
Flexible Proteins at the Origin of Life


Abstract
:1. Introduction
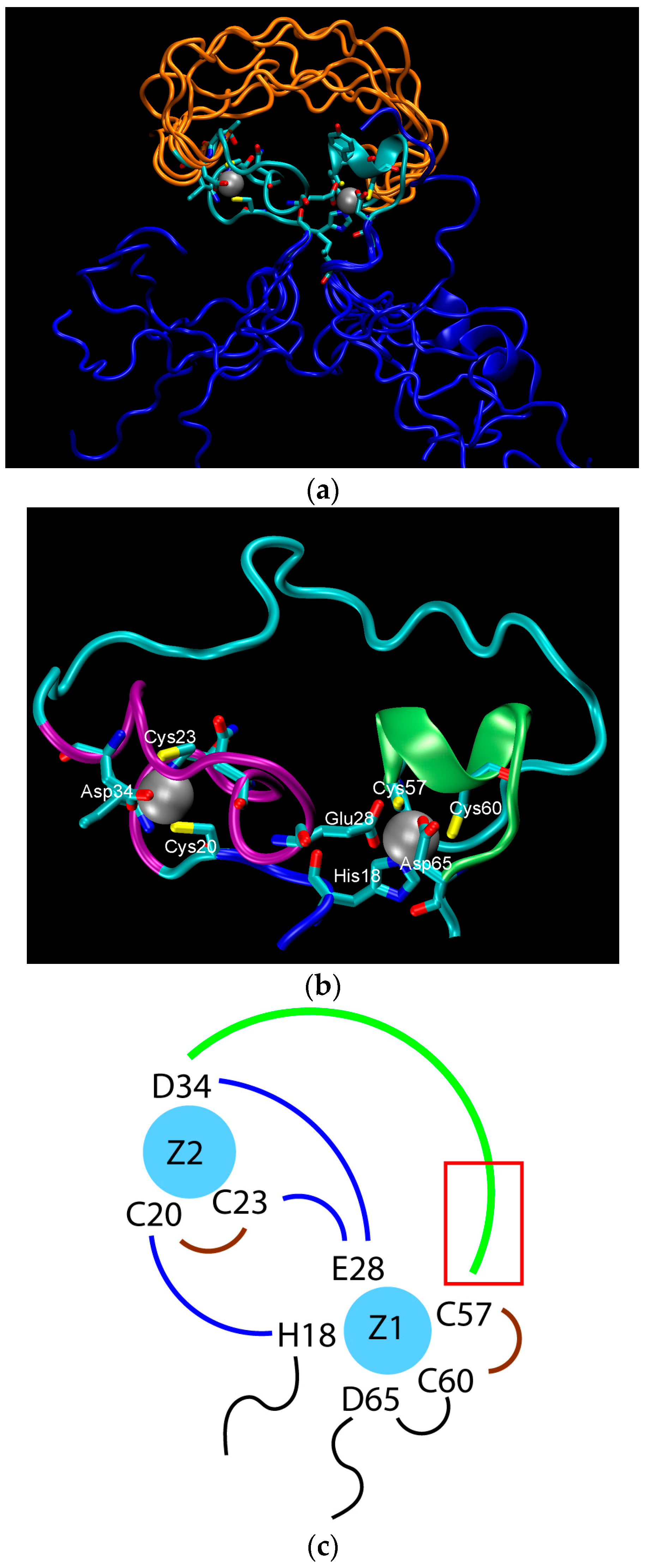
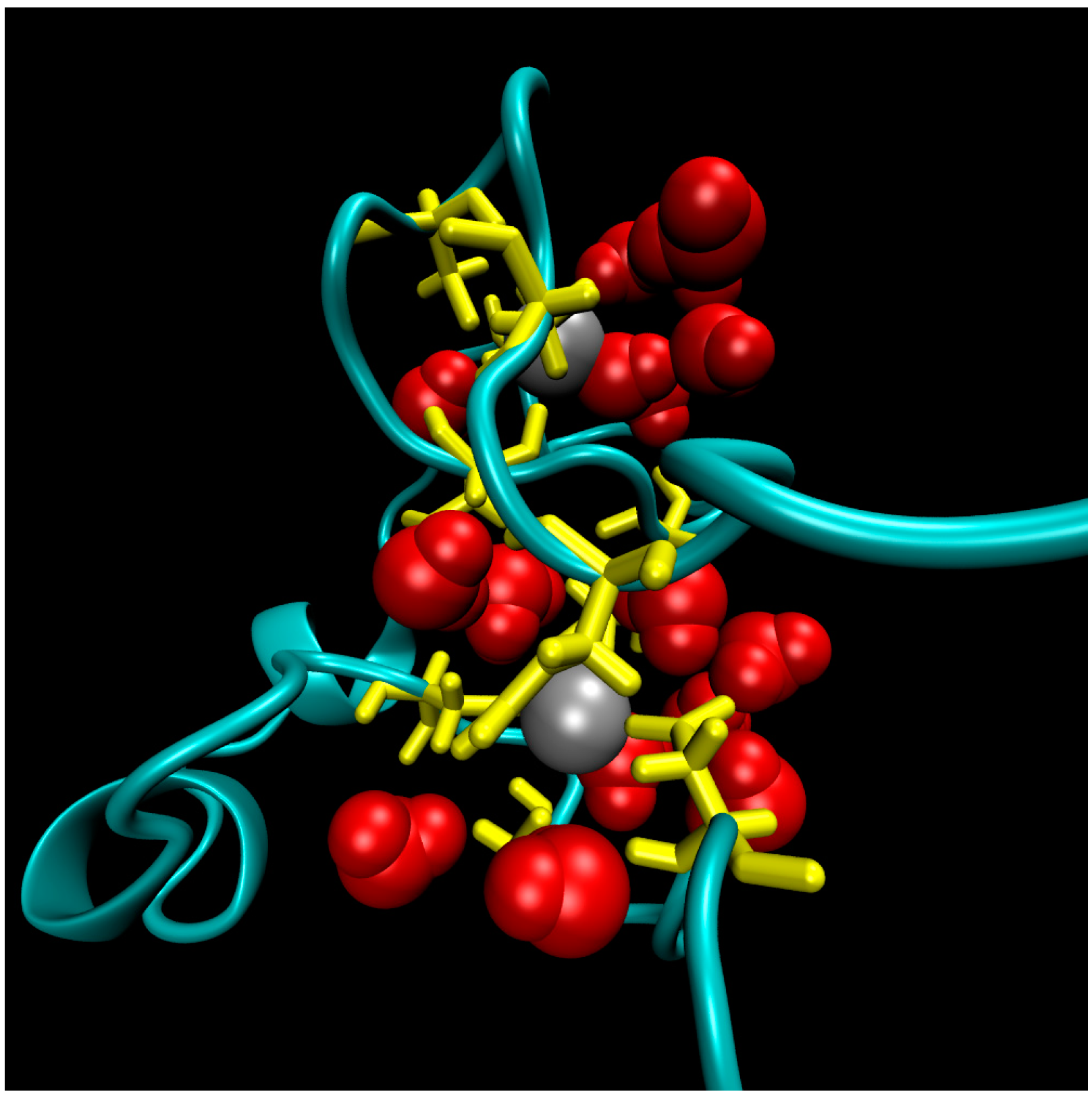
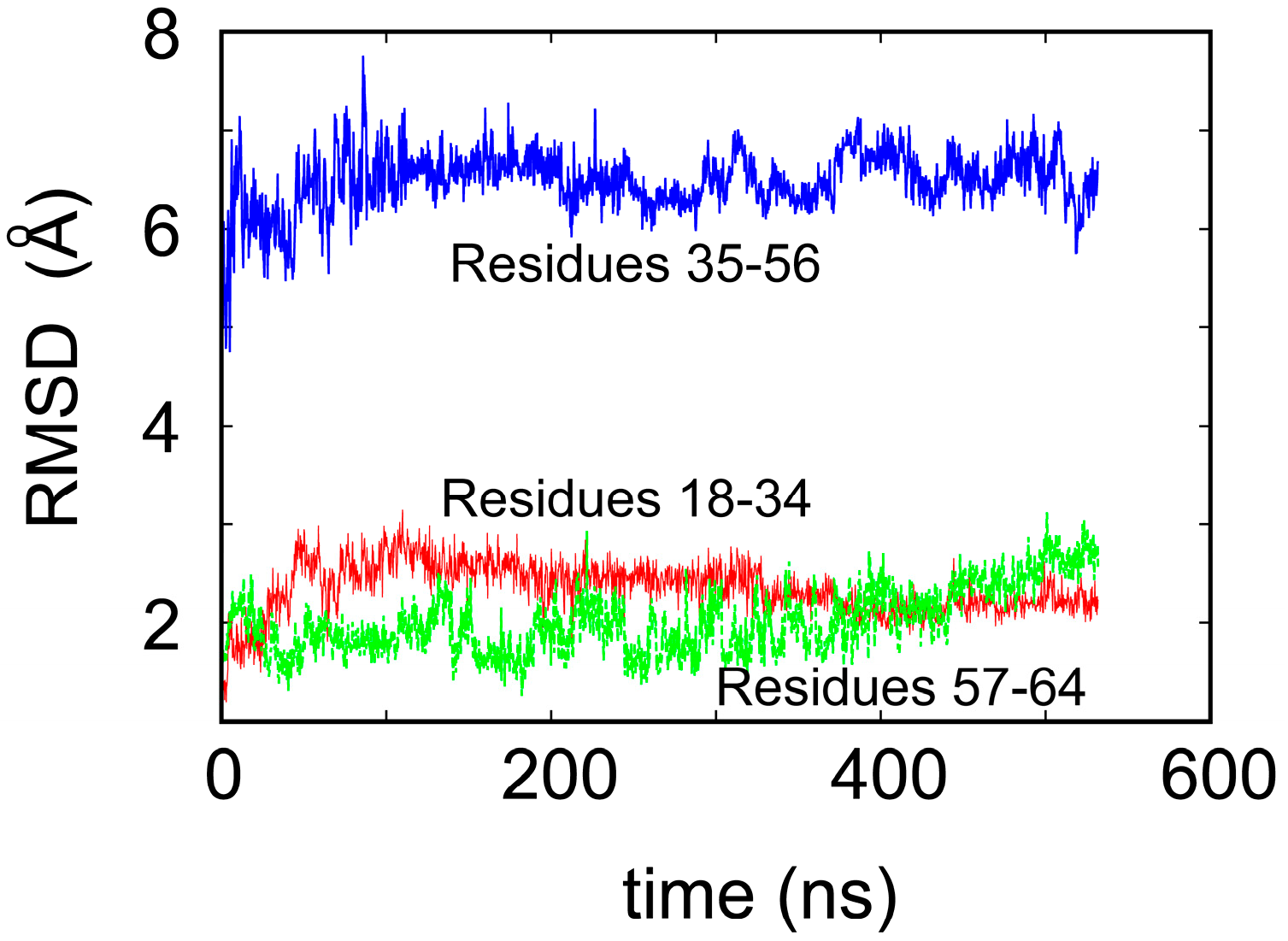
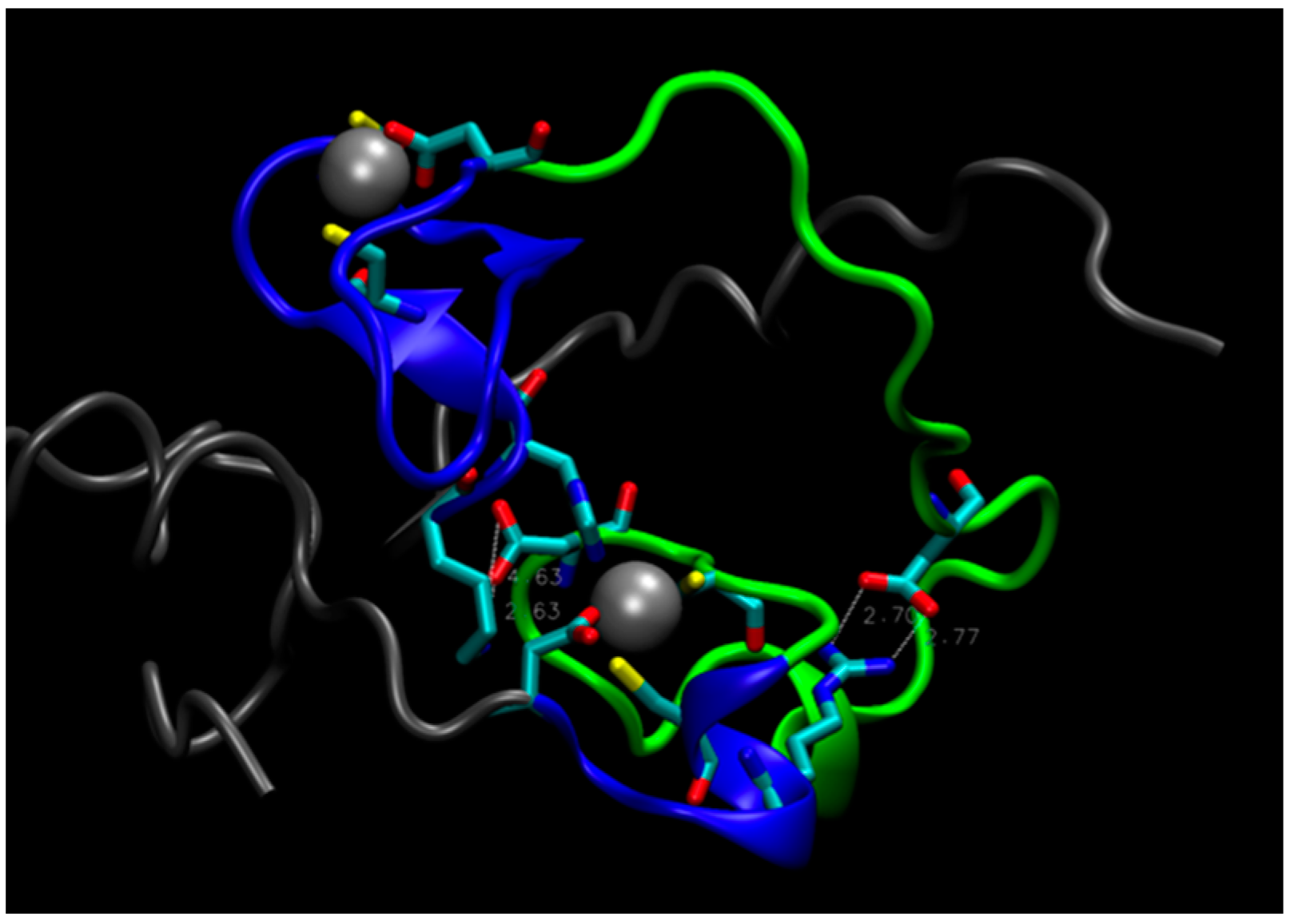
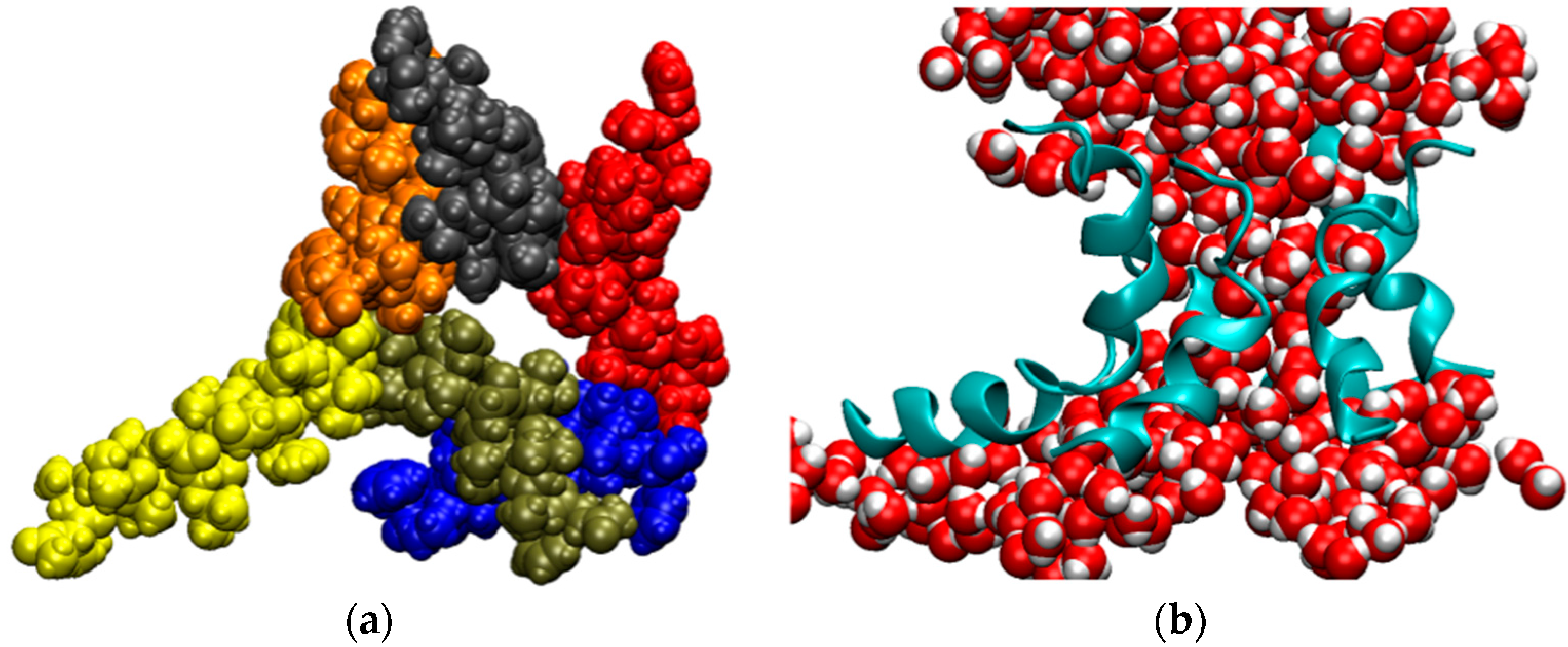
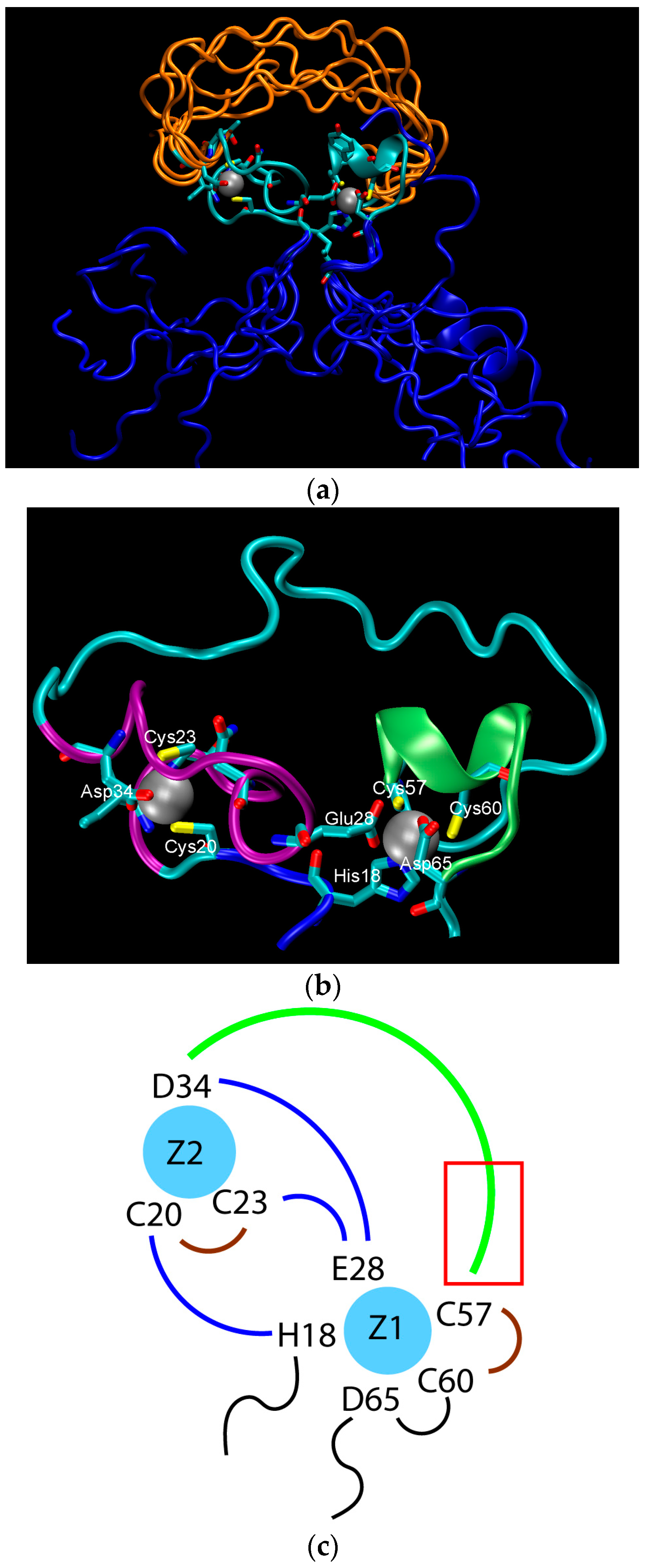
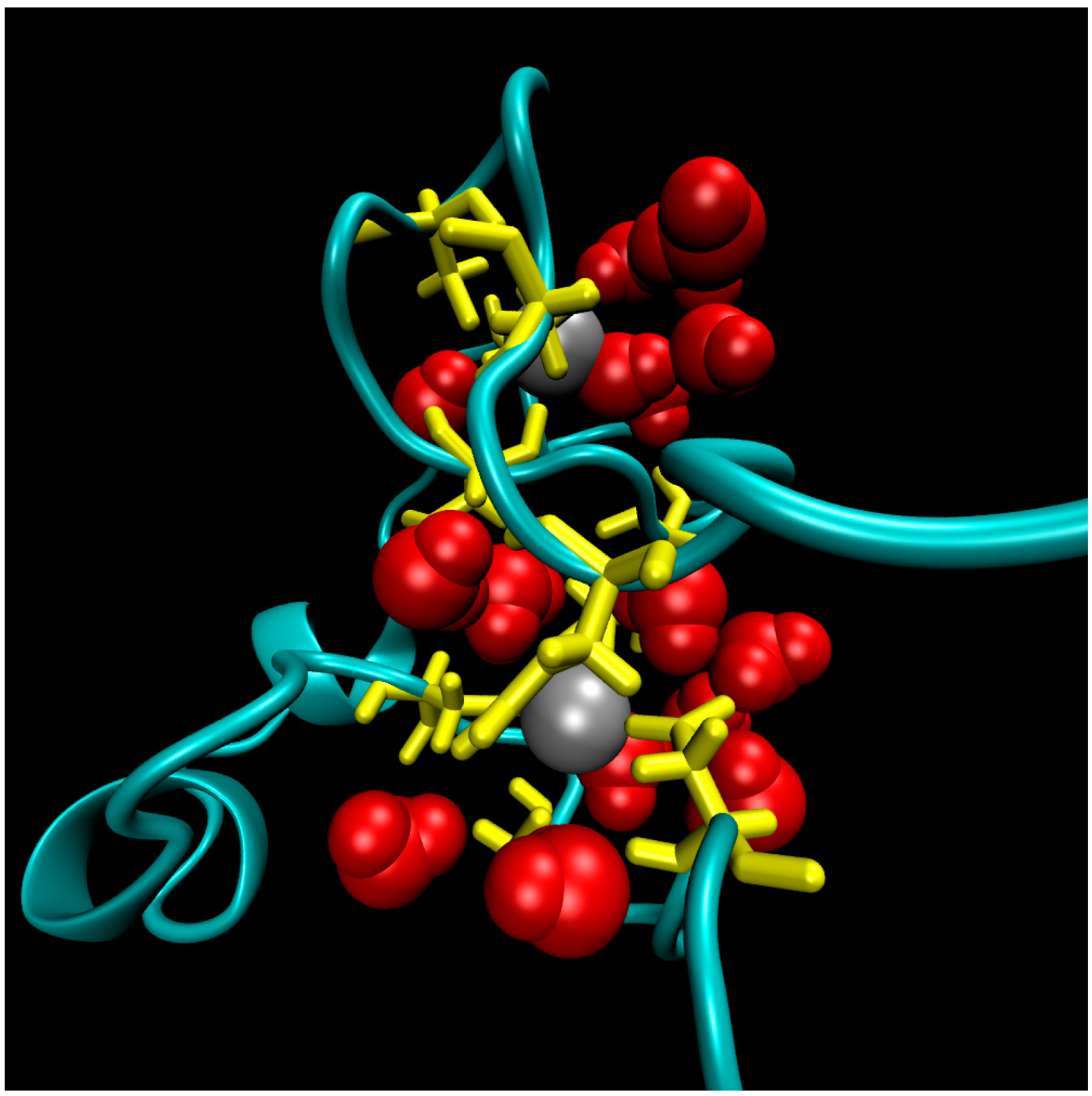
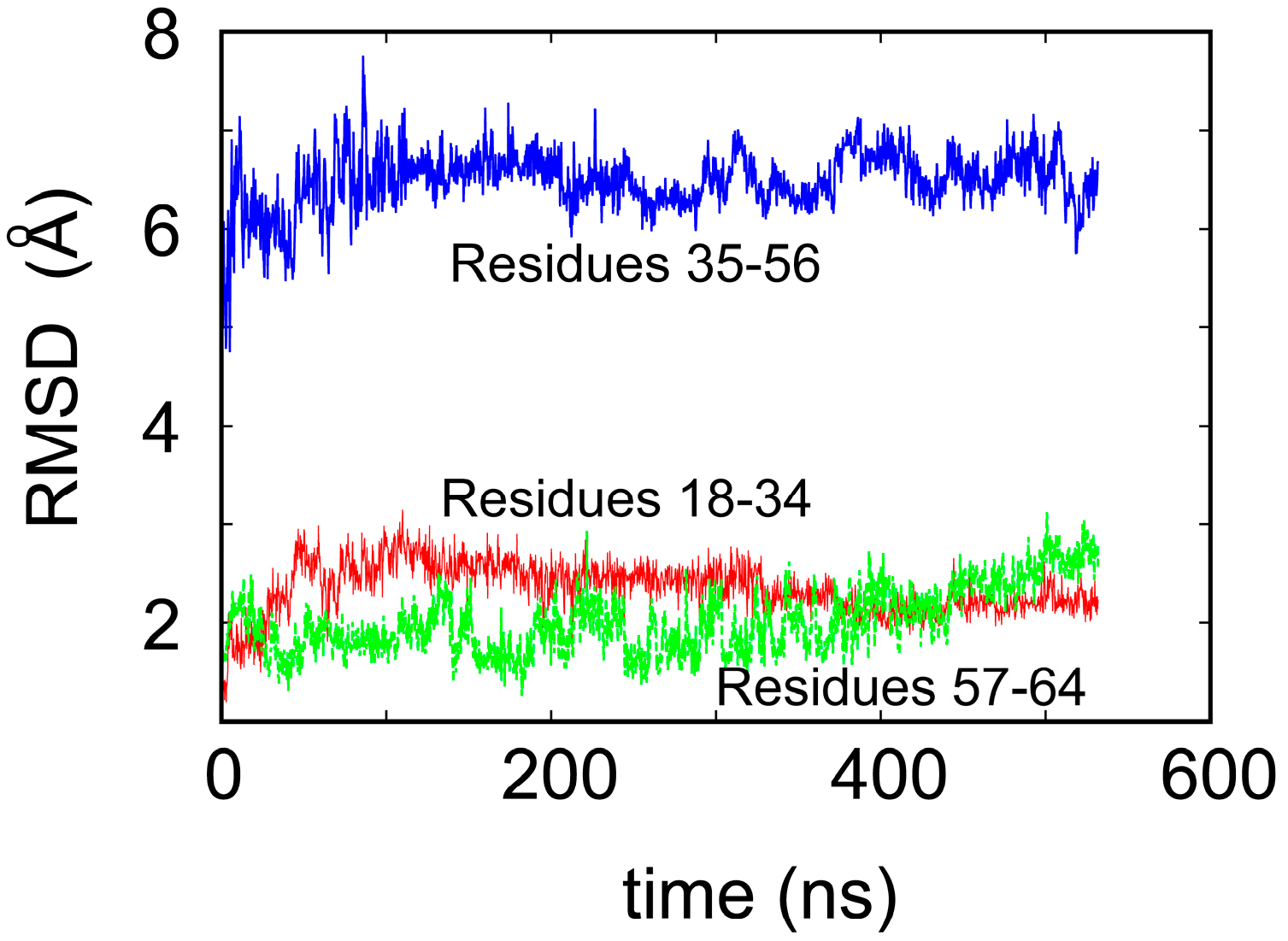
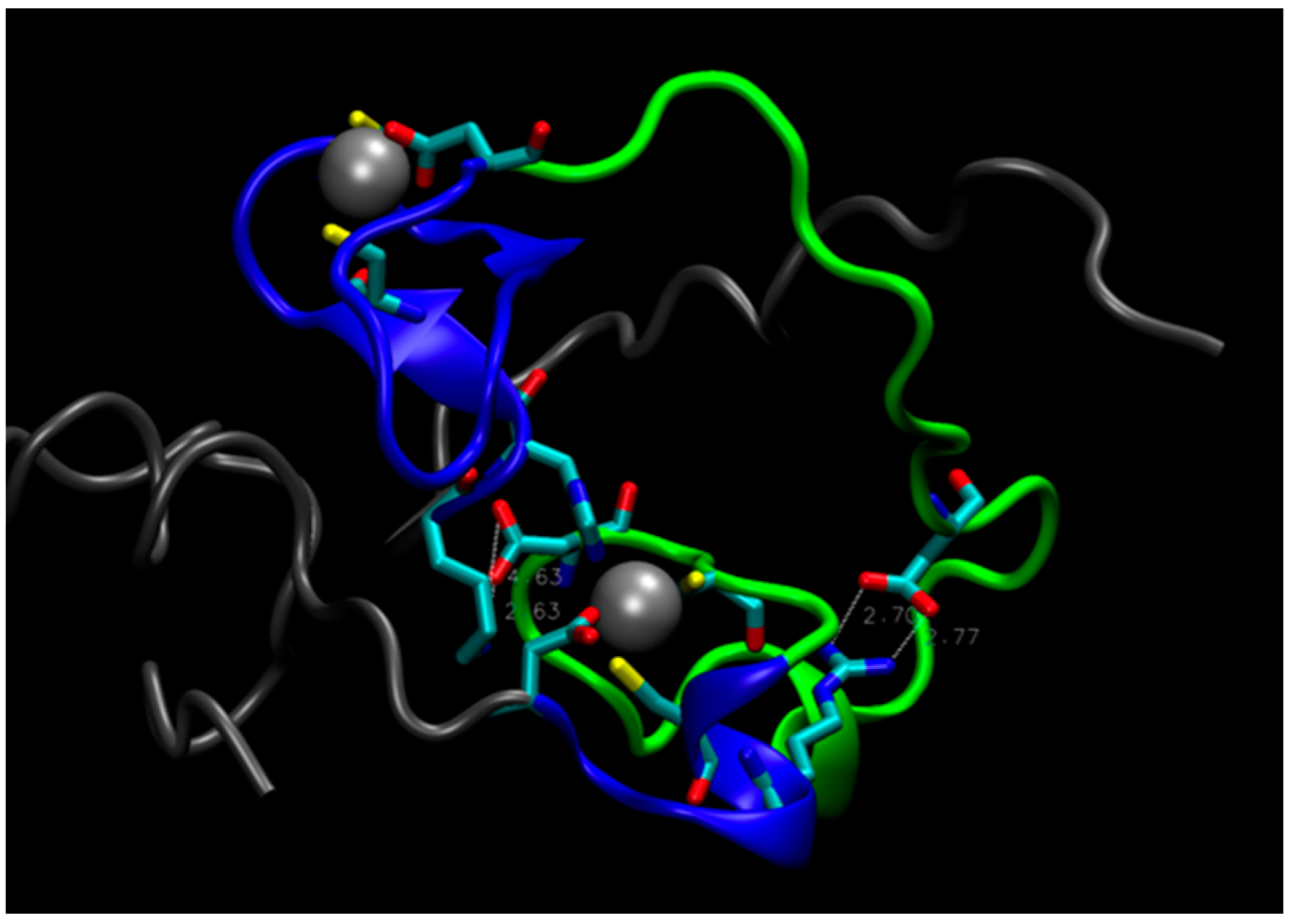
2. Water-Soluble Proteins
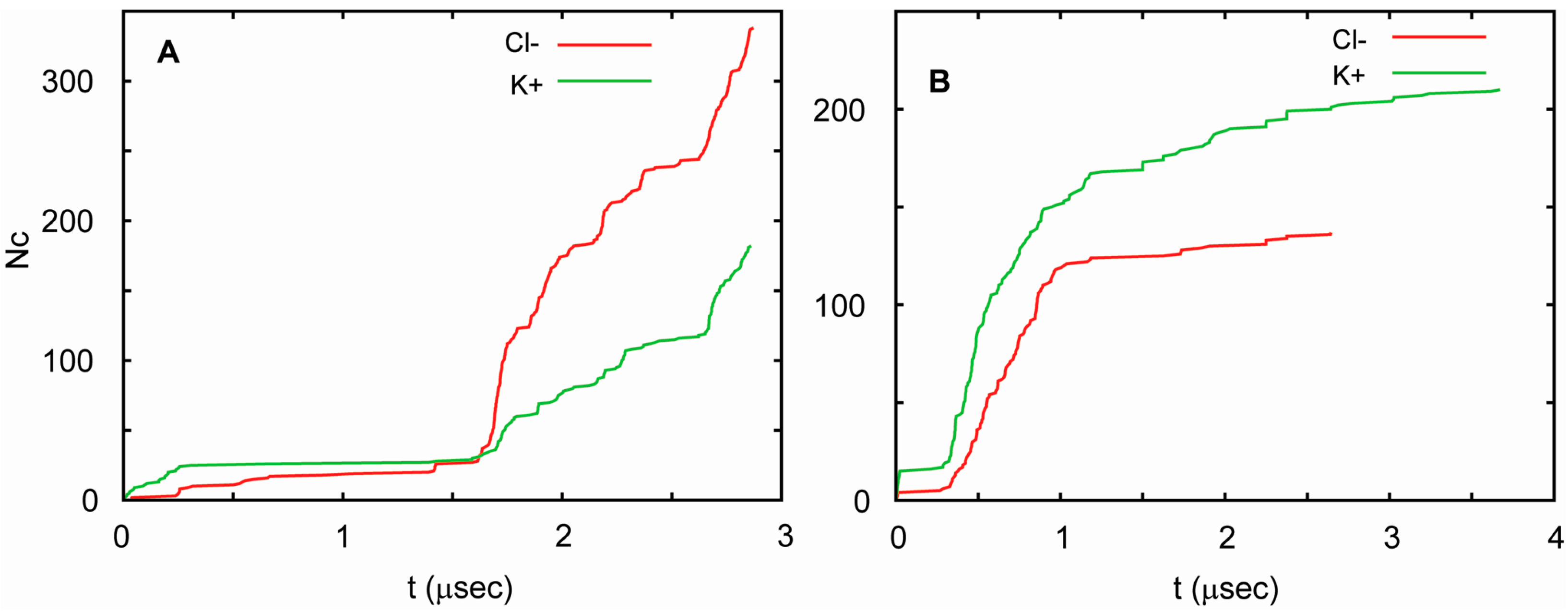
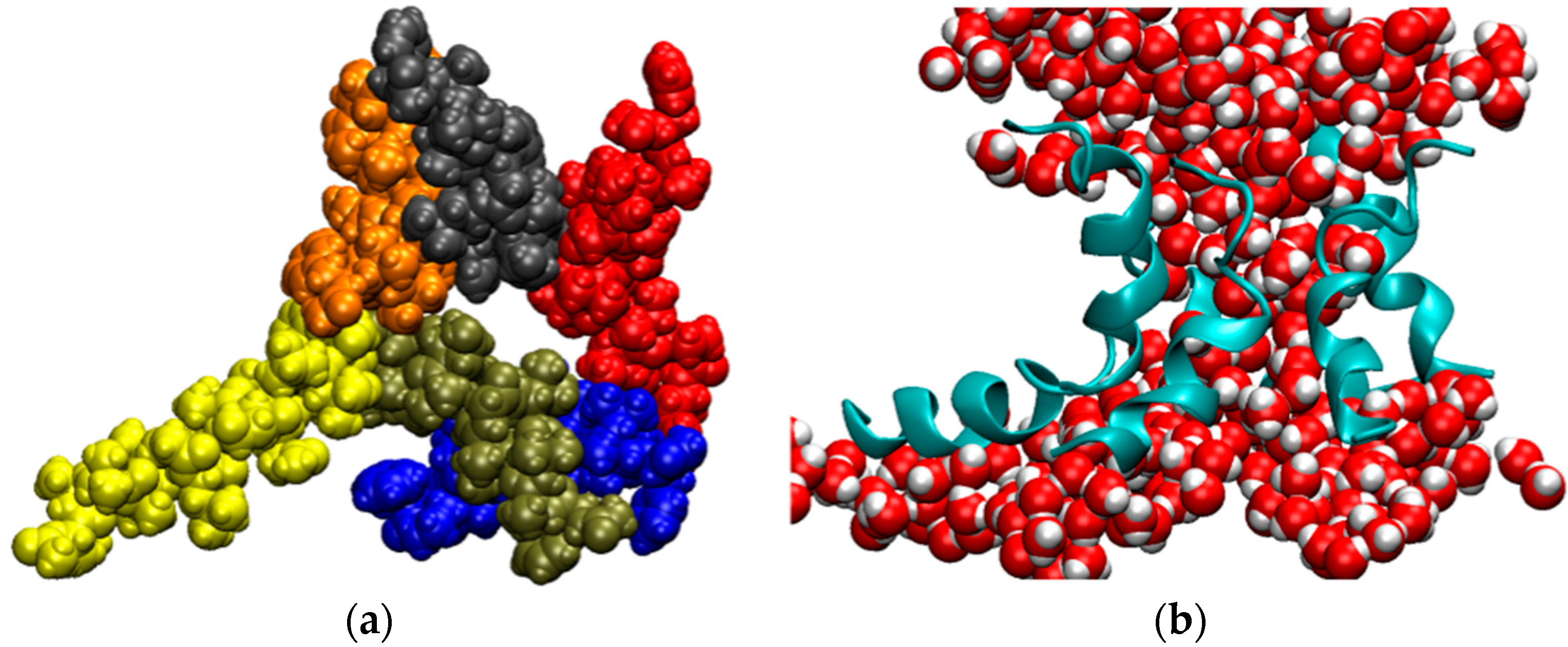
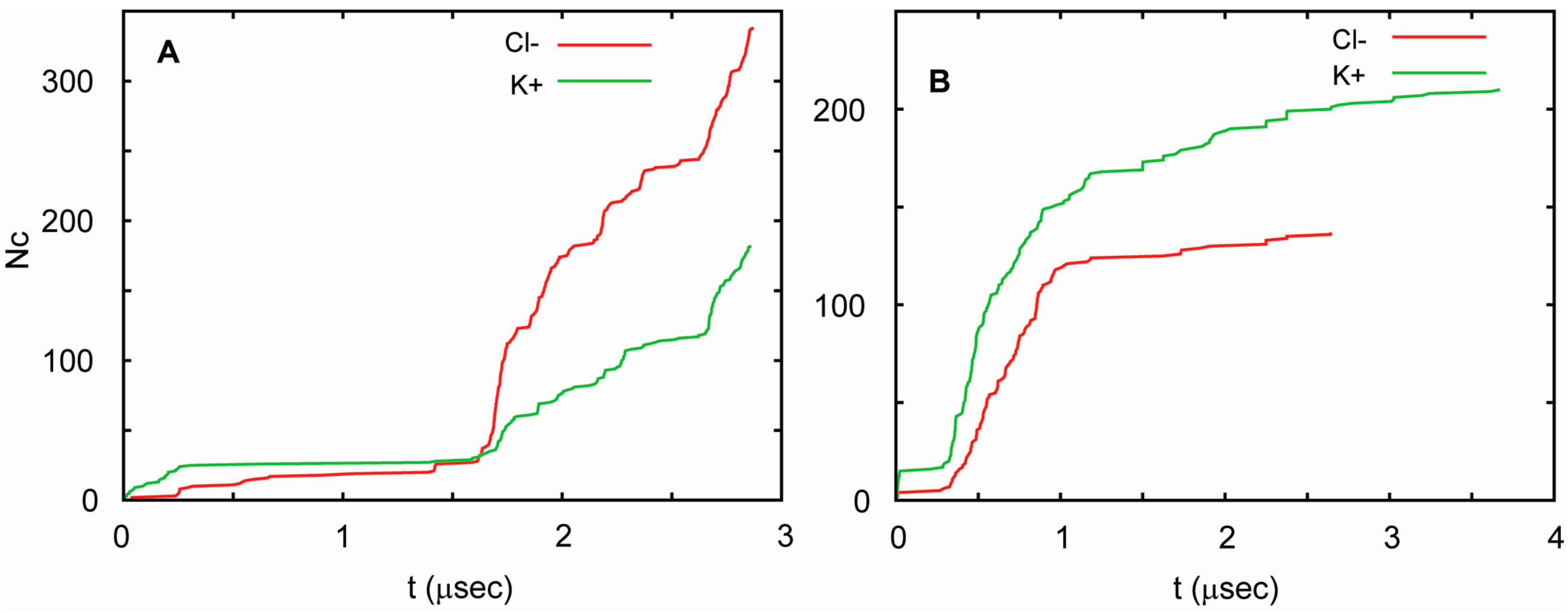
3. Membrane Proteins
4. Methods
4.1. Molecular Dynamics Simulations of Water-Soluble Proteins
4.2. Molecular Dynamics Simulations of Antiamoebin
5. Summary and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pohorille, A.; Schweighofer, K.; Wilson, M.A. The origin and early evolution of membrane channels. Astrobiology 2005, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tiessen, A.; Pérez-Rodríguez, P.; Delaye-Arredondo, L.J. Mathematical modeling and comparison of protein size distribution in different plant, animal, fungal and microbial species reveals a negative correlation between protein size and protein number, thus providing insight into the evolution of proteomes. BMC Res. Notes 2012, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Szostak, J.W. Functional proteins from a random-sequence library. Nature 2001, 410, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Mulligan, V.K.; Bahl, C.D.; Gilmore, J.M.; Harvey, P.J.; Cheneval, O.; Buchko, G.W.; Pulavarti, S.V.S.R.K.; Kaas, Q.; Eletsky, A.; et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Granja, J.R.; Martinez, J.A.; Severin, K.; Ghadiri, M.R. A self-replicating peptide. Nature 1996, 382, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Severin, K.; Lee, D.H.; Kennan, A.J.; Ghadiri, M.R. A synthetic peptide ligase. Nature 1997, 389, 706–709. [Google Scholar] [PubMed]
- Chan, H.S.; Dill, K.A. Origins of structure in globular proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 6388–6392. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C. One thousand families for the molecular biologist. Nature 1992, 357, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S.; Benkovic, S.J. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006, 75, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Malabanan, M.M.; Amyes, T.L.; Richard, J.P. A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, C.; Hilvert, D. Protein conformational disorder and enzyme catalysis. Top Curr. Chem. 2013, 337, 41–67. [Google Scholar] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Vamvaca, K.; Vögeli, B.; Kast, P.; Pervushin, K.; Hilvert, D. An enzymatic molten globule: Efficient coupling of folding and catalysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12860–12864. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J. Proteome Res. 2006, 5, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [PubMed]
- Joyce, G.F. RNA evolution and the origins of life. Nature 1989, 338, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. Molecular evolution: Booting up life. Nature 2002, 420, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.M.; Blaber, M. Prebiotic protein design supports a halophile origin of foldable proteins. Front. Microbiol. 2014, 4, 418. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Weber, A.L. Bioenergetics and Life’s Origins. Cold Spring Harbor Perspect. Biol. 2010, 2, a004929. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J.; Fresco, J.R.; Lesk, A.M.; Singh, M. Evolution of amino acid frequencies in proteins over deep time: Inferred order of introduction of amino acids into the genetic code. Mol. Biol. Evol. 2002, 19, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. The triplet code from first principles. J. Biomol. Struct. Dyn. 2004, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leman, L.; Orgel, L.; Ghadiri, M.R. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Rode, B.M. Peptides and the origin of life. Peptides 1999, 20, 773–786. [Google Scholar] [CrossRef]
- Adamala, K.; Anella, F.; Wieczorek, R.; Stano, P.; Chiarabelli, C.; Luisi, P.L. Open questions in origin of life: Experimental studies on the origin of nucleic acids and proteins with specific and functional sequences by a chemical synthetic biology approach. Comput. Struct. Biotechnol. J. 2014, 9, e201402004. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Davis, F.P.; Sali, A. The optimal size of a globular protein domain: A simple sphere-packing model. Chem. Phys. Lett. 2005, 405, 224–228. [Google Scholar] [CrossRef]
- Dill, K.A. Theory for the folding and stability of globular proteins. Biochemistry 1985, 24, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.K.; Kubelka, J.; Herbst-Irmer, R.; Eaton, W.A.; Hofrichter, J.; Davies, D.R. High-resolution X-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7517–7522. [Google Scholar] [CrossRef] [PubMed]
- Yoda, T.; Sugita, Y.; Okamoto, Y. Hydrophobic core formation and dehydration in protein folding studied by generalized-ensemble simulations. Biophys. J. 2010, 99, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Vamvaca, K.; Vögeli, B.; Hilvert, D. Structure and dynamics of a molten globular enzyme. Nat. Struct. Mol. Biol. 2007, 14, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Stojanovski, B.M.; Breydo, L.; Hunter, G.A.; Uversky, V.N.; Ferreira, G.C. Catalytically active alkaline molten globular enzyme: Effect of pH and temperature on the structural integrity of 5-aminolevulinate synthase. Biochim. Biophys. Acta 2014, 1844, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- James, L.C.; Tawfik, D.S. Conformational diversity and protein evolution—A 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.D.; Seelig, B. Advances in the directed evolution of proteins. Curr. Opin. Chem. Biol. 2014, 22, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Dodevski, I.; Markou, G.C.; Sarkar, C.A. Conceptual and methodological advances in cell-free directed evolution. Curr. Opin. Struct. Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Severin, K.; Yokobayashi, Y.; Ghadiri, M.R. Emergence of symbiosis in peptide self-replication through a hypercyclic network. Nature 1997, 390, 591–594. [Google Scholar] [PubMed]
- Issac, R.; Chmielewski, J. Approaching exponential growth with a self-replicating peptide. J. Am. Chem. Soc. 2002, 124, 6808–6809. [Google Scholar] [CrossRef] [PubMed]
- Dadon, Z.; Wagner, N.; Ashkenasy, G. The road to non-enzymatic molecular networks. Angew. Chem. Int. Ed. Engl. 2008, 47, 6128–6136. [Google Scholar] [CrossRef] [PubMed]
- Dadon, Z.; Wagner, N.; Cohen-Luria, R.; Ashkenasy, G. Reaction Networks. In Supramolecular Chemistry; John Wiley & Sons, Ltd.: New York, NY, USA, 2012. [Google Scholar]
- Li, X.; Chmielewski, J. Peptide self-replication enhanced by a proline kink. J. Am. Chem. Soc. 2003, 125, 11820–11821. [Google Scholar] [CrossRef] [PubMed]
- Saghatelian, A.; Yokobayashi, Y.; Soltani, K.; Ghadiri, M.R. A chiroselective peptide replicator. Nature 2001, 409, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Kim, S.; Fela, D.; Baum, J.; Hecht, M.H. Solution structure of a de novo protein from a designed combinatorial library. Proc. Natl. Acad. Sci. USA 2003, 100, 13270–13273. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Hecht, M.H. Enzyme-like proteins from an unselected library of designed amino acid sequences. Protein Eng. Des. Sel. 2004, 17, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Hecht, M.H. Novel proteins: From fold to function. Curr. Opin. Chem. Biol. 2011, 15, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.C.; Hecht, M.H. Directed evolution of the peroxidase activity of a de novo-designed protein. Protein Eng. Des. Sel. 2012, 25, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Mularz, A.E.; Hecht, M.H. Divergent evolution of a bifunctional de novo protein. Protein Sci. 2015, 24, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.S.; Greisman, J.B.; Hecht, M.H. De Novo Proteins with Life-Sustaining Functions Are Structurally Dynamic. J. Mol. Biol. 2016, 428 2 Pt A, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.J.; Thomson, A.R.; Dawson, W.M.; Brady, R.L.; Woolfson, D.N. Installing hydrolytic activity into a completely de novo protein framework. Nat. Chem. 2016, 8, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.A.; Caetano-Anollés, G. Origin and evolution of protein fold designs inferred from phylogenomic analysis of CATH domain structures in proteomes. PLoS Comput. Biol. 2013, 9, e1003009. [Google Scholar] [CrossRef] [PubMed]
- Rufo, C.M.; Moroz, Y.S.; Moroz, O.V.; Stöhr, J.; Smith, T.A.; Hu, X.; Degrado, W.F.; Korendovych, I.V. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem. 2014, 6, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Riek, R. On the possible amyloid origin of protein folds. J. Mol. Biol. 2012, 421, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Seelig, B.; Szostak, J.W. Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature 2007, 448, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Chao, F.A.; Morelli, A.; Haugner, J.C.; Churchfield, L.; Hagmann, L.N.; Shi, L.; Materson, L.R.; Sarangi, R.; Veglia, G.; Seelig, B. Structure and dynamics of a primordial catalytic fold generated by in vitro evolution. Nat. Chem. Biol. 2013, 9, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Churchfield, L. Probing Structural and Catalytic Features of a Test Tube-Evolved RNA Ligase Enzyme by Site-Directed Mutagenesis; University of Minnesota: Saint Paul, MN, USA, 2011. [Google Scholar]
- Williams, R.J. 16th Sir Hans Krebs lecture. The symbiosis of metal and protein functions. Eur. J. Biochem. 1985, 150, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Schut, G.J.; Boyd, E.S.; Mulder, D.W.; Shepard, E.M.; Broderick, J.B.; King, P.W.; Adams, M.W. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 2015, 1853, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Lo Surdo, P.; Walsh, M.A.; Sollazzo, M. A novel ADP- and zinc-binding fold from function-directed in vitro evolution. Nat. Struct. Mol. Biol. 2004, 11, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.; Meyer, R.; Sommergruber, W. Spectroscopic characterization of rhinoviral protease 2A: Zn is essential for the structural integrity. Protein. Sci. 1995, 4, 2526–2531. [Google Scholar] [CrossRef] [PubMed]
- Abian, O.; Vega, S.; Neira, J.L.; Velazquez-Campoy, A. Conformational stability of hepatitis C virus NS3 protease. Biophys. J. 2010, 99, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Adamala, K.; Szostak, J.W. Competition between model protocells driven by an encapsulated catalyst. Nat. Chem. 2013, 5, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Gorlero, M.; Wieczorek, R.; Adamala, K.; Giorgi, A.; Schininà, M.E.; Stano, P.; Luisi, P.L. Ser-His catalyses the formation of peptides and PNAs. FEBS Lett. 2009, 583, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, R.; Dörr, M.; Chotera, A.; Luisi, P.L.; Monnard, P.A. Formation of RNA phosphodiester bond by histidine-containing dipeptides. ChemBioChem 2013, 14, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002, 321, 741–765. [Google Scholar] [CrossRef]
- Goldman, A.D.; Beatty, J.T.; Landweber, L.F. The TIM Barrel Architecture Facilitated the Early Evolution of Protein-Mediated Metabolism. J. Mol. Evol. 2016, 82, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M. Osmosensing by bacteria: Signals and membrane-based sensors. Microbiol. Mol. Biol. Rev. 1999, 63, 230–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.A.; Roberts, R.W.; Szostak, J.W. The emergence of competition between model protocells. Science 2004, 305, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Pohorille, A.; Deamer, D. Self-assembly and function of primitive cell membranes. Res. Microbiol. 2009, 160, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.C.; Breaker, R.R.; Joyce, G.F.; Deamer, D.W. Production of RNA by a polymerase protein encapsulated within phospholipid vesicles. J. Mol. Evol. 1994, 39, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.A.; Luptak, A.; Deamer, D.W. Models of primitive cellular life: Polymerases and templates in liposomes. Philos. Trans. R Soc. Lond B Biol. Sci. 2007, 362, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Gurtovenko, A.A.; Anwar, J.; Vattulainen, I. Defect-mediated trafficking across cell membranes: Insights from in silico modeling. Chem. Rev. 2010, 110, 6077–6103. [Google Scholar] [CrossRef] [PubMed]
- Przybylski, A.T.; Stratten, W.P.; Syren, R.M.; Fox, S.W. Membrane, action, and oscillatory potentials in simulated protocells. Naturwissenschaften 1982, 69, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Przybylski, A.T.; Fox, S.W. Excitable artificial cells of proteinoid. Appl. Biochem. Biotechnol. 1984, 10, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Zhang, L.; Rozek, A.; Hancock, R.E. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A. How was Membrane Permeability Produced in an RNA World? Orig. Life Evolut. Biosph. 2005, 35, 135–149. [Google Scholar] [CrossRef]
- Hladky, S.B.; Gruen, D.W. Thickness fluctuations in black lipid membranes. Biophys. J. 1982, 38, 251–258. [Google Scholar] [CrossRef]
- Bobone, S.; Gerelli, Y.; De Zotti, M.; Bocchinfuso, G.; Farrotti, A.; Orioni, B.; Sebastiani, F.; Latter, E.; Penfold, J.; Senesi, R.; et al. Membrane thickness and the mechanism of action of the short peptaibol trichogin GA IV. Biochim. Biophys. Acta 2013, 1828, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Duclohier, H.; Snook, C.F.; Wallace, B.A. Antiamoebin can function as a carrier or as a pore-forming peptaibol. Biochim. Biophys. Acta 1998, 1415, 255–260. [Google Scholar] [CrossRef]
- Langosch, D.; Heringa, J. Interaction of transmembrane helices by a knobs-into-holes packing characteristic of soluble coiled coils. Proteins 1998, 31, 150–159. [Google Scholar] [CrossRef]
- Dunker, A.K.; Marvin, D.A. A model for membrane transport through alpha-helical protein pores. J. Theor. Biol. 1978, 72, 9–16. [Google Scholar] [CrossRef]
- Dunker, A.K.; Jones, T.C. Proposed knobs-into-holes packing for several membrane proteins. Membr. Biochem. 1978, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.R.; Prestegard, J.H.; Engelman, D.M. A transmembrane helix dimer: Structure and implications. Science 1997, 276, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Jasti, J.; Furukawa, H.; Gonzales, E.B.; Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 2007, 449, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.D.; Wasserman, Z.R.; DeGrado, W.F. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.D.; Schneider, J.P.; Kienker, P.K.; DeGrado, W.F. Electrostatic Effects on Ion Selectivity and Rectification in Designed Ion Channel Peptides. J. Am. Chem. Soc. 1997, 119, 3212–3217. [Google Scholar] [CrossRef]
- Boheim, G. Statistical analysis of alamethicin channels in black lipid membranes. J. Membr. Biol. 1974, 19, 277–303. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.; Mueller, P. A molecular model of membrane excitability. J. Supramol. Struct. 1974, 2, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Thøgersen, L.; Schiøtt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Hjørringgaard, C.U.; Vad, B.S.; Matchkov, V.V.; Nielsen, S.B.; Vosegaard, T.; Nielsen, N.C.; Otzen, D.E.; Skrydstrup, T. Cyclodextrin-scaffolded alamethicin with remarkably efficient membrane permeabilizing properties and membrane current conductance. J. Phys. Chem. B 2012, 116, 7652–7659. [Google Scholar] [CrossRef] [PubMed]
- Chugh, J.K.; Wallace, B.A. Peptaibols: Models for ion channels. Biochem. Soc. Trans. 2001, 29 Pt 4, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Pizzarello, S.; Cooper, G.; Flynn, G.J. The Nature and Distribution of the Organic Material in Carbonaceous Chondrites and Interplanetary Dust Particles. In Meteorites and the Early Solar System II; University of Arizona Press: Tucson, AZ, USA, 2006. [Google Scholar]
- Wilson, M.A.; Wei, C.; Bjelkmar, P.; Wallace, B.A.; Pohorille, A. Molecular dynamics simulation of the antiamoebin ion channel: Linking structure and conductance. Biophys. J. 2011, 100, 2394–2402. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Pohorille, A. Activation and proton transport mechanism in influenza A M2 channel. Biophys. J. 2013, 105, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.X.; Sharpe, S.; Ghirlando, R.; Yau, W.M.; Tycko, R. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 2010, 19, 1877–1896. [Google Scholar] [CrossRef] [PubMed]
- Lingueglia, E. Acid-sensing ion channels in sensory perception. J. Biol. Chem. 2007, 282, 17325–17329. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, C.C.; Minchin, R.F.; Le Dain, A.C.; Martinac, B. Estimation of the pore size of the large-conductance mechanosensitive ion channel of Escherichia coli. Biophys. J. 1997, 73, 1925–1931. [Google Scholar] [CrossRef]
- Meller, A.; Branton, D. Single molecule measurements of DNA transport through a nanopore. Electrophoresis 2002, 23, 2583–2591. [Google Scholar] [CrossRef]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; Mackinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cuello, L.G.; Romero, J.G.; Cortes, D.M.; Perozo, E. pH-Dependent Gating in the Streptomyces lividans K+ Channel. Biochemistry 1998, 37, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Meuser, D.; Splitt, H.; Wagner, R.; Schrempf, H. Exploring the open pore of the potassium channel from Streptomyces lividans. FEBS Lett. 1999, 462, 447–452. [Google Scholar] [CrossRef]
- Heginbotham, L.; LeMasurier, M.; Kolmakova-Partensky, L.; Miller, C. Single streptomyces lividans K(+) channels: Functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 1999, 114, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2014, 47, 5.6.1–5.6.32. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Kucerka, N.; Tristram-Nagle, S.; Nagle, J.F. Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J. Membr. Biol. 2005, 208, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Aksimentiev, A.; Schulten, K. Imaging alpha-hemolysin with molecular dynamics: Ionic conductance, osmotic permeability, and the electrostatic potential map. Biophys. J. 2005, 88, 3745–3761. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Gaidukov, L.; Khersonsky, O.; McQ Gould, S.; Roodveldt, C.; Tawfik, D.S. The ’evolvability’ of promiscuous protein functions. Nat. Genet. 2005, 37, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Raggi, L.; Bada, J.L.; Lazcano, A. On the lack of evolutionary continuity between prebiotic peptides and extant enzymes. Phys. Chem. Chem. Phys. 2016, 18, 20028–20032. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trajectory | T1 | T2 | T3 |
|---|---|---|---|
| Simulation time (µs) | 3.0 | 3.8 | 3.8 |
| Box size x, y, z (Å) | 86.3, 84.1, 93.9 | 82.3, 84.3, 108.2 | 83.66, 83.66, 106.62 |
| No. of lipid/water molecules | 222/13053 | 200/16228 | 198/16228 |
| Protein potentials | CHARMM22 | AMBER99 | CHARMM22 |
| Lipid potentials | CHARMM27 | CHARMM36 | CHARMM36 |
| Water potentials | TIP3P | SPC/E | TIP3P |
| Ionic strength | 1 M | 1 M | 1 M |
| Voltage, V (mV) | 150 | 150 | 150 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pohorille, A.; Wilson, M.A.; Shannon, G. Flexible Proteins at the Origin of Life. Life 2017, 7, 23. https://doi.org/10.3390/life7020023
Pohorille A, Wilson MA, Shannon G. Flexible Proteins at the Origin of Life. Life. 2017; 7(2):23. https://doi.org/10.3390/life7020023
Chicago/Turabian StylePohorille, Andrew, Michael A. Wilson, and Gareth Shannon. 2017. "Flexible Proteins at the Origin of Life" Life 7, no. 2: 23. https://doi.org/10.3390/life7020023
APA StylePohorille, A., Wilson, M. A., & Shannon, G. (2017). Flexible Proteins at the Origin of Life. Life, 7(2), 23. https://doi.org/10.3390/life7020023