Formaldehyde—A Key Monad of the Biomolecular System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
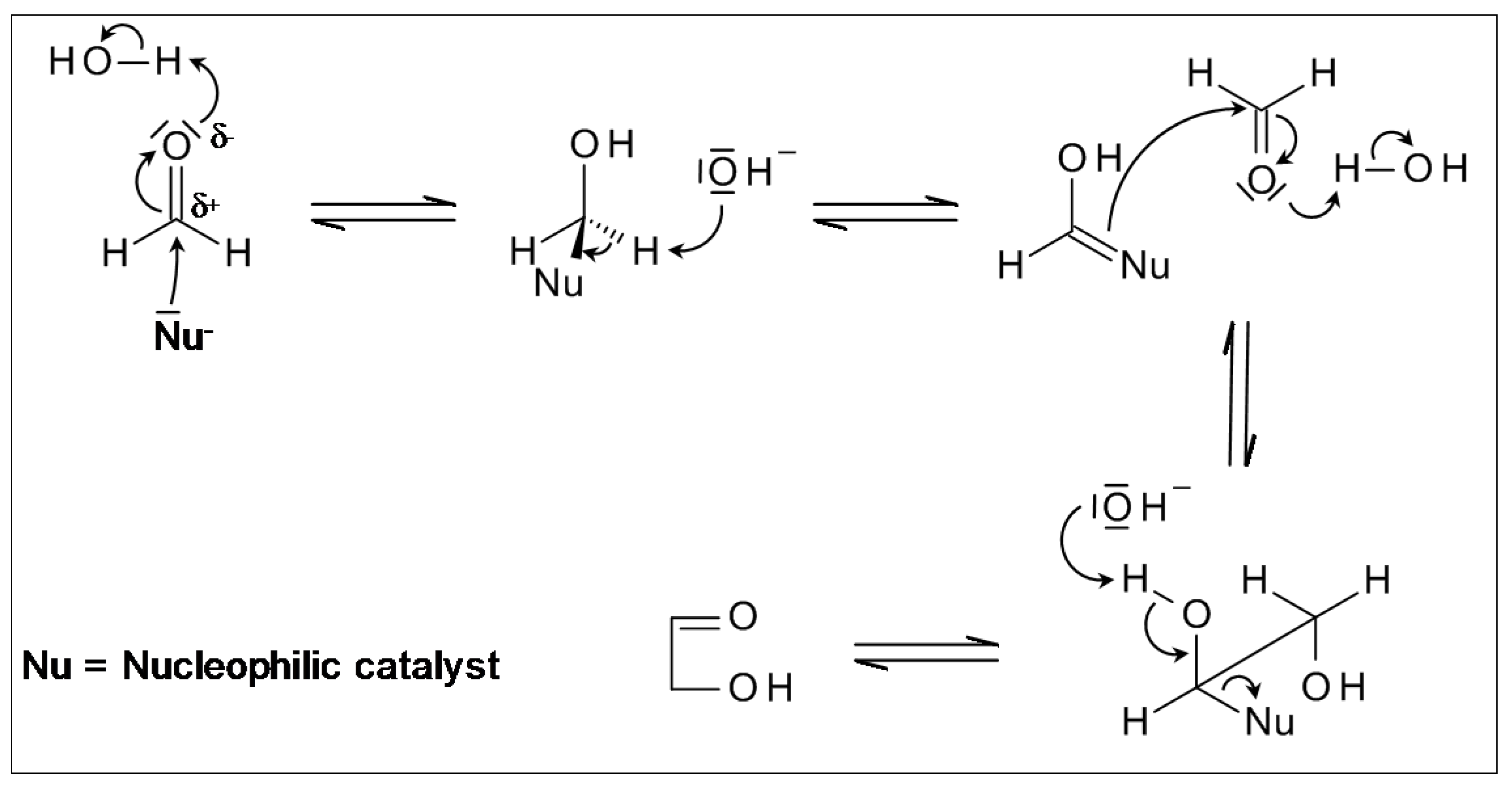
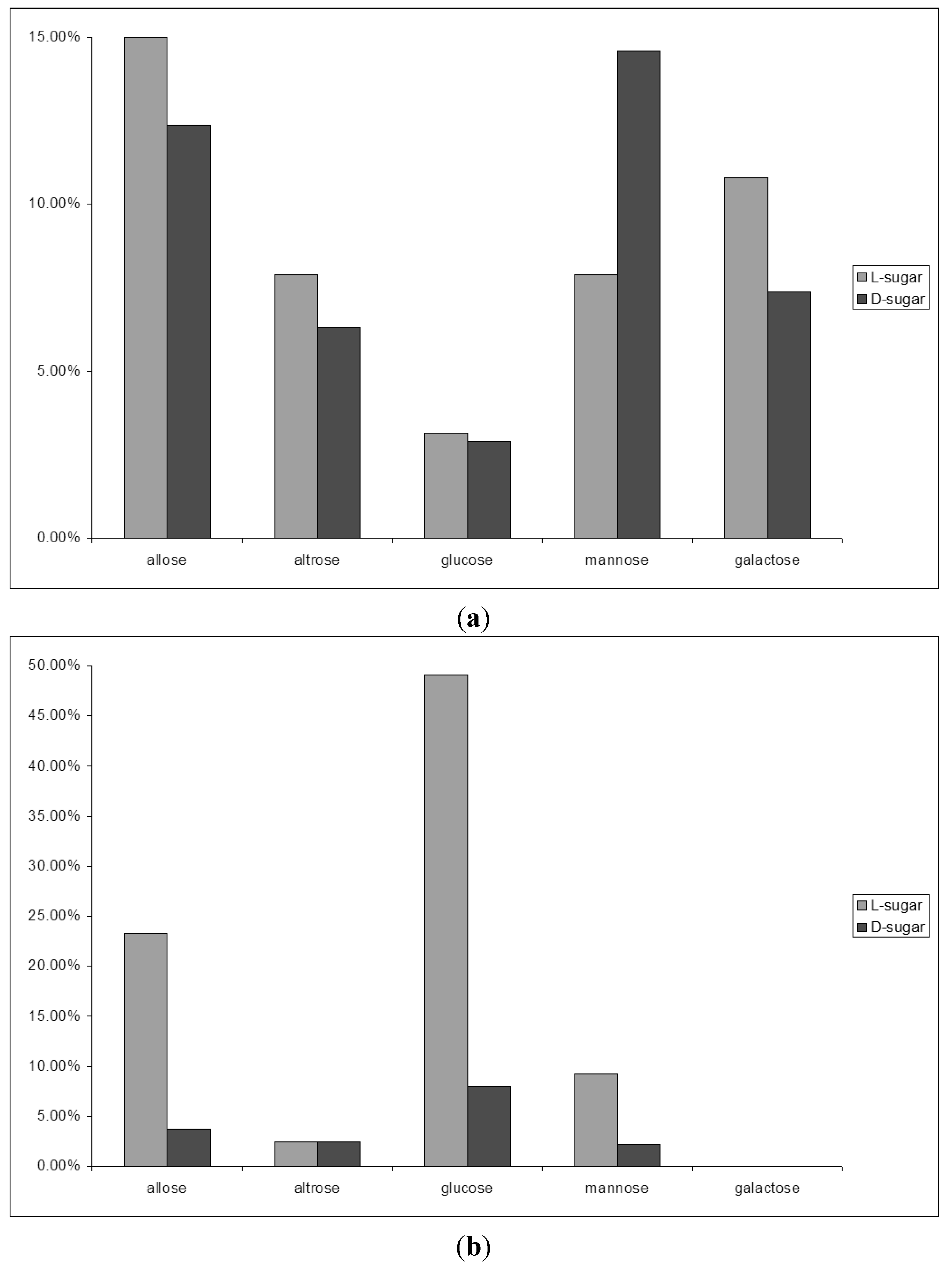
2.1. From Formaldehyde to Glycolaldehyde, and on to Sugars
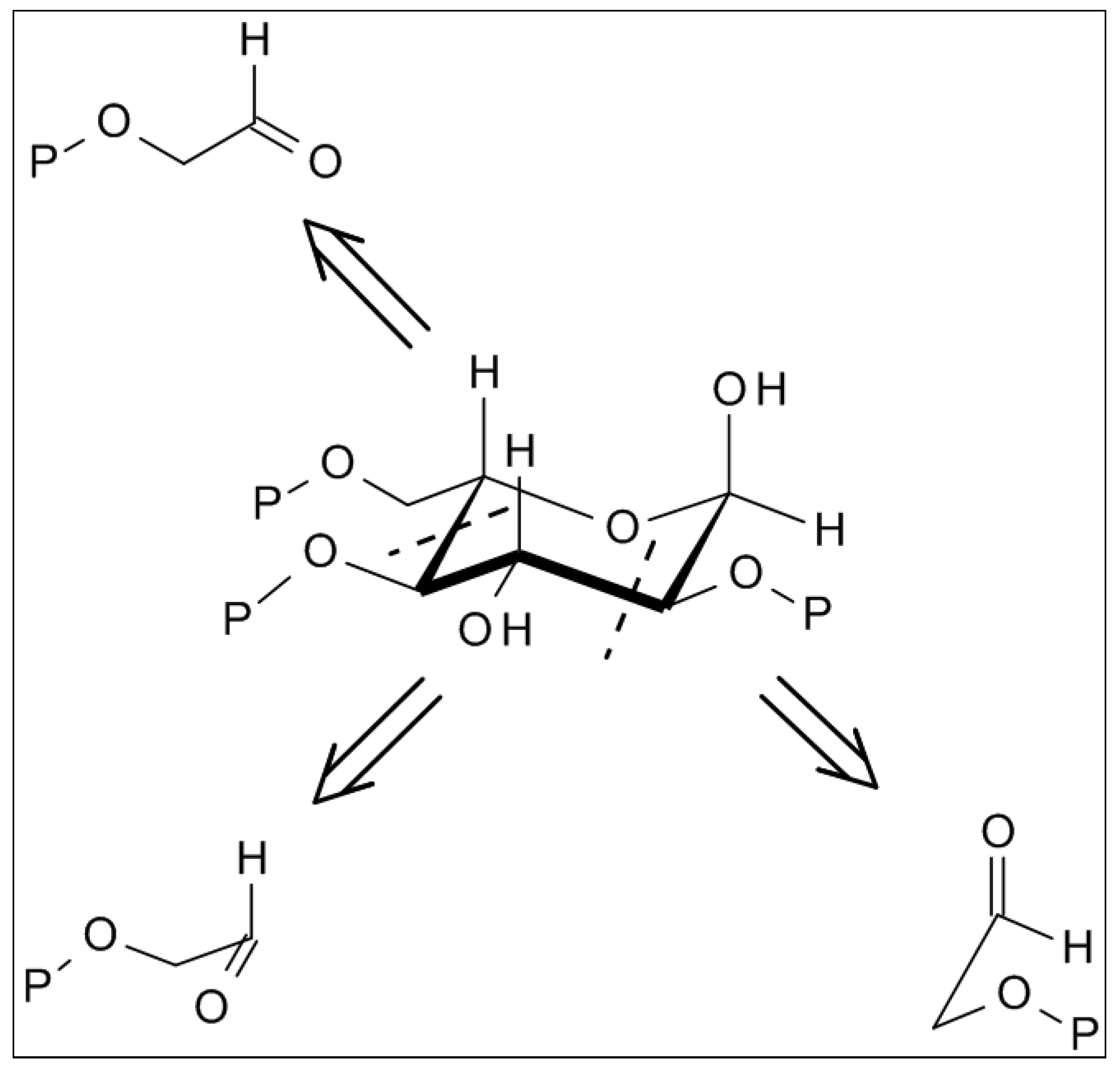
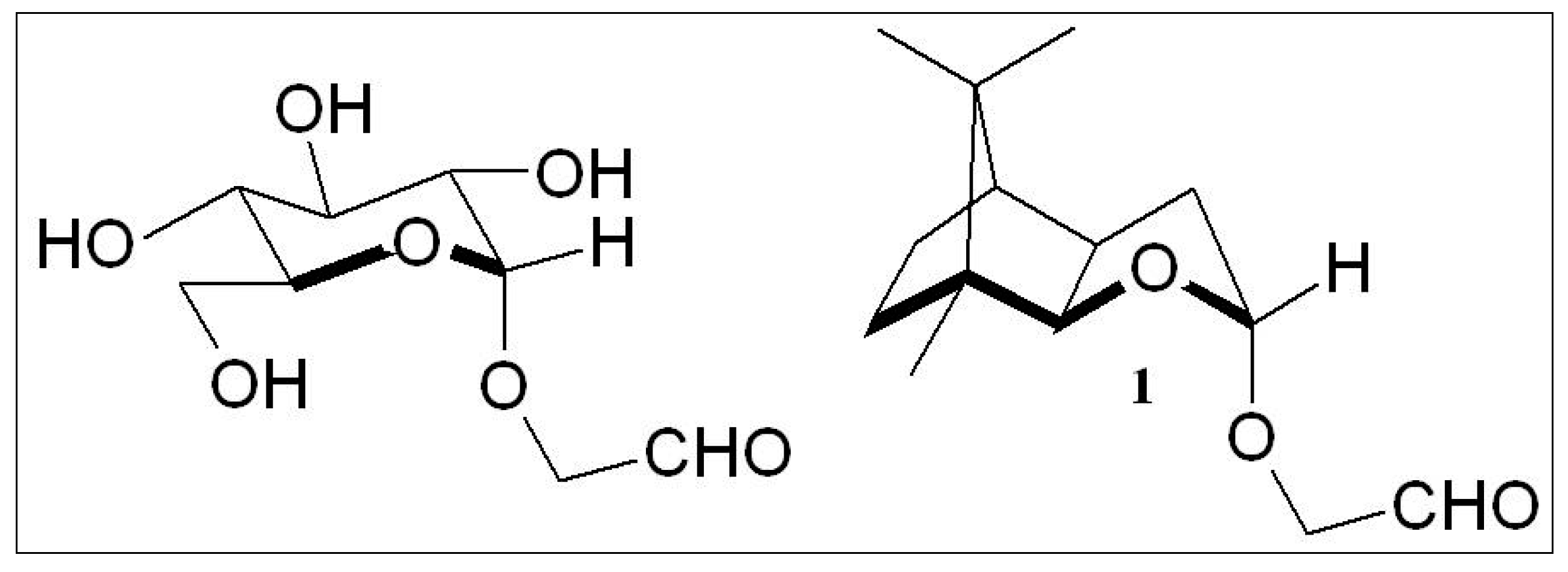
2.2. “Stereoelectronic Effects”—A Key Contributor to the Amplification of Chirality of Biomolecules
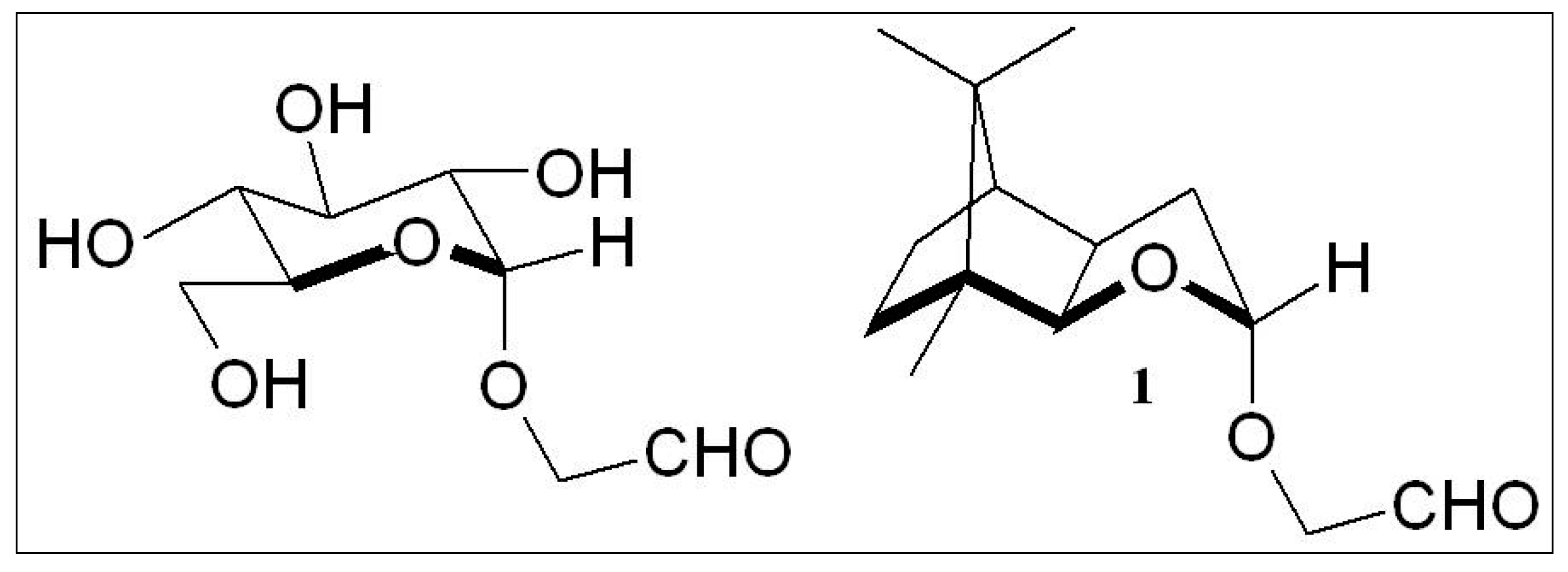
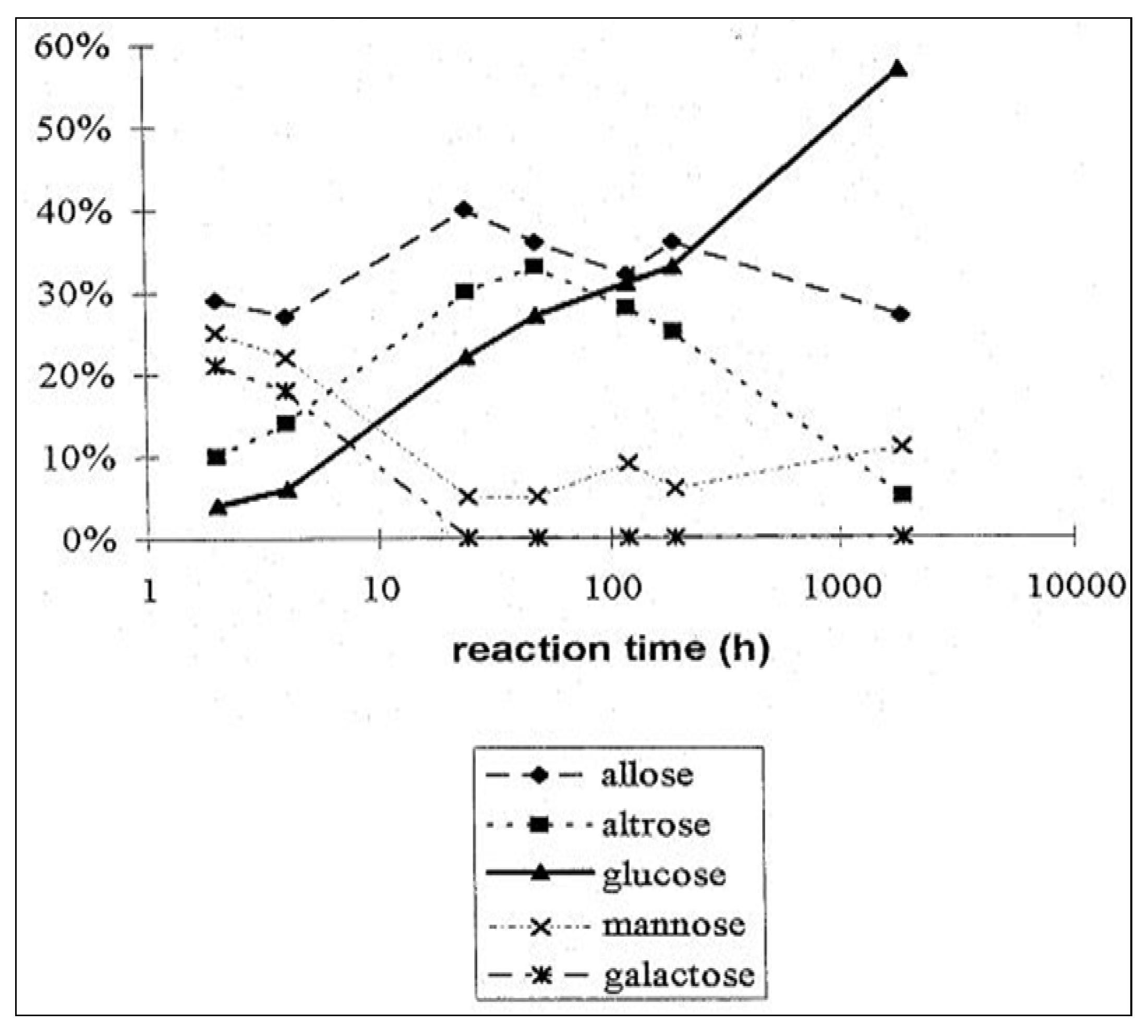
2.3. D-Glucose—A Product of Molecular Self-Constitution
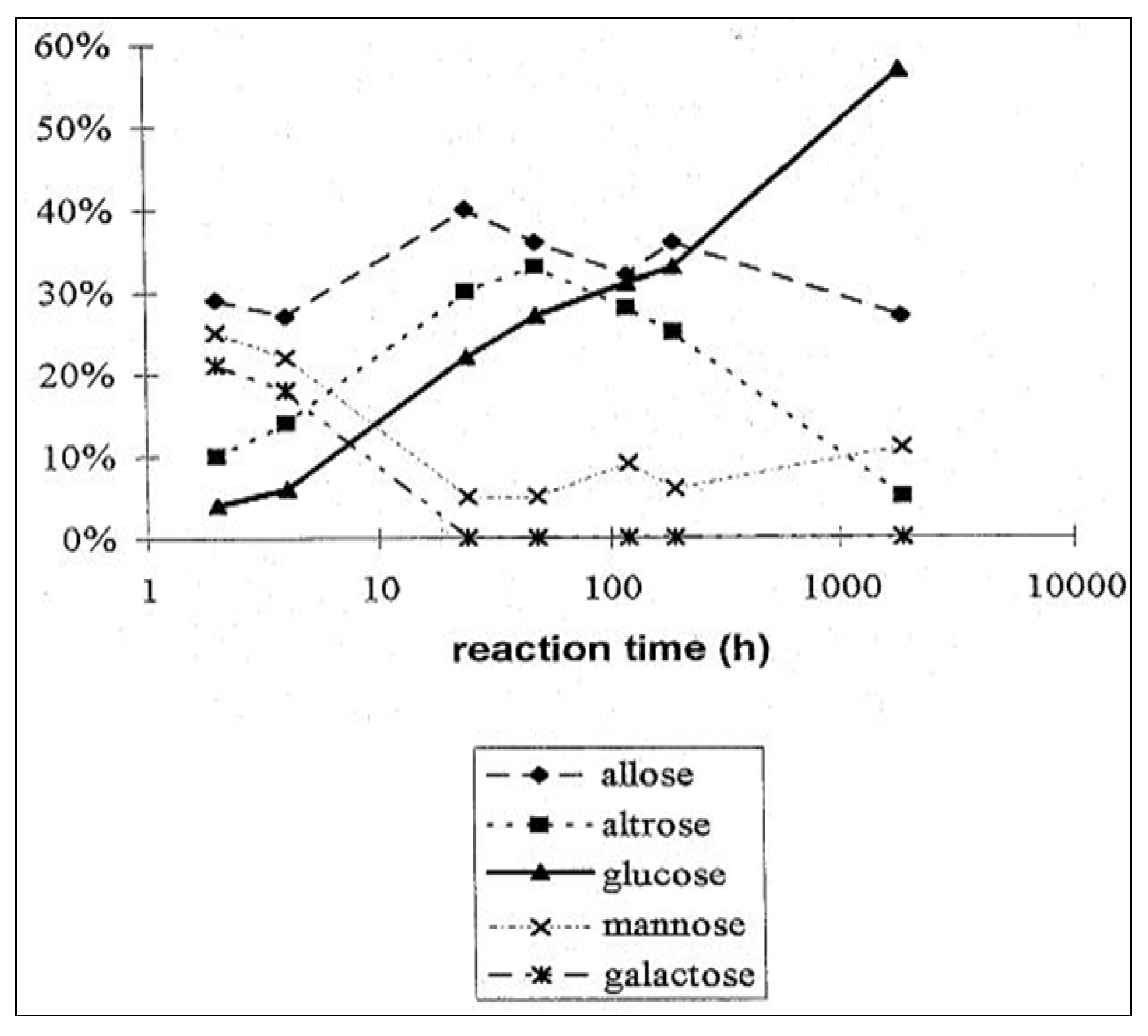
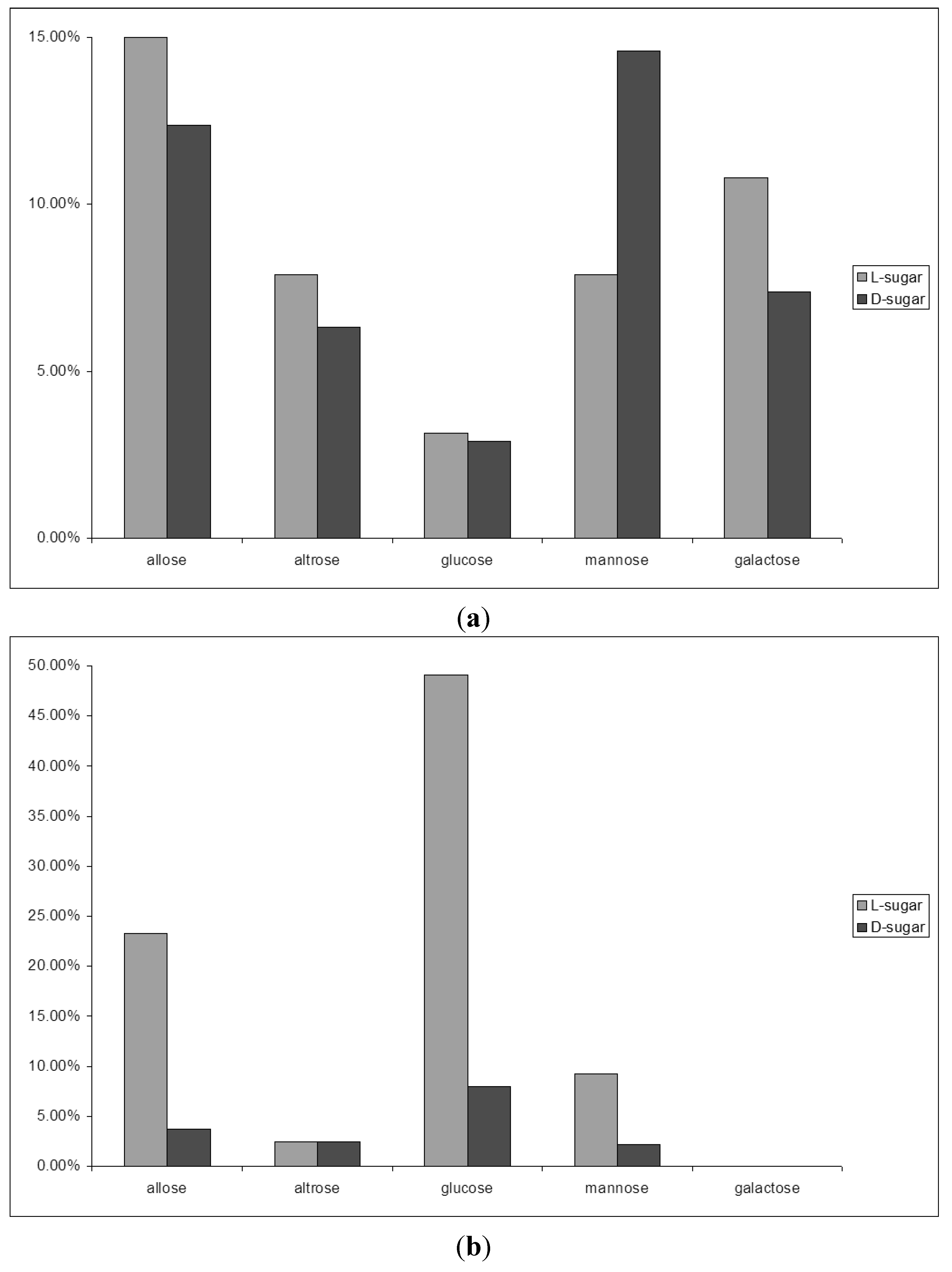
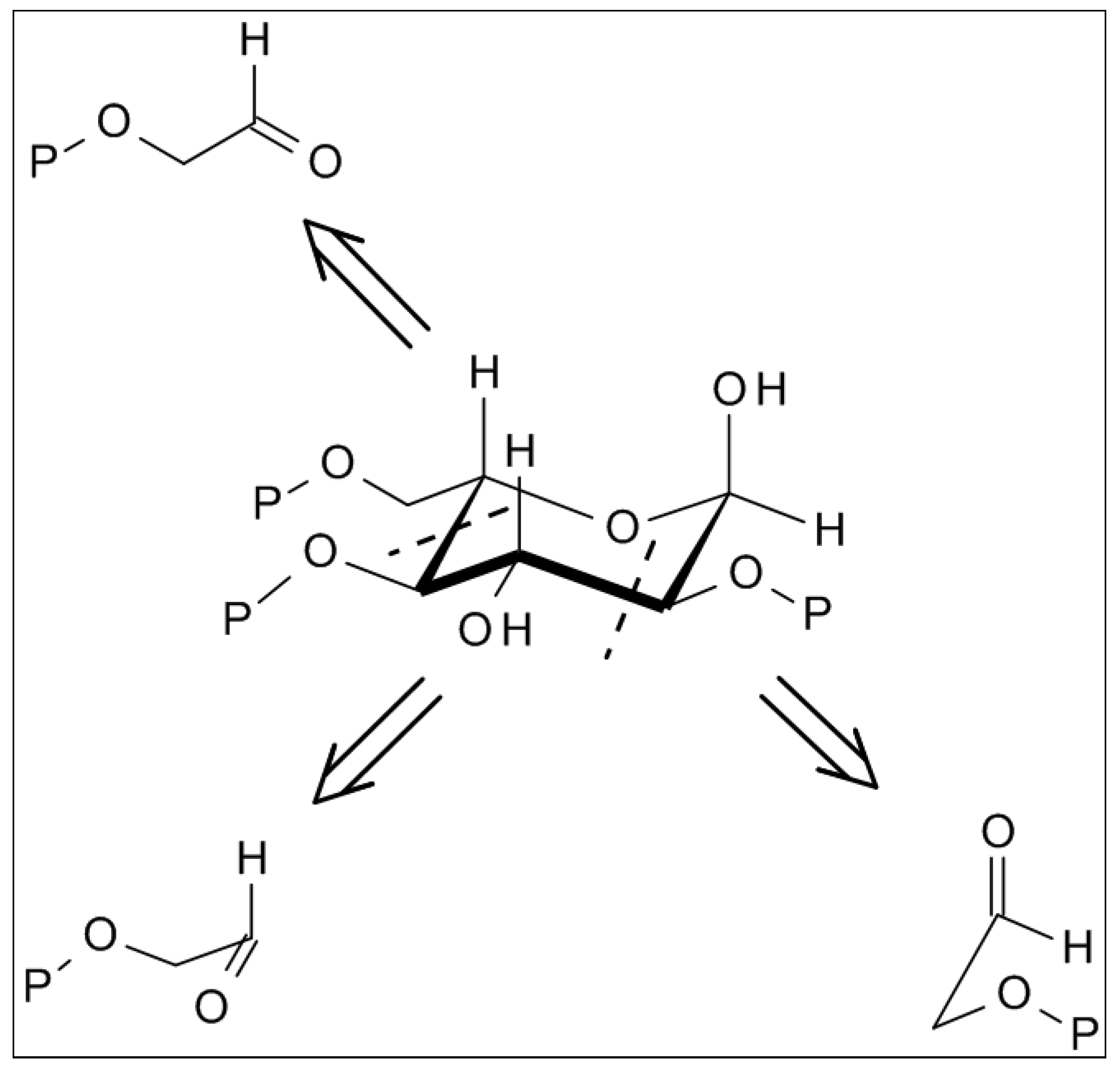
2.4. From Glycolaldehyde Phosphate to RNA
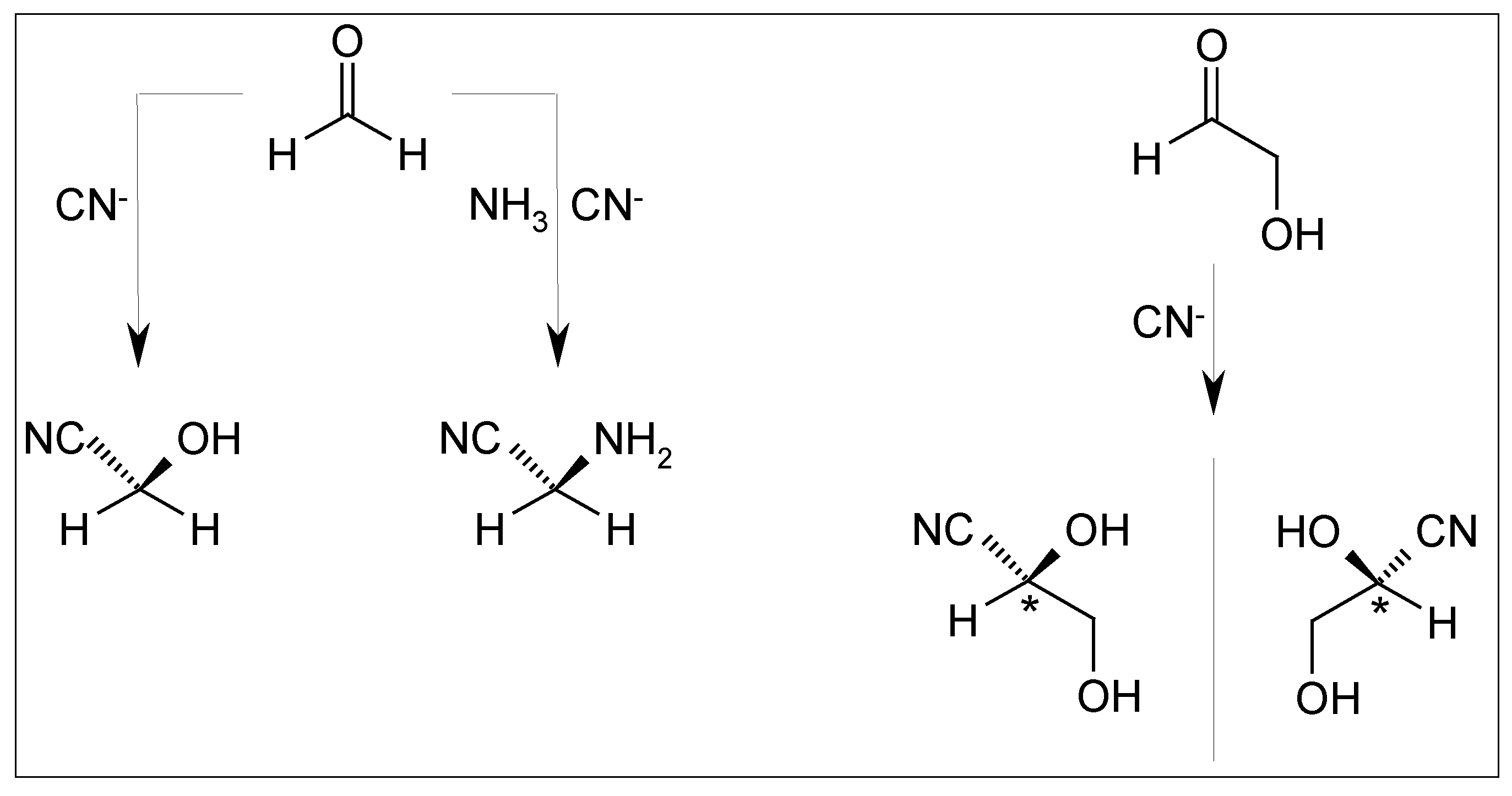
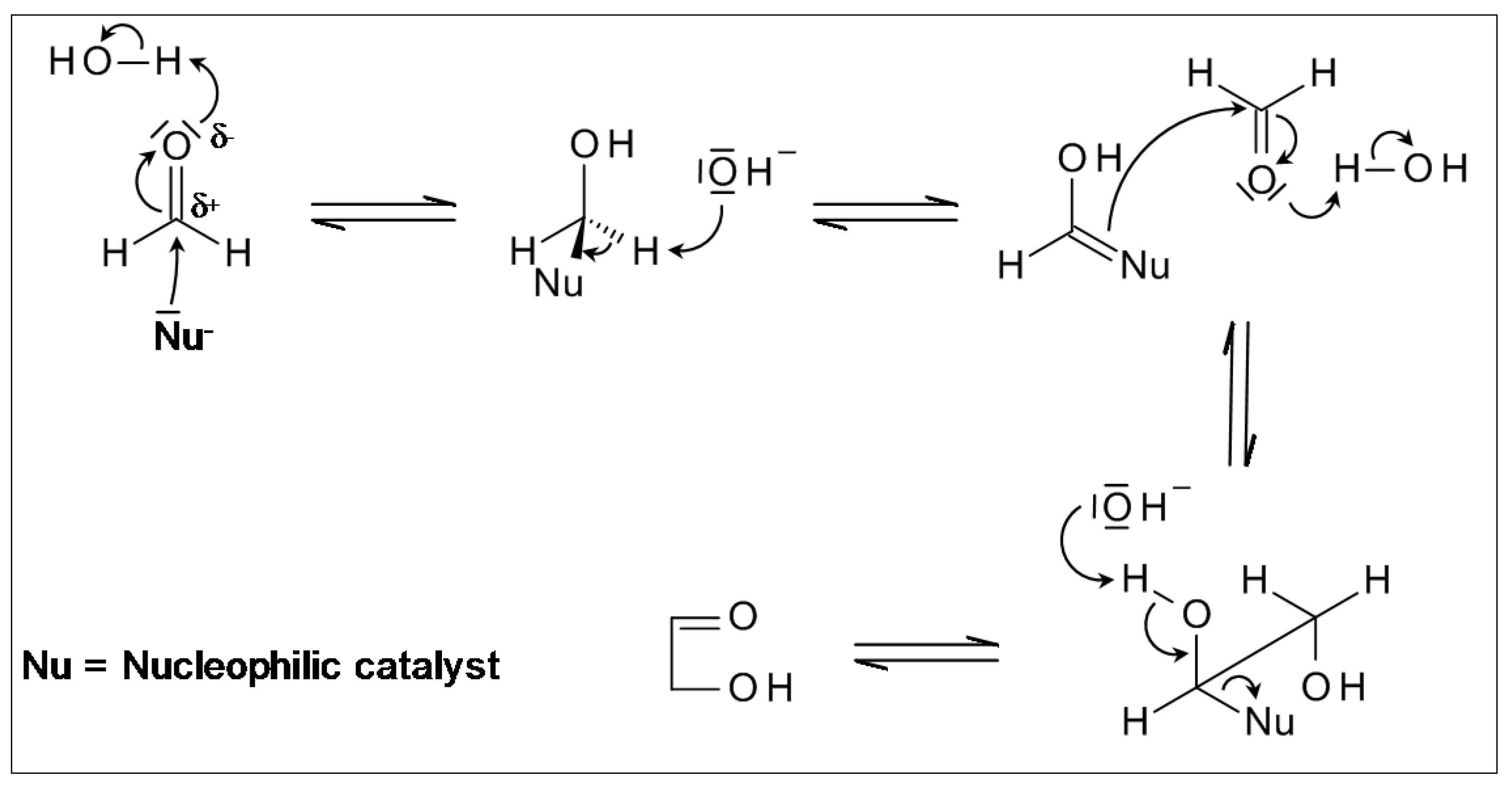
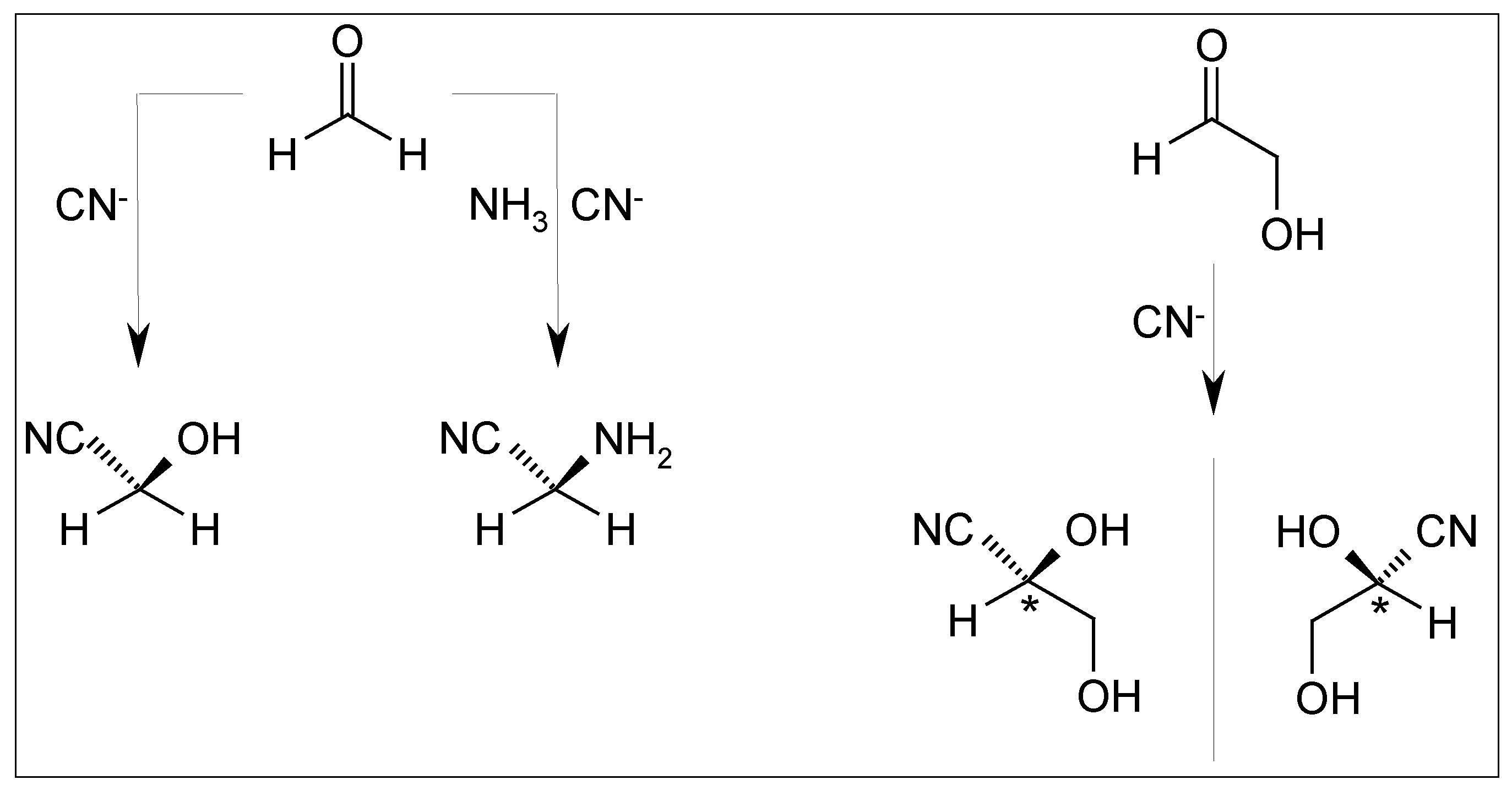
2.5. Cyanhydrine Formation as Potential Prebiotic Source of Hydroxy and Amino Acids and Their Polymers
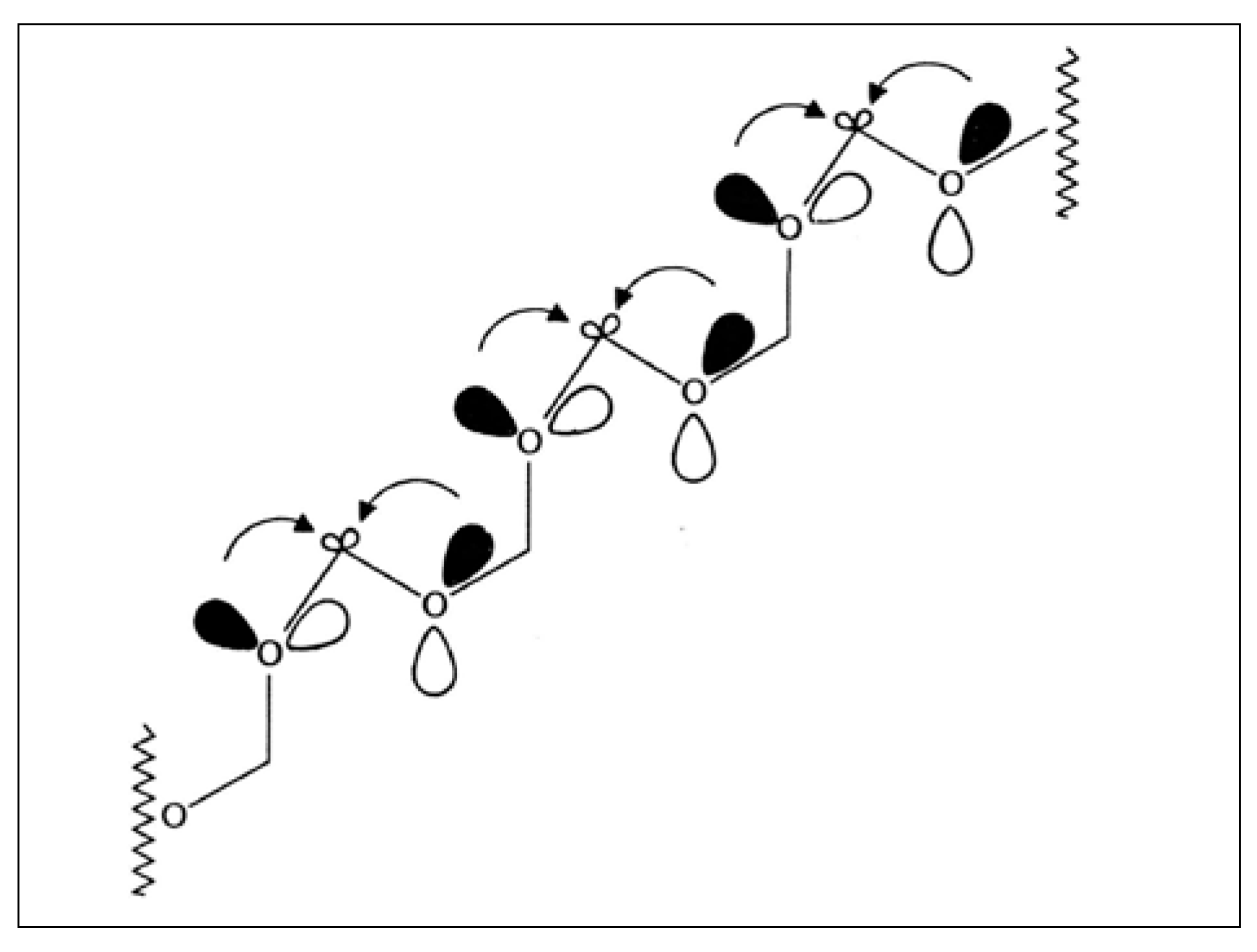
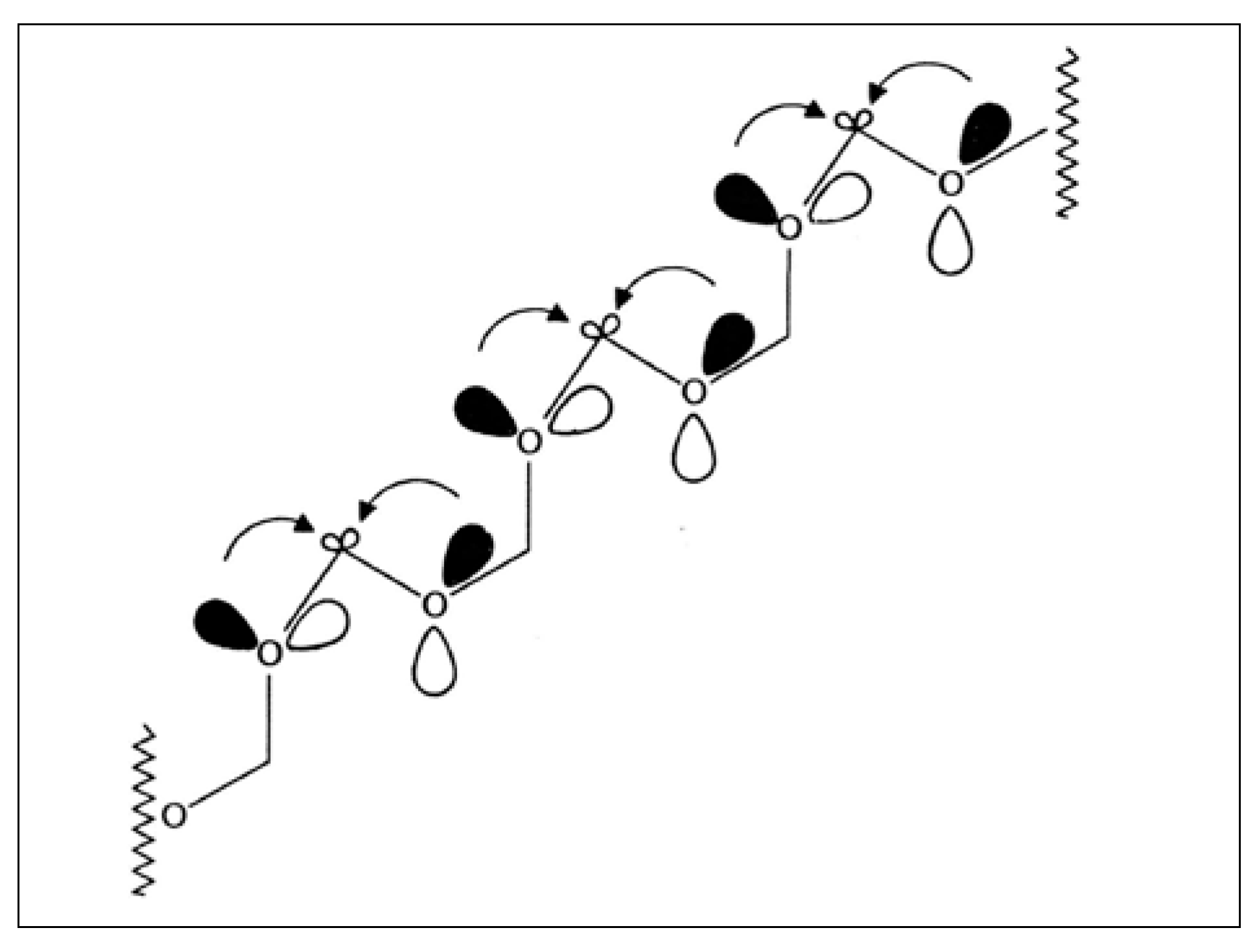
2.6. Back to Formaldehyde: Polyformaldehyde—Centrochirality to Helical Chirality
3. Conclusions—“From Accedentia to Essentia”
Acknowledgments
Conflict of Interest
References
- Saladino, R.; Botta, G.; Pino, S.; Costanzo, G.; di Mauro, E. Genetics first or metabolism first? The formamide clue. Chem. Soc. Rev. 2012, 41, 5526–5565. [Google Scholar] [CrossRef]
- Mizuno, T.; Weiss, A.H. Synthesis and utilization of formose sugars. Adv. Carbohydr. Chem. Biochem. 1974, 29, 173–227. [Google Scholar] [CrossRef]
- Socha, R.F.; Weiss, A.H.; Sakharov, M.M. Homogeneously catalyzed condensation of formaldehyde to carbohydrates: VII. An overall formose reaction model. J. Catal. 1981, 67, 207–217. [Google Scholar] [CrossRef]
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar]
- Miller, S.L.; Urey, H.C. Organic compound synthesis on the primitive earth. Science 1959, 130, 245–251. [Google Scholar]
- Miller, S.L.; Urey, H.C. Origin of life. Science 1959, 130, 1622–1624. [Google Scholar]
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196. [Google Scholar] [CrossRef]
- Decker, P.; Schweer, H.; Pohlmann, R.; Bioids, X. Identification of formose sugars, presumable prebiotic metabolites, using capillary gas chromatography/gas chromatography—Mass spectrometry of n-butoxime trifluoroacetates on OV-225. J. Chromatogr. 1982, 244, 281–291. [Google Scholar] [CrossRef]
- Oro, J. Synthesis of adenine from ammonium cyanide. Biochem. Biophys. Res. Commun. 1960, 2, 407–412. [Google Scholar] [CrossRef]
- Ferris, J.P.; Orgel, L.E. An unusual photochemical rearrangement in the synthesis of adenine from hydrogen cyanide. J. Am. Chem. Soc. 1966, 88, 1074. [Google Scholar] [CrossRef]
- Ferris, J.P.; Kuder, J.E. Chemical evolution. III. Photochemical conversion of enaminonitriles to imidazoles. J. Am. Chem. Soc. 1970, 92, 2527–2533. [Google Scholar] [CrossRef]
- Eschenmoser, A. Chemistry of potentially prebiological natural products. Orig. Life Evol. Biosph. 1994, 24, 389–423. [Google Scholar] [CrossRef]
- Larralde, R.; Robertson, M.P.; Miller, S. Rates of decomposition of ribose and other sugars: Implications for chemical evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 8158–8160. [Google Scholar] [CrossRef]
- Hollis, J.M.; Lovas, F.J.; Jewell, P.R. Interstellar glycolaldehyde: The first sugar. Astrophys. J. 2000, 540, L107–L110. [Google Scholar] [CrossRef]
- Cleaves, H.J. The origin of the biologically coded amino acids. J. Theor. Biol. 2010, 263, 490–498. [Google Scholar] [CrossRef]
- Cleaves, H.J.; Lazcano, A. The origin of biomolecules. ACS Symp. Ser. 2009, 1025, 17–43. [Google Scholar] [CrossRef]
- Yanagawa, H.; Kobayashi, Y.; Egami, F. Genesis of amino acids in the primeval sea: Formation of amino acids from sugars and ammonia in a modified sea medium. J. Biochem. 1980, 87, 359–362. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Ettmayer, P. Chiral lactols, VIII. A way for the asymmetric induction in the formation of sugars. Liebigs Ann. Chem. 1989, 7, 637–643. [Google Scholar]
- Müller, D.; Pitsch, S.; Kittaka, A.; Wagner, E.; Wintner, C.E.; Eschenmoser, A. Chemie von α-aminonitrilen. aldomerisierung von glycolaldehyd-phosphat zu racemischen hexose-2,4,6-triphosphaten und (in Gegenwart von Fromaldehyd) racemischen pentose-2,4-diphosphaten: rac-Allose-2,4,6-triphosphat und rac. Ribose-2,4-diphosphat sind die reaktionshauptprodukte. Helv. Chim. Acta 1990, 73, 1410–1468. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Ettmayer, P. Chiral Lactols, X. Allose as the main product of glycolaldehyde trimerization. Liebigs Ann. Chem. 1991, 5, 417–424. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Ettmayer, P.; Freissmuth, J. Chiral lactols, XII. Studies on the aldolizaton of glycolaldehyde catalyzed by bases. Liebigs Ann. Chem. 1994, 6, 611–613. [Google Scholar]
- Richter, P. Diastereoselektivität und Asymmetrische Induktion der Monosaccharidbildung aus 2-Hydroxyethanalderivaten; Shaker Verlag: Aachen, Germany, 1997. [Google Scholar]
- Noe, C.R.; Freissmuth, J.; Lachmann, B.; Richter, P.J.W. Goethe University: Frankfurt, Germany, Unpublished work. 2013.
- Eschenmoser, A. Vitamin B12: Experimente zur Frage nach dem Ursprung seiner molekularen Struktur. Angew. Chem. 1988, 100, 5–40. [Google Scholar] [CrossRef]
- Eschenmoser, A. Etiology of potentially primordial biomolecular structures: From Vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: A retrospective. Angew. Chem. Int. Ed. Engl. 2011, 50, 12412–12472. [Google Scholar] [CrossRef]
- Noe, C.R. Chirale Lactole, I. Die 2,3,3a,4,5,6,7,7a-Octahydro-7,8,8,-trimethyl-4,5,-methanobenzofuran-2-yl Schutzgruppe. Chem. Ber. 1982, 115, 1576–1590. [Google Scholar]
- Noe, C.R. Chirale Lactole, II. Racematspaltung und enantioselektive Acetalisierung mit der 2,3,3a,4,5,6,7,7a-Octahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2-yl Schutzgruppe. Chem. Ber. 1982, 115, 1591–1606. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Wagner, E.; Völlenkle, H. Chirale lactole, IV. Selektivitäten bei acetalisierungsreaktionen enantiomerenreiner lactole am beispiel von octahydro-7,8,8-trimethyl-5,8-methano-2H-1-benzopyran-2-ol. Chem. Ber. 1985, 118, 1733–1745. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Steinbauer, G.; Jangg, E.; Völlenkle, H. Chirale Lactole, VII. O,O- und O,N-Acetalbildungsreaktionen der enantiomerenreinen exo-anellierten Octahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2-yl Schutzgruppe. Chem. Ber. 1988, 121, 1231–1239. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Gärtner, P.; Mereiter, K.; Steinbauer, G. Chiral Lactols, XIV. Stereoselective fusion of five-membered ring lactols to the bornane ring system. Liebigs Ann. 1996, 6, 1015–1021. [Google Scholar]
- Noe, C.R. Chirale Lactole, III. Eine enantioslektive alkylierung der mercaptoessigsäure. Chem. Ber. 1982, 115, 1607–1616. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Steinbauer, G.; Völlenkle, H. Chirale Lactole, V. Synthese von benzoin aus meso-hydrobenzoin. Chem. Ber. 1985, 118, 4453–4458. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Ettmayer, P.; Gärtner, P.; Letschnig, M. The asymmetrical potential oft the glycoside bond. Österr. Chem. Z 1990, 91, 36–41. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Göstl, G.; Gärtner, P. Aminoalkohole. 1. Mitt.: Ein verfahren zur synthese enantiomerenreiner 1,2-aminoalkohole mit erythro-konfiguration. Monatsh. Chem. 1991, 122, 283–290. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Kürner, H.; Steinbauer, G.; Koberg, H.; Gärtner, P. Pheromone I, (+)-cis-disparlure: Synthese und feldtests. Monatsh. Chem. 1991, 122, 101–110. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Dungler, K.; Miculka, C.; Gärtner, P. Ein verfahren zur synthese enantiomerenreiner alkanole durch reduktive entschwefelung aus thiophenalkoholen. Monatsh. Chem. 1991, 122, 705–718. [Google Scholar] [CrossRef]
- Mäurer, M.; Stegmann, H.B. Chiral Recognition of diastereomeric esters and acetals by EPR and NMR investigations. Chem. Ber. 1990, 123, 1679B–1685B. [Google Scholar] [CrossRef]
- Kirby, A.J. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen; Springer: Berlin, Germany, 1983. [Google Scholar]
- Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry, Organic Chemistry Series; Baldwin, J.F., Ed.; Pergamon Press: Oxford, UK, 1983. [Google Scholar]
- Thibaudeau, C.; Acharya, P.; Chattopadhyaya, J. Stereoelectronic Effects in Nucleosides and Nucleotides and Their Structural Implications, 2nd ed.; Uppsala University Press: Uppsala, Sweden, 2005. [Google Scholar]
- Knollmüller, M.; Noe, C.R.; Oberhauser, B. Die acetalgruppe, 1. Mitt. acetale von halogenmethyl-arylcarbinolen. Monatsh. Chem. 1986, 117, 407–419. [Google Scholar]
- Knollmüller, M.; Noe, C.R.; Steinbauer, G.; Dungler, K. Enantiomer-selektive acetalbildung, ein verfahren zur reinherstellung bzw. Anreicherung von enantiomeren. Synthesis 1986, 1986, 501–505. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Oberhauser, B.; Steinbauer, G.; Wagner, E. Chirale lactole, VI. Eine methode zur bestimmung der absolutkonfiguration chiraler α-hydroxysubstitierter nitrile, alkine und aldehyde. Chem. Ber. 1986, 119, 729–743. [Google Scholar]
- Schönauer, K.J.; Walter, P.; Noe, C.R. Absolute configuration of secondary alcohols determined by gas chromatography. Monatsh. Chem. 1986, 117, 127–130. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Dungler, K.; Miculka, C. Stereoelektronische effekte und chirale erkennung, I. Diastereoselektive etherbildungen aus arylcarbinolen. Chem. Ber. 1994, 127, 359–365. [Google Scholar] [CrossRef]
- Noe, C.R.; Miculka, C.; Völlenkle, H. Konformationen diastereoisomerer bis-(alkylaryl)ether. Monatsh. Chem. 1994, 125, 983–990. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Ziebarth-Schroth, I.; Letschnig, M. Stereoelectronic effects and chiral recognition. II. Kinetic und thermodynamic control in the formation of chiral thioacetals and chiral thioethers. Liebigs Ann. 1996, 1996, 1009–1013. [Google Scholar]
- Knollmüller, M.; Gaischin, L.; Ferencic, M.; Noe-Letschnig, M.; Girreser, U.; Gärtner, P.; Mereiter, K.; Noe, C.R. Addition von enantiomerenreinen Aminen an aktivierte Olefine, 1. Mitt. Über die Addition an w-Nitrostyrol. Monatsh. Chem. 1998, 129, 1025–1033. [Google Scholar]
- Knollmüller, M.; Ferencic, M.; Gärtner, P.; Girreser, U.; Klinge, M.; Gaischin, L.; Mereiter, K.; Noe, C.R. Addition von enantiomerenreinen Aminen an aktivierte Olefine, 2. Mitt. Über die Addition an (E)-4-Oxo-4phenyl-2-butensäure-ethylester. Monatsh. Chem. 1999, 130, 769–782. [Google Scholar]
- Koppenhoefer, B.; Schwierskott, M.; Brendle, H.-G.; Noe, C.R.; Jangg, E.; Völlenkle, H. Highly Regioselective Bromination of [3aR-(3aα,4β,7β,7aα]-Hexahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2(3H)-one via Nonclassical Bornyl Cations. J. Org. Chem. 1996, 61, 4476–4479. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Jangg, E.; Gmeiner, G.P.; Urban, E.; Eppacher, S. Increasing chiral recognition in acetal formation. Chirality 2009, 21, 428–435. [Google Scholar] [CrossRef]
- Mehraban, K.; Lachmann, B.; Urban, E.; Mereiter, K.; Eppacher, S.; Noe, C.R. Synthesis of (S)-α-hydroxy acids. Helv. Chim. Acta 2013. submitted for publication. [Google Scholar]
- Noe, C.R.; Knollmüller, M.; Göstl, G.; Oberhauser, B.; Völlenkle, H. Stereoelectronic Effects and Chiral Recognition: A Natural System of Relationships between Chiral Compounds based on Selectivities in the Formation of Acetals. Angew. Chem. Int. Ed. Engl. 1987, 26, 442–444. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Wagner, E.; Völlenkle, H. Kohlenhydrat-Modelle, I. Kinetische und thermodynamische Effekte bei Acetalisierungsreaktionen enantiomerenreiner Thiolactole. Chem. Ber. 1985, 118, 3299–3310. [Google Scholar] [CrossRef]
- Dungler, K. Synthese und Reaktionen Enantiomerenreiner Thiolactole. MS.c. Thesis, University of Technology Vienna, Vienna, Austria, 1984. [Google Scholar]
- Ziebart-Schroth, I. Kinetische und Thermodynamische Effekte bei der Bildung Chiraler Thioacetale und Thioether. MS.c. Thesis, University of Technology Vienna, Vienna, Austria, 1990. [Google Scholar]
- Freissmuth, J. Zucker aus Glykolaldehyd-Synthese und Enantiomeranalytik. Ph.D. Thesis, University of Technology Vienna, Vienna, Austria, 1995. [Google Scholar]
- Noe, C.R.; Lachmann, B.; Eppacher, S.; Richter, P.; Freissmuth, J.J.W. Goethe University: Frankfurt, Germany, Unpublished work. 2013.
- Noe, C.R.; Freissmuth, J. Capillary zone electrophoresis of aldose enantiomers: Separation after derivatization with S-(−)-1-phenylethylamine. J. Chromat. A 1995, 704, 503–512. [Google Scholar] [CrossRef]
- Noe, C.R.; Freissmuth, J.; Rothley, D.; Lachmann, B.; Richter, P. Kapillarelektrophoretische analytik komplexer kohlenhydratgemische. Pharmazie 1996, 51, 868–873. [Google Scholar]
- Noe, C.R.; Freissmuth, J.; Eppacher, S.; Lachmann, B.; Richter, P.J.W. Goethe University: Frankfurt, Germany, Unpublished work. 2013.
- Weber, A.L.; Pizzarello, S. The peptide-catalyzed stereospecific synthesis of tetroses: A possible model for prebiotic molecular evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 12713–12717. [Google Scholar] [CrossRef]
- Pizzarello, S.; Weber, A.L. Stereoselective syntheses of pentose sugars under realistic prebiotic conditions. Orig. Life Evol. Biosph. 2010, 40, 3–10. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef]
- Benner, S.A.; Kim, H.-J.; Carrigan, M.A. Asphalt, water and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc. Chem. Res. 2012, 45, 2025–2034. [Google Scholar] [CrossRef]
- Eschenmoser, A. The search for the chemistry of life’s origin. Tetrahedron 2007, 63, 12821–12844. [Google Scholar] [CrossRef]
- Pitsch, S.; Wendeborn, S.; Juan, B.; Eschenmoser, A. Why pentose- and not hexose-nucleic acids? Part VII. Pyranosyl-RNA (“p-RNA”). Preliminary communication. Helv. Chim. Acta 1993, 76, 2161–2183. [Google Scholar]
- Choudhary, A.; Kamer, K.J.; Powner, M.W.; Sutherland, J.D.; Raines, R.T. A stereoelectronic effect in prebiotic nucleotide synthesis. ACS Chem. Biol. 2010, 5, 655–657. [Google Scholar] [CrossRef]
- Drenkard, S.; Ferris, J.; Eschenmoser, A. Chemie von α-aminonitrilen. aziridin-2-carbonitril, ein vorläufer von rca-O3Phosphoserinnitril und glycolaldehyd-phosphat. Helv. Chim. Acta 1990, 73, 1373–1390. [Google Scholar]
- Eschenmoser, A. Searching for nucleic acid alternatives. In Chemical Synthetic Biology; Luisi, P.L., Chiarabelli, C., Eds.; John Wiley & Sons: Chichester, UK, 2011; pp. 4–47. [Google Scholar]
- Powner, M.W.; Zheng, S.L.; Szostak, J.W. Multicomponent assembly of proposed DNA precursors in water. J. Am. Chem. Soc. 2012, 134, 13889–13895. [Google Scholar] [CrossRef]
- Majumdar, L.; Das, A.; Chakrabarti, S.K.; Chakrabarti, S. Hydro-chemical study of the evolution of interstellar pre-biotic molecules during the collapse of molecular clouds. Res. Astron. Astrophys. 2012, 12, 1613–1624. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Gärtner, P.; Katikarides, E.; Gaischin, L.; Völlenkle, H. Chiral Lactols, XIII. On the determination of the absolute configuration of aromatic cyanohydrins and structurally related compounds. Liebigs Ann. 1995, 1995, 1353–1360. [Google Scholar] [CrossRef]
- Jangg, E. Stereoelektronische Effekte und chirale Erkennung. Ph.D. Thesis, University of Vienna, Vienna, Austria, 1990. [Google Scholar]
- Girreser, U.; Haberhauer, G.; Noe, C.R. O,O-acetal formation of exo-annelated octahydro-7,8,8-trimethyl-4,7-methanobenzofuran-2-ol with lactic acid and phenyllactic acid derivates. Monatsh. Chem. 1998, 129, 281–289. [Google Scholar]
- Letschnig, M. Untersuchungen über den Einfluss stereoelektronischer Effekte auf die Bildung chiraler Acylale und Ester. MS.c. Thesis, University of Technology Vienna, Vienna, Austria, 1987. [Google Scholar]
- Noe, C.R.; Miculka, C.; Bats, J.W. Helicity of oligomeric formaldehyde. Angew. Chem. Int. Ed. Engl. 1994, 33, 1476–1478. [Google Scholar] [CrossRef]
- Bats, J.W.; Miculka, C.; Noe, C.R. 1,17-Diphenyl-2,4,6,8,10,12,14,16-octaoxaheptadecane. Acta Crystallogr. 2007, 63, o190–o192. [Google Scholar] [CrossRef]
- Eppacher, S.; Giester, G.; Bats, J.W.; Noe, C.R. Enantiomerically pure poly(oxymethylene)helices: Correlating helicity with centrochirality. Helv. Chim. Acta 2008, 91, 581–597. [Google Scholar] [CrossRef]
- Noe, C.R.; Knollmüller, M.; Ettmayer, P. Paraformaldehyde as possible chiral amplifier. Angew. Chem. Int. Ed. Engl. 1988, 27, 1379–1380. [Google Scholar] [CrossRef]
- Miculka, C.; Noe, C.R.; Eppacher, S. Chirality transfer in the formation of poly(oxymethylene) helices by anionic polymerization. Helv. Chim. Acta 2012, 95, 845–851. [Google Scholar] [CrossRef]
- Lambert, J.B.; Lu, G.; Singer, S.R.; Kolb, V.M. Silicate complexes of sugars in aqueous solutions. J. Am. Chem. Soc. 2004, 126, 9611–9625. [Google Scholar] [CrossRef]
- Benner, S.A.; Kim, H.J.; Kim, M.J.; Ricardo, A. Planetary organic chemistry and the origins of biomolecules. Cold Spring Harb. Perspect. Biol. 2010, 2, a003467. [Google Scholar] [CrossRef]
- De Spinoza, B. Ethica Ordine Geometrico Demonstrata, 3rd ed.; Bartuschat, W., Ed.; Felix Meiner Verlag: Hamburg, Germany, 2010; p. 70. [Google Scholar]
- Oparin, A.I. Genesis and Evolutionary Development of Life; Academic Press: New York, NY, USA, 1968; pp. 9–40. [Google Scholar]
- Perry, R.S.; Kolb, V.M. On the applicability of darwinian principles to chemical evolution that led to life. Int. J. Astrobiol. 2004, 3, 45–53. [Google Scholar] [CrossRef]
- Kolb, V.M. On the applicability of the principle of the quantity-to-quality transition to chemical evolution that led to life. Int. J. Astrobiol. 2005, 4, 227–232. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Noe, C.R.; Freissmuth, J.; Richter, P.; Miculka, C.; Lachmann, B.; Eppacher, S. Formaldehyde—A Key Monad of the Biomolecular System. Life 2013, 3, 486-501. https://doi.org/10.3390/life3030486
Noe CR, Freissmuth J, Richter P, Miculka C, Lachmann B, Eppacher S. Formaldehyde—A Key Monad of the Biomolecular System. Life. 2013; 3(3):486-501. https://doi.org/10.3390/life3030486
Chicago/Turabian StyleNoe, Christian R., Jerome Freissmuth, Peter Richter, Christian Miculka, Bodo Lachmann, and Simon Eppacher. 2013. "Formaldehyde—A Key Monad of the Biomolecular System" Life 3, no. 3: 486-501. https://doi.org/10.3390/life3030486
APA StyleNoe, C. R., Freissmuth, J., Richter, P., Miculka, C., Lachmann, B., & Eppacher, S. (2013). Formaldehyde—A Key Monad of the Biomolecular System. Life, 3(3), 486-501. https://doi.org/10.3390/life3030486