Dissecting the Repertoire of DNA-Binding Transcription Factors of the Archaeon Pyrococcus furiosus DSM 3638

 ,
,
Abstract
1. Introduction
2. Methods
2.1. Identification of DNA-Binding TFs
2.2. Structural Diversity Associated with TFs
2.3. Functional Classes of the Regulated Genes
2.4. Identification of DNA-Binding Sites
2.5. Enrichment Analysis of Functional Annotations
3. Results
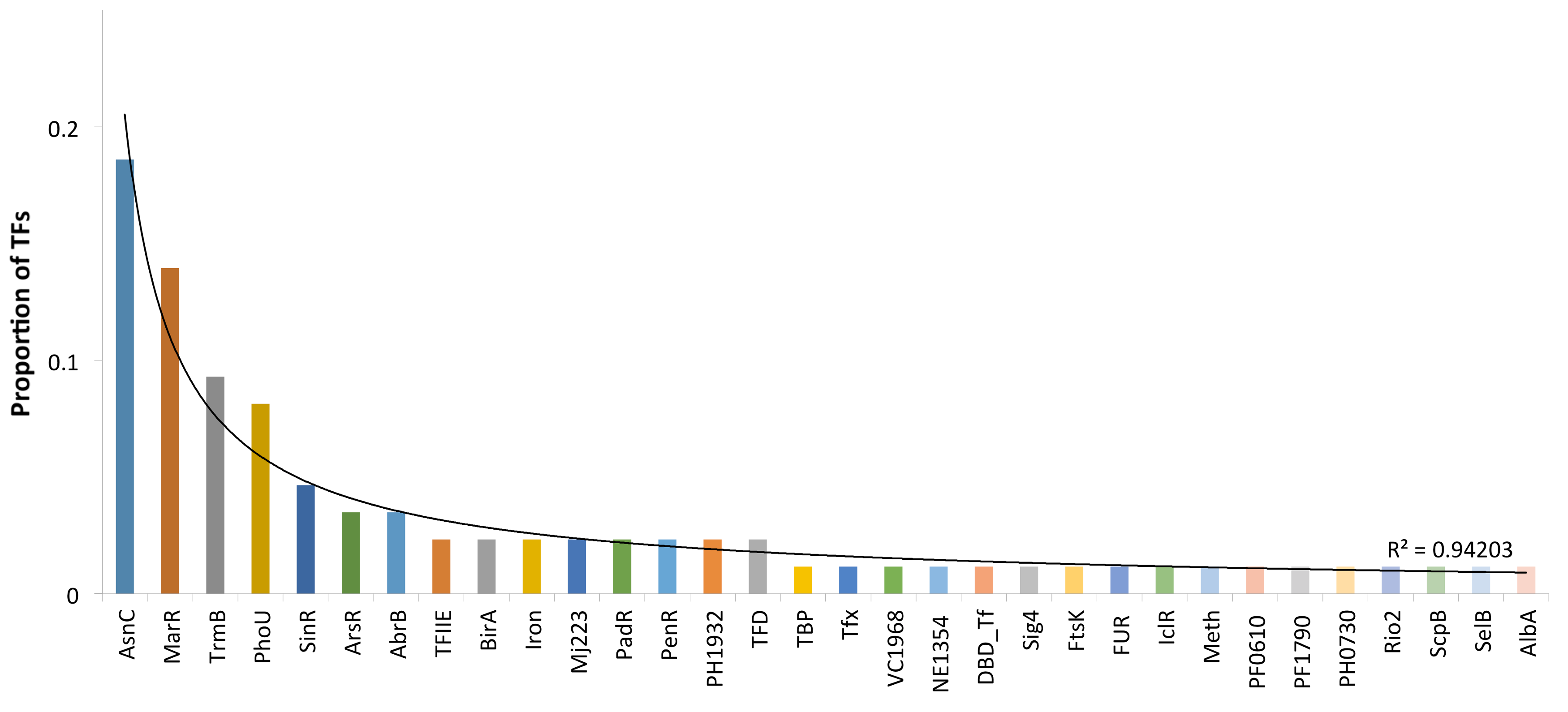
3.1. Distribution and Domain Architecture of TFs
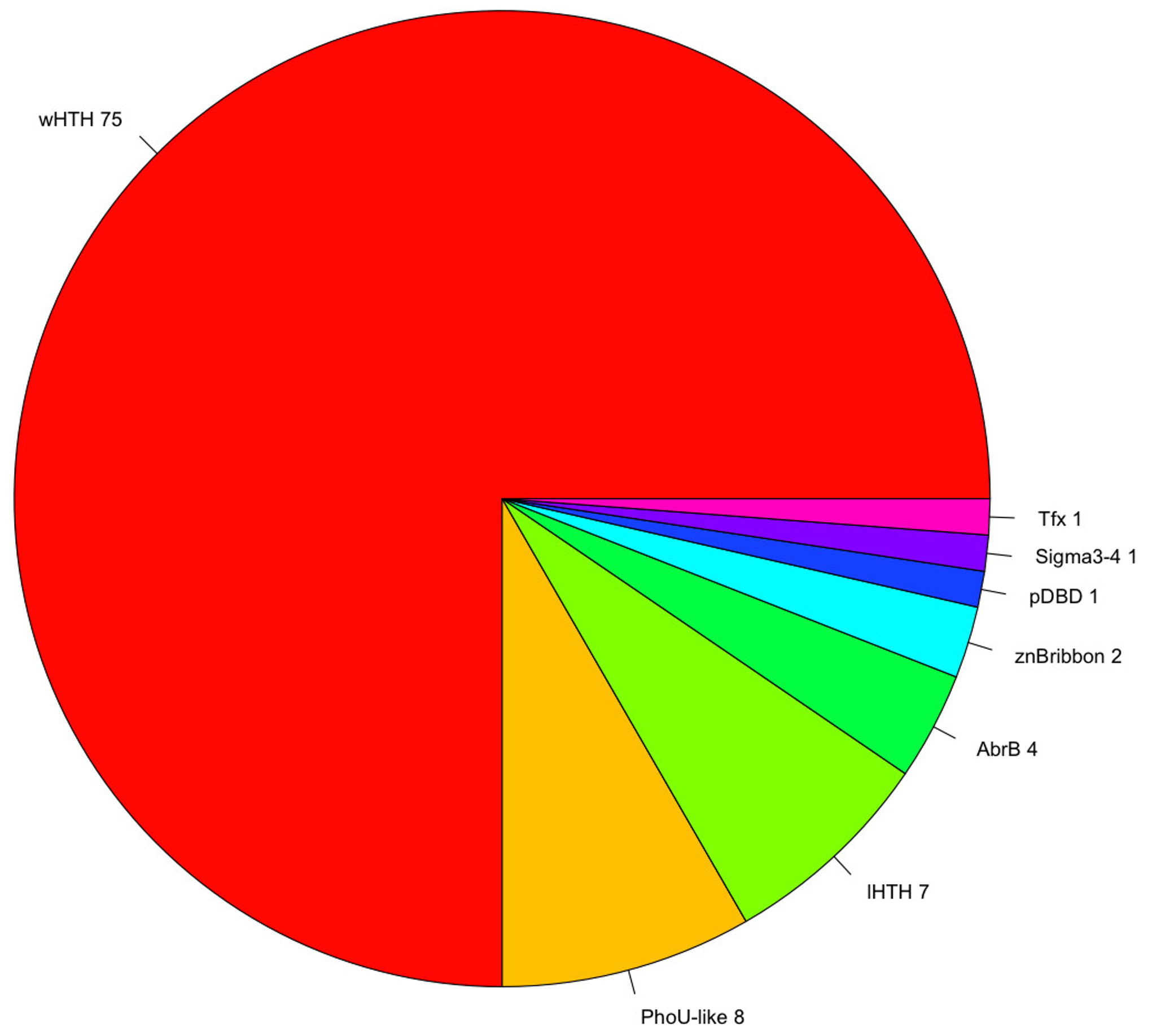
3.2. Protein Domain and Regulatory Families Associated with the Repertoire of TFs
3.3. Regulons Identified
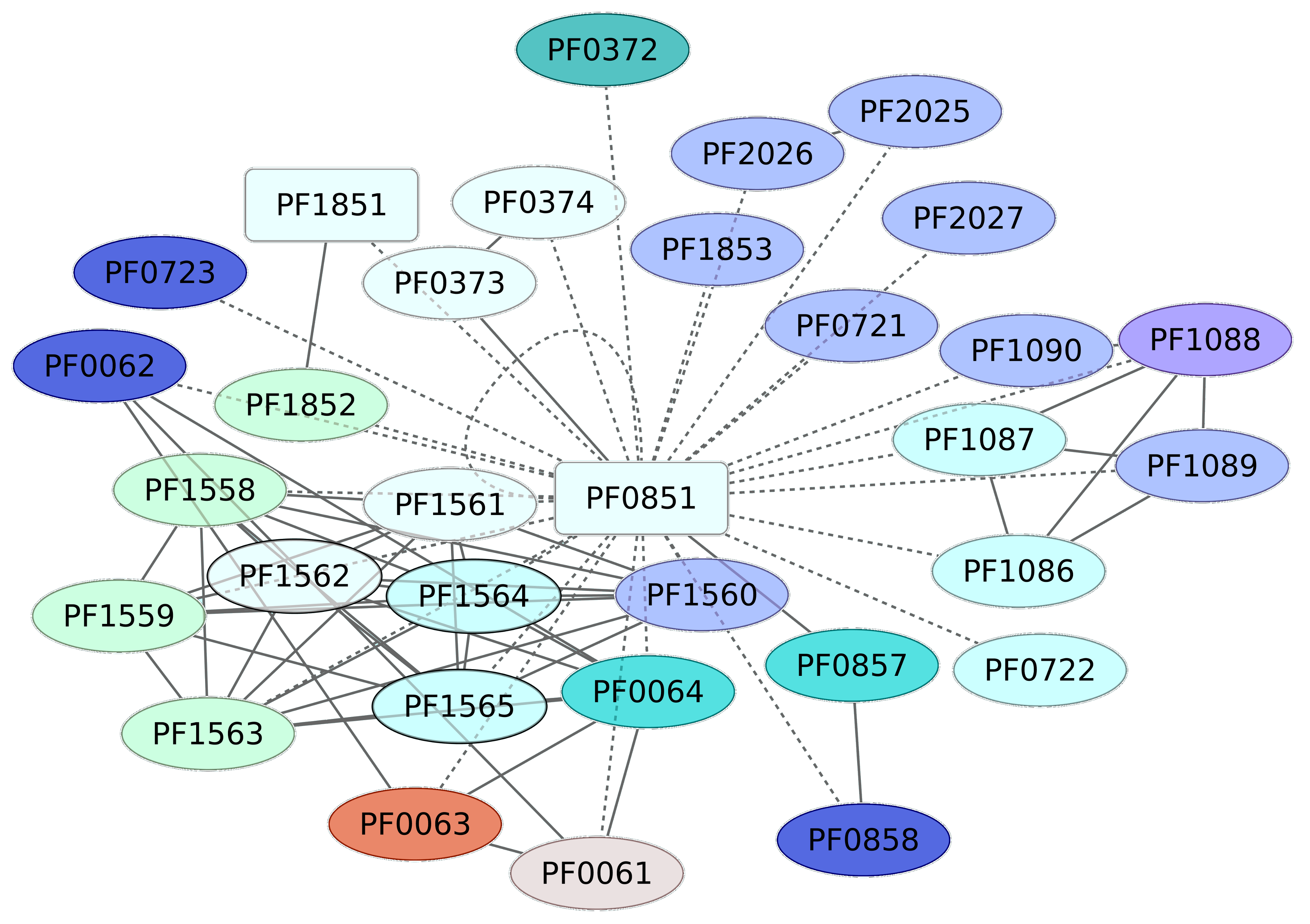
3.4. Iron-Dependent Repressor Regulon (PF0851)
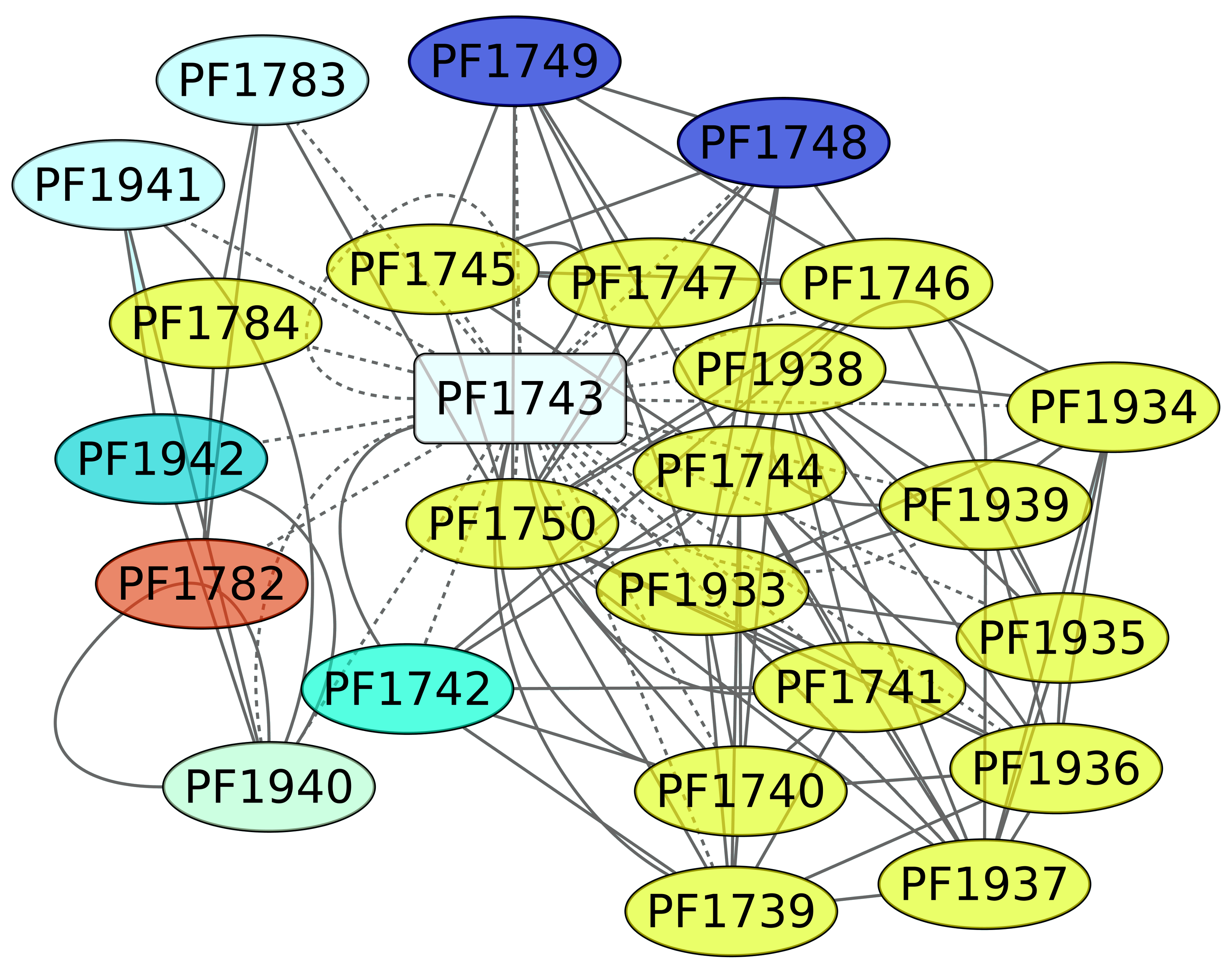
3.5. Maltose and Trehalose Metabolism (PF1743)
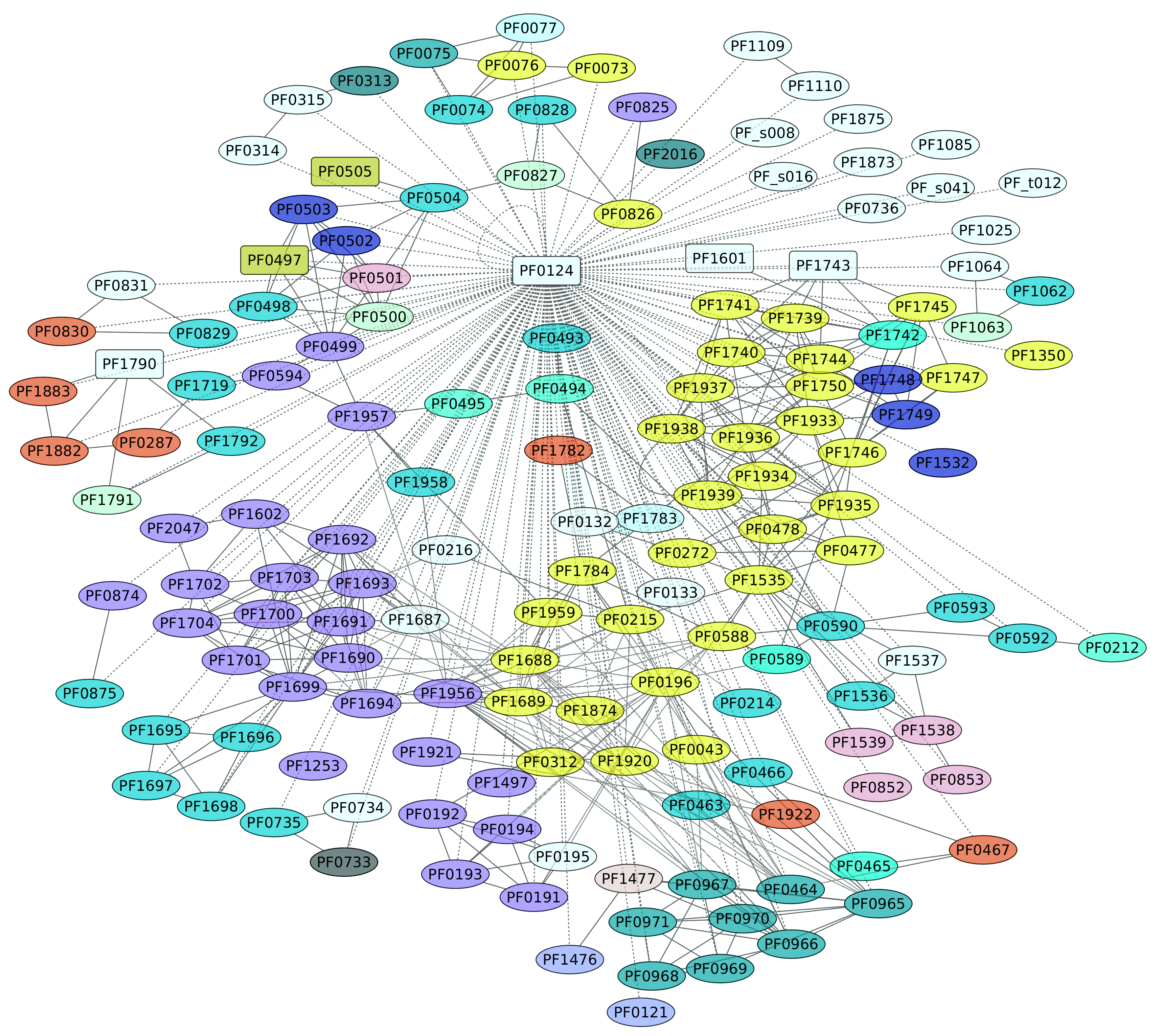
3.6. TrmBL1 Regulon (PF0124)
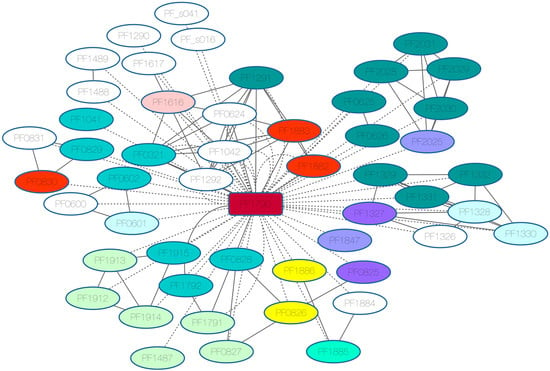
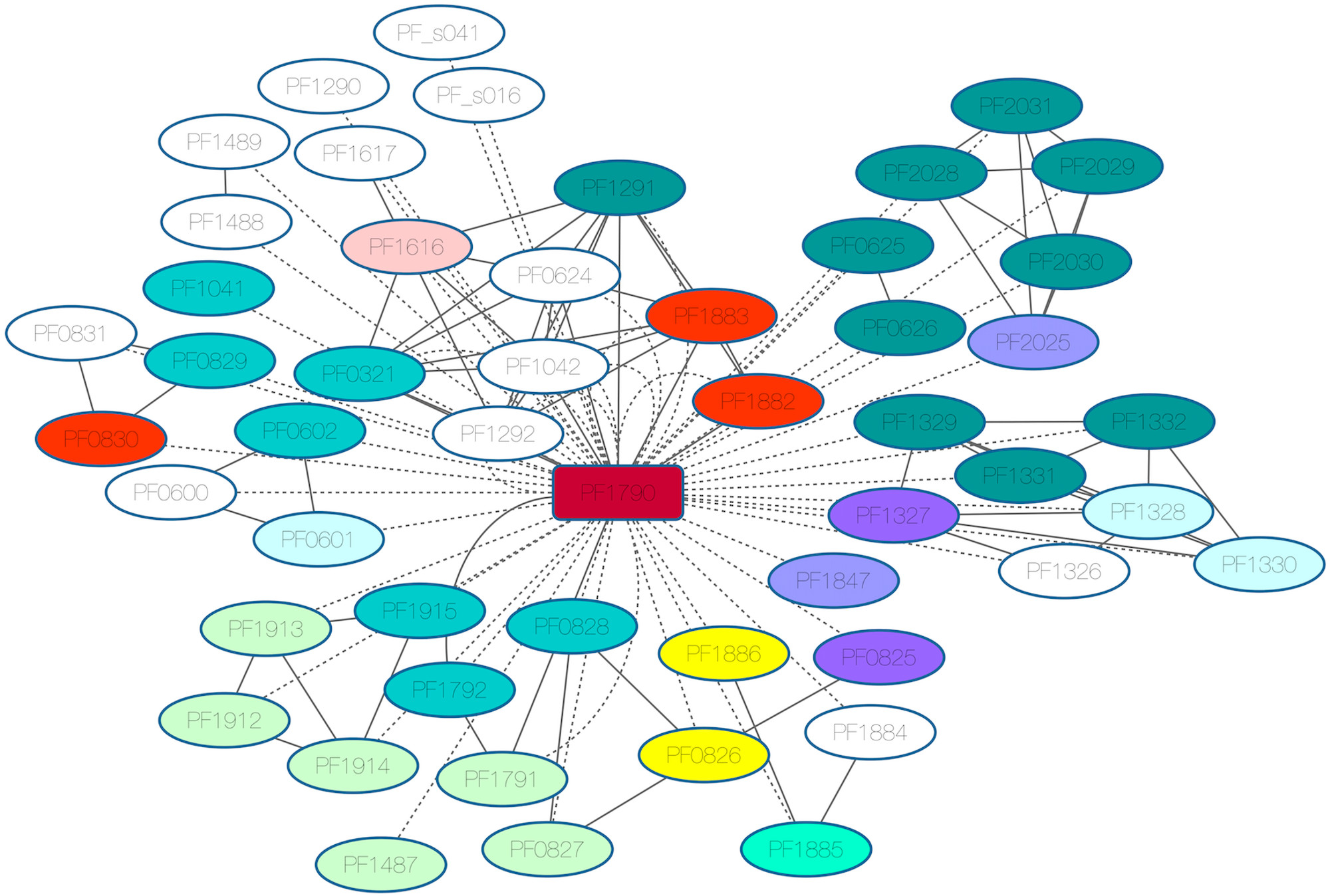
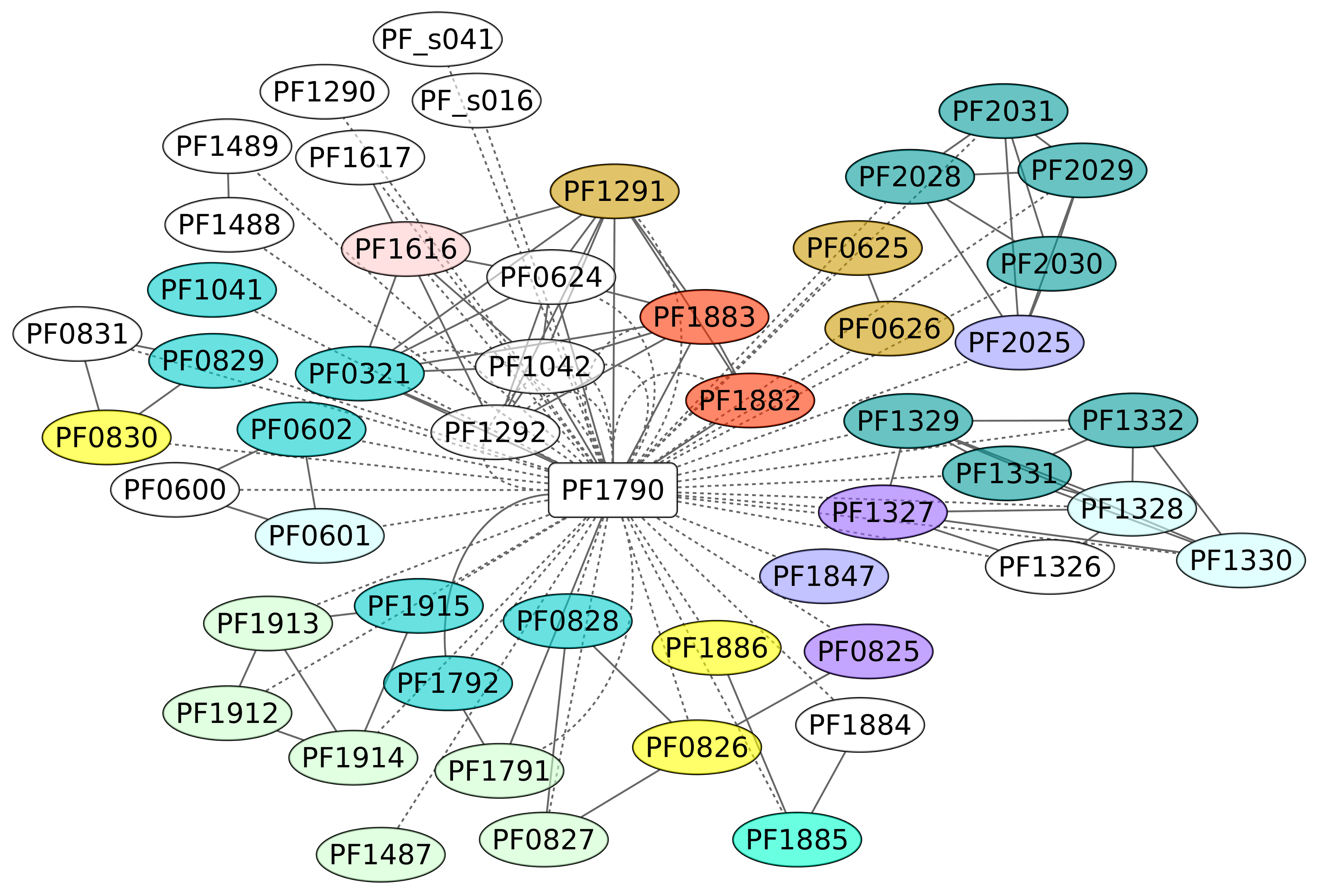
3.7. Phr Regulon (PF1790)
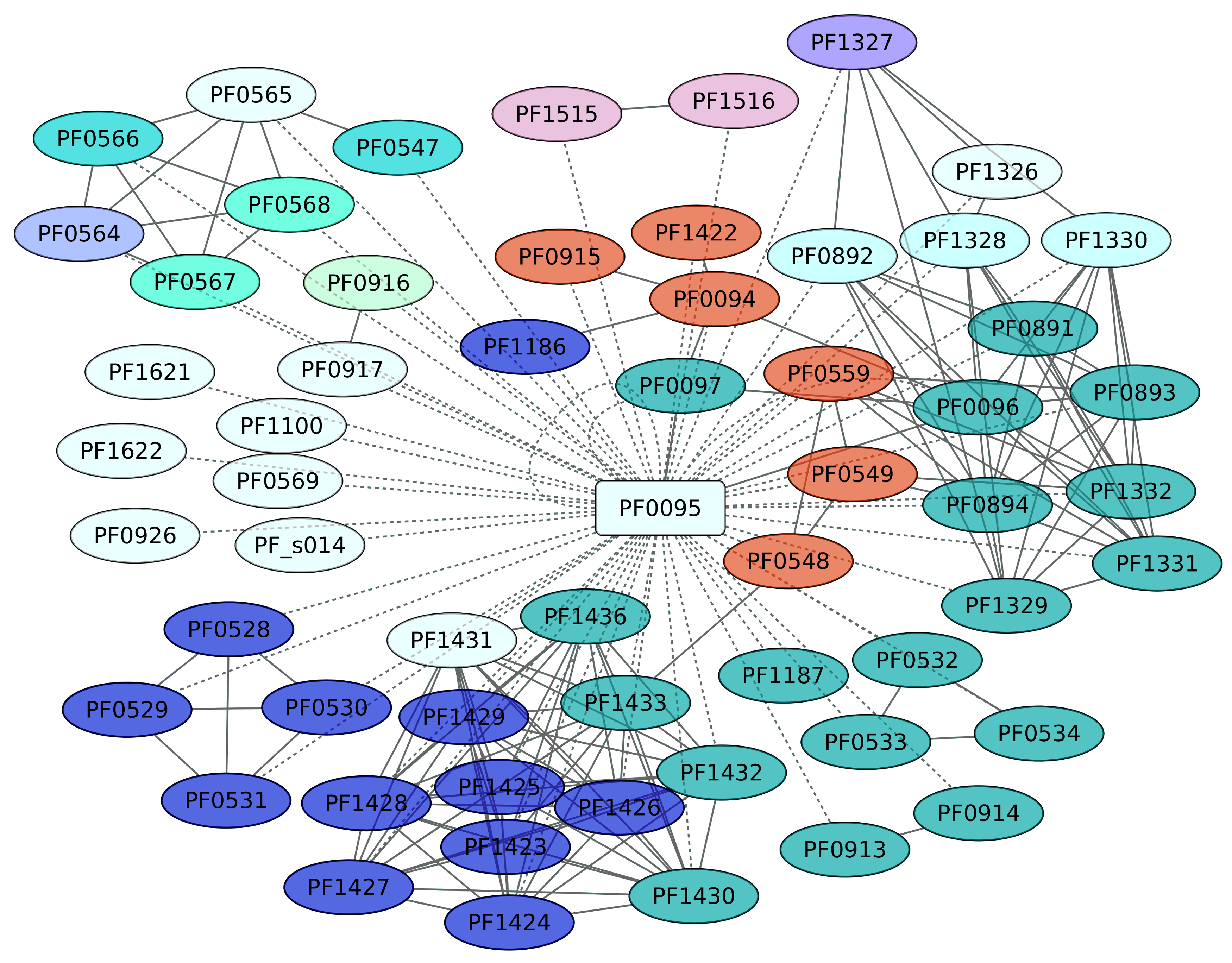
3.8. Sur Regulon (PF0095)
3.9. LrpA Regulon (PF1601)
3.10. TBP (PF1295)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Auguet, J.C.; Barberan, A.; Casamayor, E.O. Global ecological patterns in uncultured Archaea. ISME J. 2010, 4, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Chaban, B.; Ng, S.Y.; Jarrell, K.F. Archaeal habitats—From the extreme to the ordinary. Can. J. Microbiol. 2006, 52, 73–116. [Google Scholar] [CrossRef] [PubMed]
- Clementino, M.M.; Fernandes, C.C.; Vieira, R.P.; Cardoso, A.M.; Polycarpo, C.R.; Martins, O.B. Archaeal diversity in naturally occurring and impacted environments from a tropical region. J. Appl. Microbiol. 2007, 103, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.D.; Chang, H.W.; Kim, K.H.; Roh, S.W.; Kim, M.S.; Jung, M.J.; Lee, S.W.; Kim, J.Y.; Yoon, J.H.; Bae, J.W. Bacterial, archaeal, and eukaryal diversity in the intestines of Korean people. J. Microbiol. 2008, 46, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.S. Determinants of transcription initiation by archaeal RNA polymerase. Curr. Opin. Microbiol. 2005, 8, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.M.; Walker, J.E.; Santangelo, T.J. Transcription Regulation in Archaea. J. Bacteriol. 2016, 198, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Soppa, J. Normalized nucleotide frequencies allow the definition of archaeal promoter elements for different archaeal groups and reveal base-specific TFB contacts upstream of the TATA box. Mol. Microbiol. 1999, 31, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- De Carlo, S.; Lin, S.C.; Taatjes, D.J.; Hoenger, A. Molecular basis of transcription initiation in Archaea. Transcription 2010, 1, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Goede, B.; Naji, S.; von Kampen, O.; Ilg, K.; Thomm, M. Protein-protein interactions in the archaeal transcriptional machinery: Binding studies of isolated RNA polymerase subunits and transcription factors. J. Biol. Chem. 2006, 281, 30581–30592. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.D.; Kosa, P.L.; Sigler, P.B.; Jackson, S.P. Orientation of the transcription preinitiation complex in archaea. Proc. Natl. Acad. Sci. USA 1999, 96, 13662–13667. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.D.; Jackson, S.P. Mechanism and regulation of transcription in archaea. Curr. Opin. Microbiol. 2011, 4, 208–213. [Google Scholar] [CrossRef]
- Bell, S.D. Archaeal transcriptional regulation—Variation on a bacterial theme? Trends Microbiol. 2005, 13, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Kyrpides, N.C.; Woese, C.R. Universally conserved translation initiation factors. Proc. Natl. Acad. Sci. USA 1998, 95, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Coulson, R.M.; Touboul, N.; Ouzounis, C.A. Lineage-specific partitions in archaeal transcription. Archaea 2007, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, S.; Bai, J.; Shi, L.; Li, D.; Xu, Z.; Niu, Y.; Lu, J.; Bao, Q. ArchaeaTF: An integrated database of putative transcription factors in Archaea. Genomics 2008, 91, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Facciotti, M.T.; Reiss, D.J.; Schmid, A.K.; Pan, M.; Kaur, A.; Thorsson, V.; Shannon, P.; Johnson, M.H.; Bare, J.C.; et al. A predictive model for transcriptional control of physiology in a free living cell. Cell 2007, 131, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Turkarslan, S.; Reiss, D.J.; Pan, M.; Burn, J.A.; Costa, K.C.; Lie, T.J.; Slagel, J.; Moritz, R.L.; Hackett, M.; et al. A systems level predictive model for global gene regulation of methanogenesis in a hydrogenotrophic methanogen. Genome Res. 2013, 23, 1839–1851. [Google Scholar] [CrossRef] [PubMed]
- Leonard, P.M.; Smits, S.H.; Sedelnikova, S.E.; Brinkman, A.B.; de Vos, W.M.; van der Oost, J.; Rice, DW.; Rafferty, J.B. Crystal structure of the Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus. EMBO J. 2001, 20, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Napoli, A.; van der Oost, J.; Sensen, C.W.; Charlebois, R.L.; Rossi, M.; Ciaramella, M. An Lrp-like protein of the hyperthermophilic archaeon Sulfolobus solfataricus which binds to its own promoter. J. Bacteriol. 1999, 181, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Machielsen, R.; Leferink, N.G.; Hendriks, A.; Brouns, S.J.; Hennemann, H.G.; Daussmann, T.; van der Oost, J. Laboratory evolution of Pyrococcus furiosus alcohol dehydrogenase to improve the production of (2S,5S)-hexanediol at moderate temperatures. Extremophiles 2008, 12, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Tenorio-Salgado, S.; Huerta-Saquero, A.; Perez-Rueda, E. New insights on gene regulation in archaea. Comput. Biol. Chem. 2011, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Ortet, P.; De Luca, G.; Whitworth, D.E.; Barakat, M. P2TF: A comprehensive resource for analysis of prokaryotic transcription factors. BMC Genom. 2012, 13, 628. [Google Scholar] [CrossRef] [PubMed]
- Kummerfeld, S.K.; Teichmann, S.A. DBD: A transcription factor prediction database. Nucleic Acids Res. 2006, 34, D74–D81. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Anderson, J.B.; Derbyshire, M.K.; DeWeese-Scott, C.; Gonzales, N.R.; Gwadz, M.; Hao, L.; He, S.; Hurwitz, D.I.; Jackson, J.D.; et al. CDD: A conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007, 35, D237–D240. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rueda, E.; Tenorio-Salgado, S.; Huerta-Saquero, A.; Balderas-Martinez, Y.I.; Moreno-Hagelsieb, G. The functional landscape bound to the transcription factors of Escherichia coli K-12. Comput. Biol. Chem. 2015, 58, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY--sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009, 37, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Gough, J.; Chothia, C. SUPERFAMILY: HMMs representing all proteins of known structure SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002, 30, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Makarova, K.S.; Yutin, N.; Koonin, E.V. Updated clusters of orthologous genes for Archaea: A complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol. Direct 2012, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Madan Babu, M.; Teichmann, S.A. Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res. 2003, 31, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Demolli, S.; Geist, M.M.; Weigand, J.E.; Matschiavelli, N.; Suess, B.; Rother, M. Development of beta -lactamase as a tool for monitoring conditional gene expression by a tetracycline-riboswitch in Methanosarcina acetivorans. Archaea 2014, 2014, 725610. [Google Scholar] [CrossRef] [PubMed]
- Kravatskaya, G.I.; Kravatsky, Y.V.; Chechetkin, V.R.; Tumanyan, V.G. Coexistence of different base periodicities in prokaryotic genomes as related to DNA curvature, supercoiling, and transcription. Genomics 2011, 98, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Madera, M.; Vogel, C.; Kummerfeld, S.K.; Chothia, C.; Gough, J. The SUPERFAMILY database in 2004: Additions and improvements. Nucleic Acids Res. 2004, 32, D235–D239. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, L.E.; Koonin, E.V.; Zhulin, I.B. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 2005, 13, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Charoensawan, V.; Wilson, D.; Teichmann, S.A. Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res. 2010, 38, 7364–7377. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.M.; Iyer, L.M. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbial. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res. 1999, 27, 4658–4670. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, J.A.; Perez-Rueda, E.; Carroll, R.K.; Shaw, L.N. Global analysis of transcriptional regulators in Staphylococcus aureus. BMC Genom. 2013, 14, 126. [Google Scholar] [CrossRef] [PubMed]
- Balderas-Martinez, Y.I.; Savageau, M.; Salgado, H.; Perez-Rueda, E.; Morett, E.; Collado-Vides, J. Transcription factors in Escherichia coli prefer the holo conformation. PLoS ONE 2013, 8, e65723. [Google Scholar] [CrossRef] [PubMed]
- Gama-Castro, S.; Salgado, H.; Santos-Zavaleta, A.; Ledezma-Tejeida, D.; Muniz-Rascado, L.; Garcia-Sotelo, J.S.; Alquicira-Hernandez, K.; Martinez-Flores, I.; Pannier, L.; Castro-Mondragon, J.A.; et al. RegulonDB version 9.0: High-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016, 44, D133–D143. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rueda, E.; Janga, S.C. Identification and genomic analysis of transcription factors in archaeal genomes exemplifies their functional architecture and evolutionary origin. Mol. Biol. Evol. 2010, 27, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Yanai, I.; Camacho, C.J.; DeLisi, C. Predictions of gene family distributions in microbial genomes: Evolution by gene duplication and modification. Phys. Rev. Lett. 2000, 85, 2641–2644. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.C.; Groisman, E.A. Evolution of transcriptional regulatory circuits in bacteria. Cell 2009, 138, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.C.; Groisman, E.A. Transcription factor function and promoter architecture govern the evolution of bacterial regulons. Proc. Natl. Acad. Sci. USA 2009, 106, 4319–4324. [Google Scholar] [CrossRef] [PubMed]
- Taboada, B.; Ciria, R.; Martinez-Guerrero, C.E.; Merinom, E. ProOpDB: Prokaryotic Operon DataBase. Nucleic Acids Res. 2012, 40, D627–D631. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Surma, M.; Seitz, S.; Hausner, W.; Thomm, M.; Boos, W. Characterization of the TrmB-like protein, PF0124, a TGM-recognizing global transcriptional regulator of the hyperthermophilic archaeon Pyrococcus furiosus. Mol. Microbiol. 2007, 65, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Van de Werken, H.J.; Verhees, C.H.; Akerboom, J.; de Vos, W.M.; van der Oost, J. Identification of a glycolytic regulon in the archaea Pyrococcus and Thermococcus. FEMS Microbial. Lett. 2006, 260, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Moulakakis, C.; Koning, S.M.; Hausner, W.; Thomm, M.; Boos, W. TrmB, a sugar sensing regulator of ABC transporter genes in Pyrococcus furiosus exhibits dual promoter specificity and is controlled by different inducers. Mol. Microbiol. 2005, 57, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Surma, M.; Seitz, S.; Hausner, W.; Thomm, M.; Boos, W. Differential signal transduction via TrmB, a sugar sensing transcriptional repressor of Pyrococcus furiosus. Mol. Microbiol. 2007, 64, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Vierke, G.; Engelmann, A.; Hebbeln, C.; Thomm, M. A novel archaeal transcriptional regulator of heat shock response. J. Biol. Chem. 2003, 278, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Vierke, G.; Wenke, A.K.; Thomm, M.; Ladenstein, R. Crystal structure of the archaeal heat shock regulator from Pyrococcus furiosus: A molecular chimera representing eukaryal and bacterial features. J. Mol. Biol. 2007, 369, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Santos-Zavaleta, A.; Gama-Castro, S.; Perez-Rueda, E. A comparative genome analysis of the RpoS sigmulon shows a high diversity of responses and origins. Microbiology 2011, 157, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Keese, A.M.; Schut, G.J.; Ouhammouch, M.; Adams, M.W.; Thomm, M. Genome-wide identification of targets for the archaeal heat shock regulator phr by cell-free transcription of genomic DNA. J. Bacteriol. 2010, 192, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lipscomb, G.L.; Keese, A.M.; Schut, G.J.; Thomm, M.; Adams, M.W.; Wang, B.C.; Scott, R.A. SurR regulates hydrogen production in Pyrococcus furiosus by a sulfur-dependent redox switch. Mol. Microbiol. 2010, 77, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, G.L.; Keese, A.M.; Cowart, D.M.; Schut, G.J.; Thomm, M.; Adams, M.W.; Scott, R.A. SurR: A transcriptional activator and repressor controlling hydrogen and elemental sulphur metabolism in Pyrococcus furiosus. Mol. Microbiol. 2009, 71, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Dahlke, I.; Tuininga, J.E.; Lammers, T.; Dumay, V.; de Heus, E.; Lebbink, J.H.; Thomm, M.; de Vos, W.M.; van Der Oost, J. An Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus is negatively autoregulated. J. Biol. Chem. 2000, 275, 38160–38169. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, I.; Thomm, M. A Pyrococcus homolog of the leucine-responsive regulatory protein, LrpA, inhibits transcription by abrogating RNA polymerase recruitment. Nucleic Acids Res. 2002, 30, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Hausner, W.; Wettach, J.; Hethke, C.; Thomm, M. Two transcription factors related with the eucaryal transcription factors TATA-binding protein and transcription factor IIB direct promoter recognition by an archaeal RNA polymerase. J. Biol. Chem. 1996, 271, 30144–30148. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
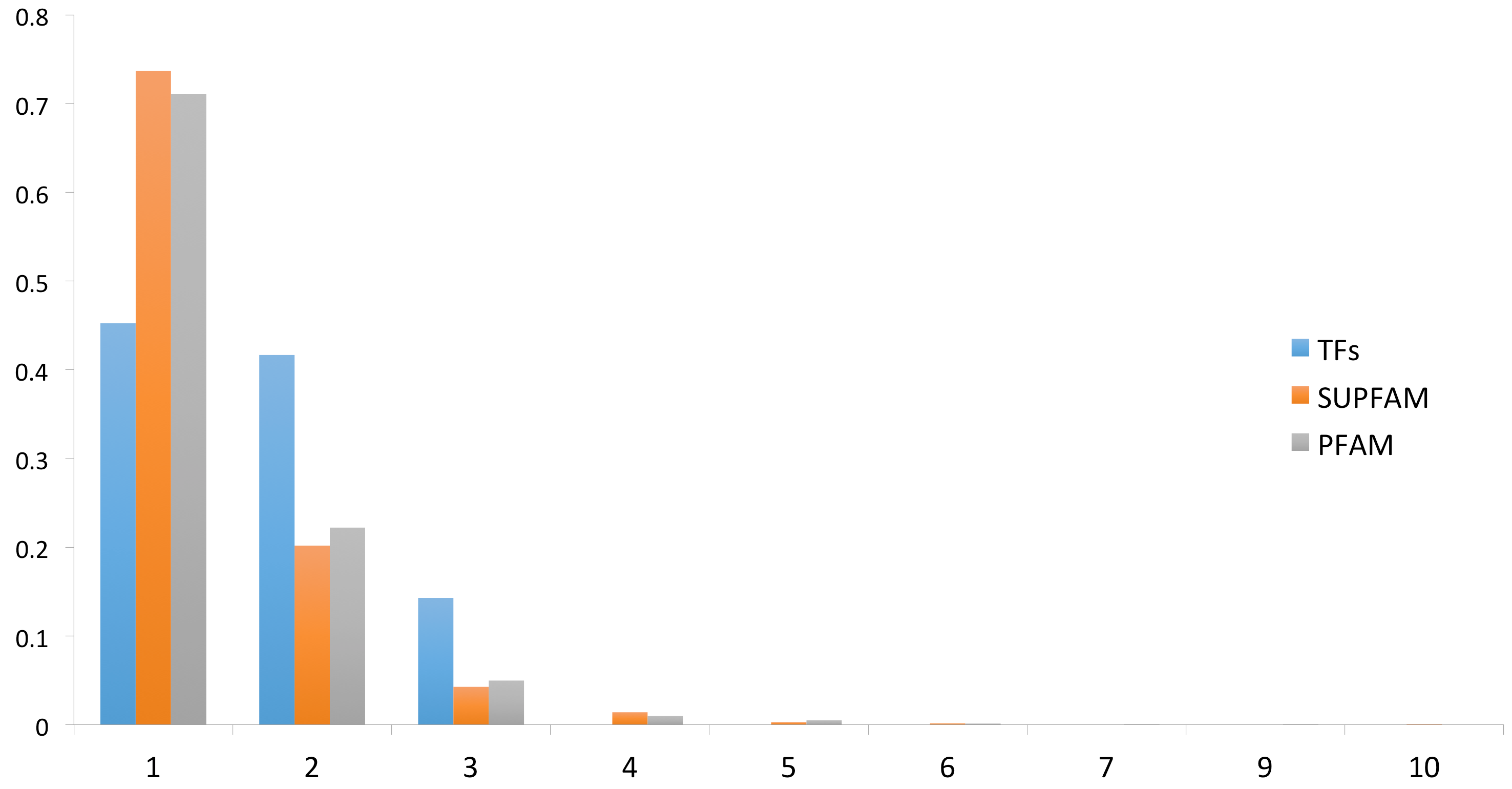{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family(s) | Total of Proteins |
|---|---|---|
| wHTH | AsnC, MarR TrmB, ArsR, TFIIE, PenR, PadR, Iron, PH1932, Mj223, BirA, TFIIE, PenR, PadR, Iron, PH1932, Mj223, BirA, IclR, ScpB, Rio2, PH0730, PF1790, PF0610, Meth, FtsK, Fur, SelB | 63 |
| PhoU-like | PhoU | 7 |
| lHTH | SinR, VC1968, NE1354 | 6 |
| AbrB | AbrB | 3 |
| znBribbon | TFD | 2 |
| pDBD | DBD_Tf | 2 |
| Tfx | Tfx | 1 |
| Sigma3,4 | Sig4 | 1 |
| AlbA | AlbA | 1 |
| TBP | TBP | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denis, A.; Martínez-Núñez, M.A.; Tenorio-Salgado, S.; Perez-Rueda, E. Dissecting the Repertoire of DNA-Binding Transcription Factors of the Archaeon Pyrococcus furiosus DSM 3638. Life 2018, 8, 40. https://doi.org/10.3390/life8040040
Denis A, Martínez-Núñez MA, Tenorio-Salgado S, Perez-Rueda E. Dissecting the Repertoire of DNA-Binding Transcription Factors of the Archaeon Pyrococcus furiosus DSM 3638. Life. 2018; 8(4):40. https://doi.org/10.3390/life8040040
Chicago/Turabian StyleDenis, Antonia, Mario Alberto Martínez-Núñez, Silvia Tenorio-Salgado, and Ernesto Perez-Rueda. 2018. "Dissecting the Repertoire of DNA-Binding Transcription Factors of the Archaeon Pyrococcus furiosus DSM 3638" Life 8, no. 4: 40. https://doi.org/10.3390/life8040040
APA StyleDenis, A., Martínez-Núñez, M. A., Tenorio-Salgado, S., & Perez-Rueda, E. (2018). Dissecting the Repertoire of DNA-Binding Transcription Factors of the Archaeon Pyrococcus furiosus DSM 3638. Life, 8(4), 40. https://doi.org/10.3390/life8040040