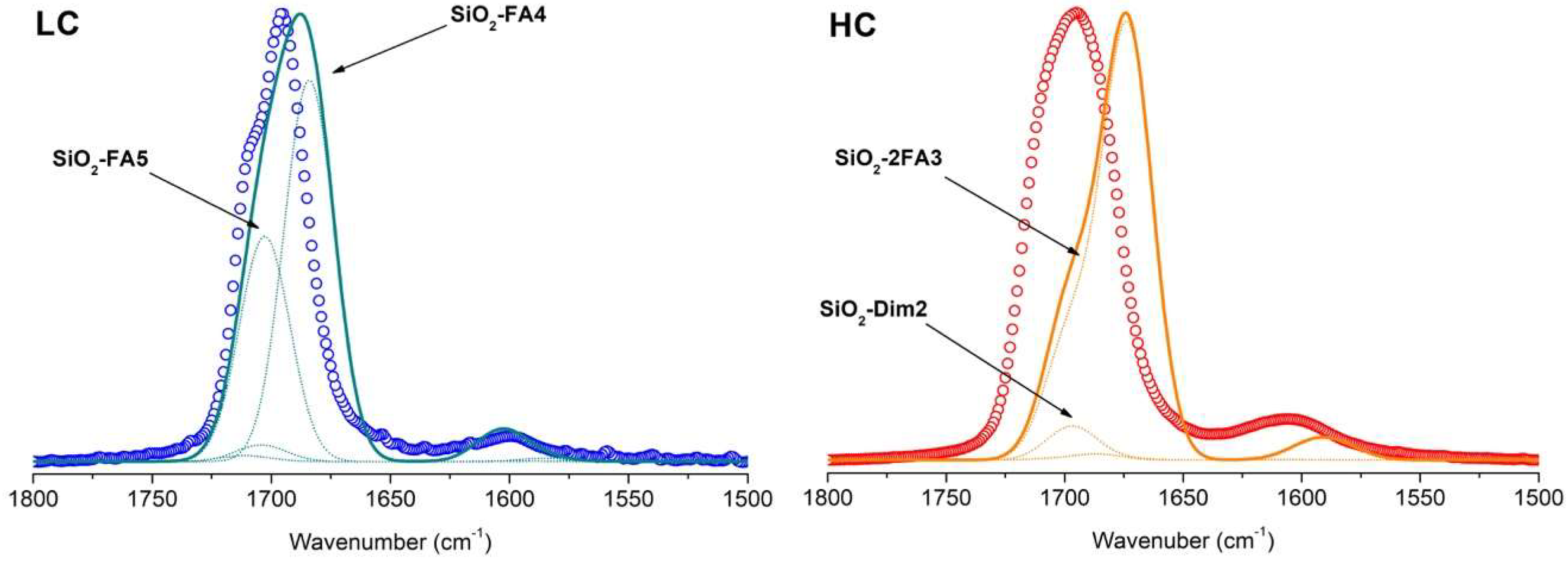
3.1. FTIR of Formamide Adsorbed on A50
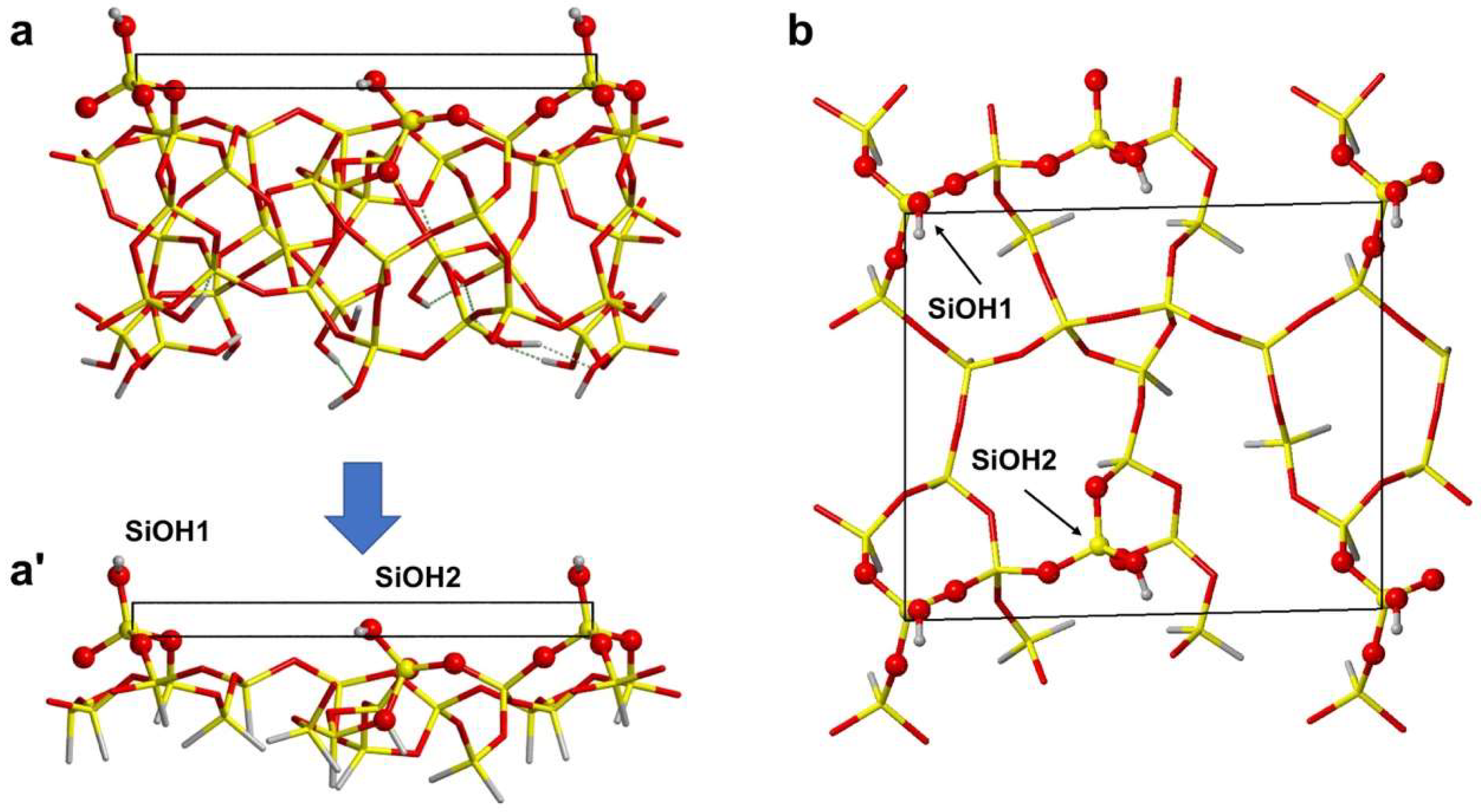
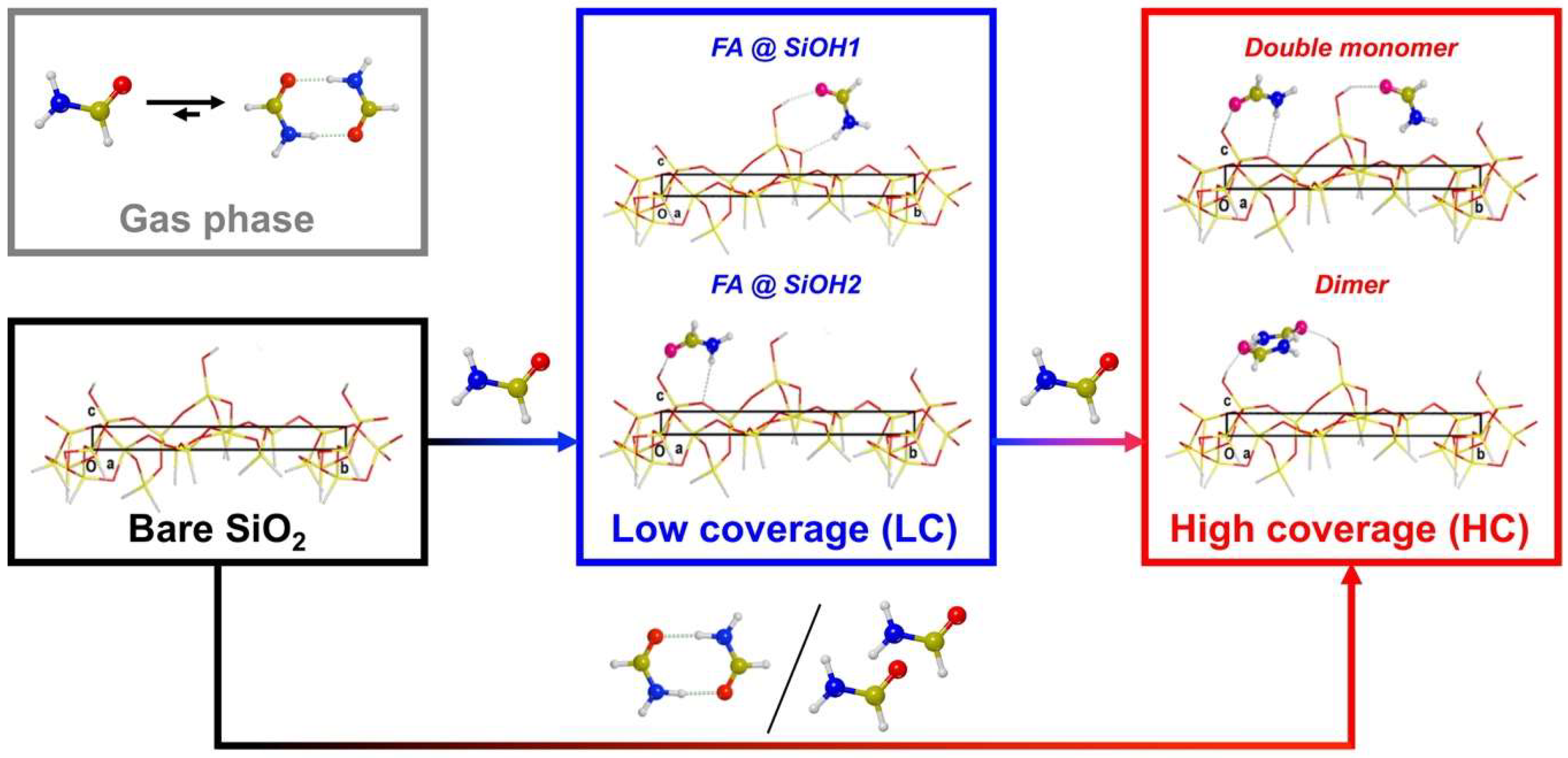
3.2. Simulation of Formamide-A50 Adducts
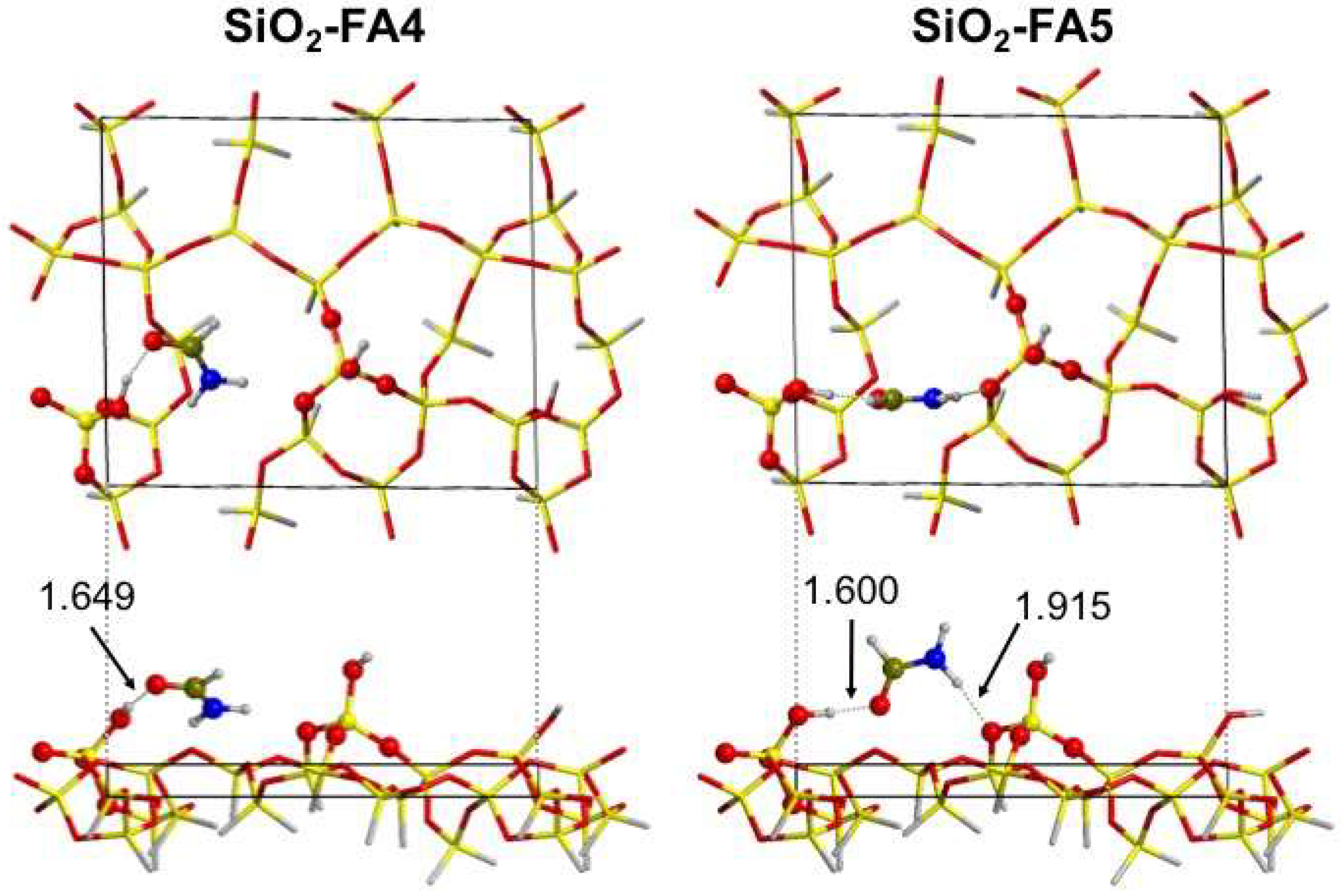
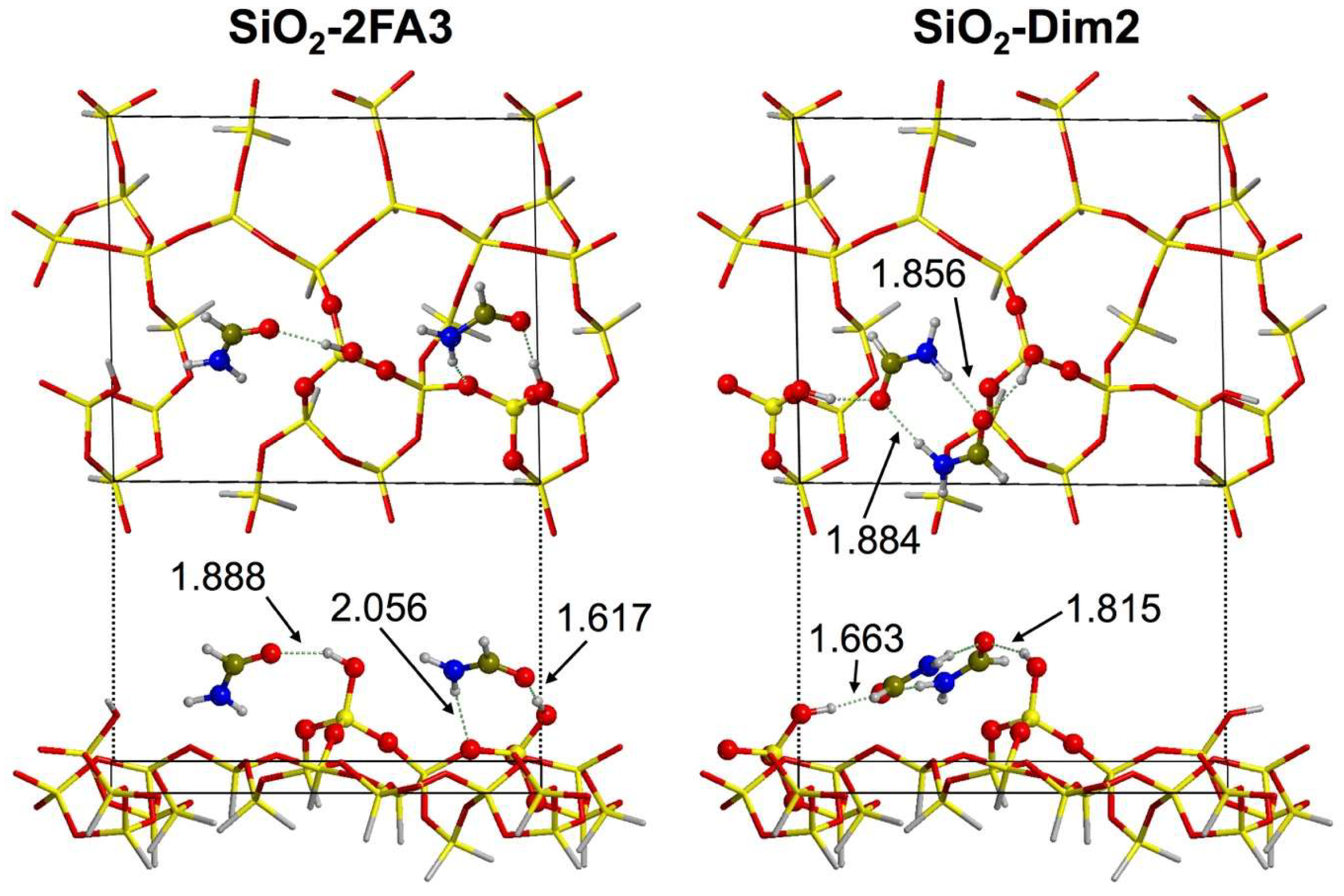
3.2.1. Low Coverage Models
3.2.2. High-Coverage Models
3.3. Simulation of FTIR Spectra of Formamide-A50 Adducts: Comparison with Experimental Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M. Reinventing chemistry. Angew. Chem. Int. Ed. 2015, 54, 3196–3209. [Google Scholar] [CrossRef] [PubMed]
- Kahane, C.; Ceccarelli, C.; Faure, A.; Caux, E. Detection of formamide, the simplest but crucial amide, in a solar-type protostar. Astrophys. J. 2013, 763, L38. [Google Scholar] [CrossRef]
- Saladino, R.; Crestini, C.; Neri, V.; Brucato, J.R.; Colangeli, L.; Ciciriello, F.; Di Mauro, E.; Costanzo, G. Synthesis and Degradation of Nucleic Acid Components by Formamide and Cosmic Dust Analogues. ChemBioChem 2005, 6, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Aladino, R.; Botta, G.; Bizzarri, B.M.; Di Mauro, E.; Garcia-Ruiz, J.M. A Global Scale Scenario for Prebiotic Chemistry: Silica-Based Self-Assembled Mineral Structures and Formamide. Biochemistry 2016, 55, 2806–2811. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Costanzo, G.; DiMauro, E. Advances in the prebiotic synthesis of nucleic acids bases: Implications for the origin of life. Curr. Org. Chem. 2004, 8, 1425–1443. [Google Scholar] [CrossRef]
- Rotelli, L.; Trigo-Rodríguez, J.M.; Moyano-Cambero, C.E.; Carota, E.; Botta, L.; Di Mauro, E.; Saladino, R. The key role of meteorites in the formation of relevant prebiotic molecules in a formamide/water environment. Sci. Rep. 2016, 6, 38888. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, S.D.; Idriss, H. Photocatalysis and the origin of life: Synthesis of nucleoside bases from formamide on TiO2(001) single surfaces. Proc. Natl. Acad. Sci. USA 2006, 103, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Neri, V.; Ciciriello, F.; Costanzo, G.; Di Mauro, E. Origin of informational polymers: The concurrent roles of formamide and phosphates. ChemBioChem 2006, 7, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Pino, S.; Costanzo, G.; Di Mauro, E. Formamide and the origin of life. Phys. Life Rev. 2012, 9, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Costanzo, G.; Negri, R.; Di Mauro, E. A possible prebiotic synthesis of purine, adenine, cytosine, and 4(3H)-pyrimidinone from formamide: Implications for the origin of life. Bioorg. Med. Chem. 2001, 9, 1249–1253. [Google Scholar] [CrossRef]
- Muir, J.M.R.; Idriss, H. Formamide reactions on rutile TiO2(011) surface. Surf. Sci. 2009, 603, 2986–2990. [Google Scholar] [CrossRef]
- Ferus, M.; Nesvorný, D.; Šponer, J.; Kubelík, P.; Michal, R.; Shestivská, V.; Sponer, J.E.; Civiš, S. High-energy chemistry of formamide: A unified mechanism of nucleobase formation. Proc. Natl. Acad. Sci. USA 2015, 112, 657–662. [Google Scholar]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef] [PubMed]
- Rimola, A.; Fabbiani, M.; Sodupe, M.; Ugliengo, P.; Martra, G. How Does Silica Catalyze the Amide Bond Formation under Dry Conditions? Role of Specific Surface Silanol Pairs. ACS Catal. 2018, 8, 4558–4568. [Google Scholar] [CrossRef]
- Lambert, J.F. Adsorption and polymerization of amino acids on mineral surfaces: A review. Orig. Life Evol. Biosph. 2008, 38, 211–242. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-F.; Jaber, M.; Georgelin, T.; Stievano, L. A comparative study of the catalysis of peptide bond formation by oxide surfaces. Phys. Chem. Chem. Phys. 2013, 15, 13371–13380. [Google Scholar] [CrossRef] [PubMed]
- Martra, G.; Deiana, C.; Sakhno, Y.; Barberis, I.; Fabbiani, M.; Pazzi, M.; Vincenti, M. The formation and self-assembly of long prebiotic oligomers produced by the condensation of unactivated amino acids on oxide surfaces. Angew. Chem. Int. Ed. 2014, 53, 4671–4674. [Google Scholar] [CrossRef] [PubMed]
- Corno, M.; Delle Piane, M.; Monti, S.; Moreno-Couranjou, M.; Choquet, P.; Ugliengo, P. Computational Study of Acidic and Basic Functionalized Crystalline Silica Surfaces as a Model for Biomaterial Interfaces. Langmuir 2015, 31, 6321–6331. [Google Scholar] [CrossRef] [PubMed]
- Musso, F.; Sodupe, M.; Corno, M.; Ugliengo, P. H-bond features of fully hydroxylated surfaces of crystalline silica polymorphs: A periodic B3LYP study. J. Phys. Chem. C 2009, 113, 17876–17884. [Google Scholar] [CrossRef]
- Garofalini, S.H. Molecular dynamics computer simulations of silica surface structure and adsorption of water molecules. J. Non-Cryst. Solids 1990, 120, 1–12. [Google Scholar] [CrossRef]
- Tosoni, S.; Civalleri, B.; Pascale, F.; Ugliengo, P. Hydroxylated crystalline edingtonite silica faces as models for the amorphous silica surface. J. Phys. Conf. Ser. 2008, 117. [Google Scholar] [CrossRef]
- Tielens, F.; Gervais, C.; Lambert, J.F.; Mauri, F.; Costa, D. Ab initio study of the hydroxylated surface of amorphous silica: A representative model. Chem. Mater. 2008, 20, 3336–3344. [Google Scholar] [CrossRef]
- Ugliengo, P.; Sodupe, M.; Musso, F.; Bush, I.J.; Orlando, R.; Dovesi, R. Realistic models of hydroxylated amorphous silica surfaces and MCM-41 mesoporous material simulated by large-scale periodic B3LYP calculations. Adv. Mater. 2008, 20, 4579–4583. [Google Scholar] [CrossRef]
- Delle Piane, M.; Corno, M.; Ugliengo, P. Does dispersion dominate over H-bonds in drug-surface interactions? The case of silica-based materials as excipients and drug-delivery agents. J. Chem. Theory Comput. 2013, 9, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Mardyukov, A.; Sánchez-Garcia, E.; Rodziewicz, P.; Doltsinis, N.L.; Sander, W. Formamide dimers: A computational and matrix isolation study. J. Phys. Chem. A 2007, 111, 10552–10561. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, S.; Puzzarini, C. Structural and Energetic Characterization of Prebiotic Molecules: The Case Study of Formamide and Its Dimer. J. Phys. Chem. A 2016, 120, 5257–5263. [Google Scholar] [CrossRef] [PubMed]
- Erba, A.; Baima, J.; Bush, I.; Orlando, R.; Dovesi, R. Large-Scale Condensed Matter DFT Simulations: Performance and Capabilities of the CRYSTAL Code. J. Chem. Theory Comput. 2017, 13, 5019–5027. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Nada, R.; Nicholas, J.B.; McCarthy, M.I.; Hess, A.C. Basis sets for ab initio periodic Hartree-Fock studies of zeolite/adsorbate interactions: He, Ne, and Ar in silica sodalite. Int. J. Quantum Chem. 1996, 60, 809–820. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Antony, J.; Grimme, S. Density functional theory including dispersion corrections for intermolecular interactions in a large benchmark set of biologically relevant molecules. Phys. Chem. Chem. Phys. 2006, 8, 5287–5293. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K. CRYSTAL17 User’s Manual. 2017. Available online: http://www.crystal.unito.it/Manuals/crystal17.pdf (accessed on 20 September 2018).
- Lendvay, G.; Mayer, I. Some difficulties in computing BSSE-corrected potential surfaces of chemical reactions. Chem. Phys. Lett. 1998, 297, 365–373. [Google Scholar] [CrossRef]
- Sugawara, Y.; Hamada, Y.; Masamichi, T. Vibration-rotation Spectra of Formamide. Bull. Chem. Soc. Jpn. 1983, 56, 1045–1050. [Google Scholar] [CrossRef]
- Crocellà, V.; Cerrato, G.; Magnacca, G.; Morterra, C. Adsorption of Acetone on Nonporous and Mesoporous Silica. J. Phys. Chem. C 2009, 113, 16517–16529. [Google Scholar] [CrossRef]
- Crocellà, V.; Cerrato, G.; Morterra, C. On the adsorption/reaction of acetone on pure and sulfate-modified zirconias. Phys. Chem. Chem. Phys. 2013, 15, 13446. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, C.; Zecchina, A.; Groppo, E.; Bordiga, S. Probing the surfaces of heterogeneous catalysts by in situ IR spectroscopy. Chem. Soc. Rev. 2010, 39, 4951. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | ΔEc (ΔE disp) | ΔHc | ΔGc | p |
|---|---|---|---|---|
| SiO2-FA1 | −69.9 (−18.8) | −62.8 | −13.3 | 0.03 |
| SiO2-FA2 | −44.0 (−28.1) | −38.1 | 8.6 | 0.00 |
| SiO2-FA3 | −68.0 (−26.4) | −60.7 | −9.6 | 0.01 |
| SiO2-FA4 | −80.5 (−27.4) | −74.2 | −21.3 | 0.67 |
| SiO2-FA5 | −63.3 (−15.6) | −56.8 | −19.3 | 0.30 |
| Model | ΔEc (ΔE disp) | ΔHc | ΔGc | p |
|---|---|---|---|---|
| SiO2-2FA1 | −57.6 (−28.4) | −50.1 | −1.7 | 0.00 |
| SiO2-2FA2 | −56.4 (−20.4) | −49.2 | −6.3 | 0.01 |
| SiO2-2FA3 | −67.3 (−16.9) | −59.9 | −17.9 | 0.91 |
| SiO2-Dim1 | −46.8 (−24.2) | −39.9 | 7.2 | 0.00 |
| SiO2-Dim2 | −73.5 (−24.1) | −64.7 | −12.0 | 0.08 |
| SiO2-Dim3 | −51.7 (−19.3) | −43.4 | 2.3 | 0.00 |
| SiO2-Dim4 | −58.5 (−21.3) | −49.4 | −1.5 | 0.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorile, M.; Salvini, C.; Zamirri, L.; Bonino, F.; Martra, G.; Sodupe, M.; Ugliengo, P. Formamide Adsorption at the Amorphous Silica Surface: A Combined Experimental and Computational Approach. Life 2018, 8, 42. https://doi.org/10.3390/life8040042
Signorile M, Salvini C, Zamirri L, Bonino F, Martra G, Sodupe M, Ugliengo P. Formamide Adsorption at the Amorphous Silica Surface: A Combined Experimental and Computational Approach. Life. 2018; 8(4):42. https://doi.org/10.3390/life8040042
Chicago/Turabian StyleSignorile, Matteo, Clara Salvini, Lorenzo Zamirri, Francesca Bonino, Gianmario Martra, Mariona Sodupe, and Piero Ugliengo. 2018. "Formamide Adsorption at the Amorphous Silica Surface: A Combined Experimental and Computational Approach" Life 8, no. 4: 42. https://doi.org/10.3390/life8040042
APA StyleSignorile, M., Salvini, C., Zamirri, L., Bonino, F., Martra, G., Sodupe, M., & Ugliengo, P. (2018). Formamide Adsorption at the Amorphous Silica Surface: A Combined Experimental and Computational Approach. Life, 8(4), 42. https://doi.org/10.3390/life8040042