
Future Comorbidities in an Aging Cystic Fibrosis Population

Abstract
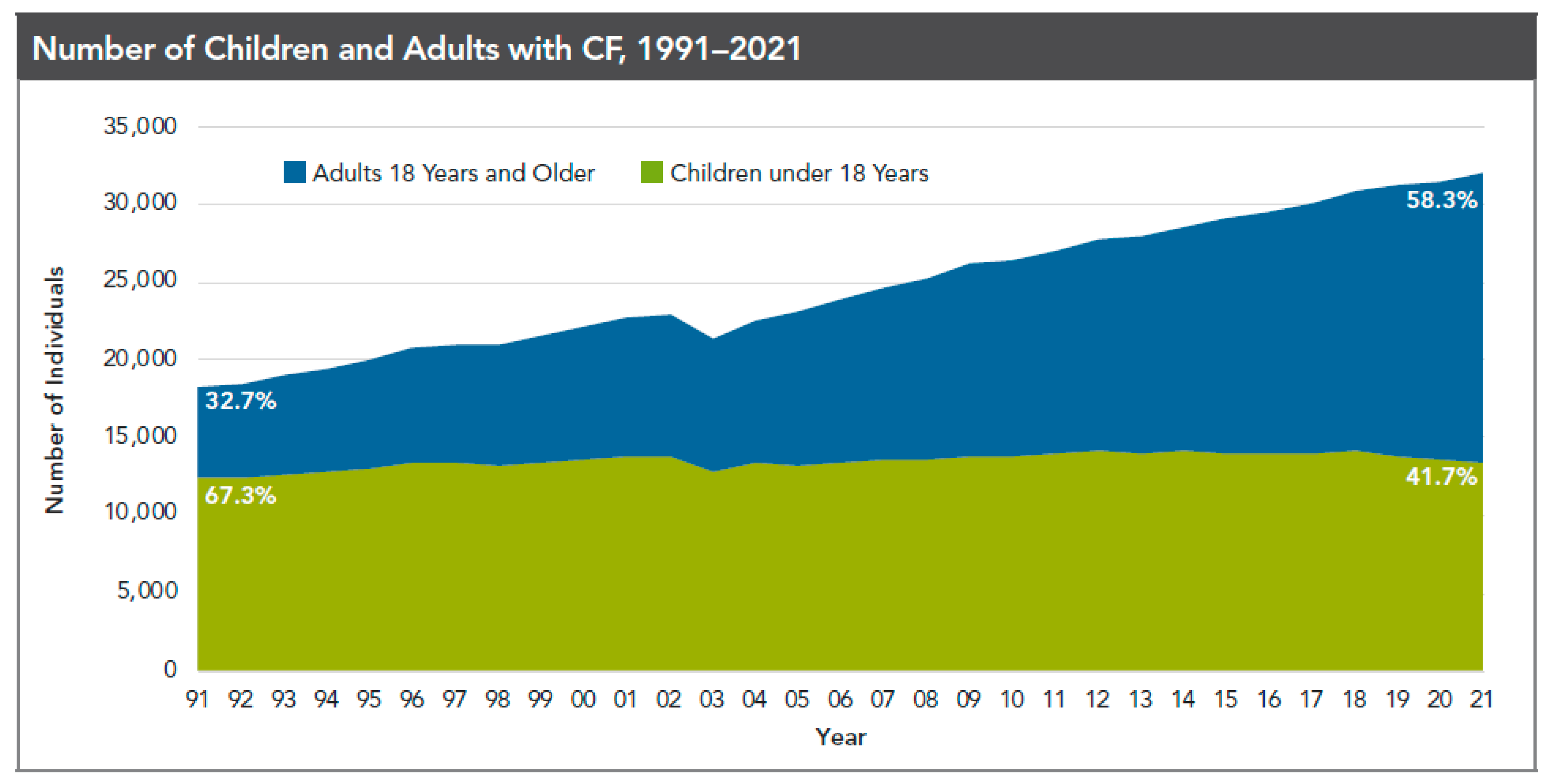
1. Background
2. Cardiovascular Disease
3. Dyslipidemia
4. Cystic Fibrosis Related Diabetes
5. Pulmonary Hypertension
6. Obstructive Sleep Apnea
7. CF Liver Disease
8. Bone Health
9. Cancer
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic Fibrosis-Related Diabetes. Front. Endocrinol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shen, Y.; Zheng, J. A review of cystic fibrosis: Basic and clinical aspects. Anim. Model Exp. Med. 2021, 4, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation. Patient Registry 2021 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2021. [Google Scholar]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Daly, C.; Vega-Hernandez, G.; MacGregor, G.; Rubin, J.L. Elexacaftor/tezacaftor/ivacaftor projected survival and long-term health outcomes in people with cystic fibrosis homozygous for F508del. J. Cyst. Fibros. 2023. [CrossRef]
- Shah, P.H.; Lee, J.H.; Salvi, D.J.; Rabbani, R.; Gavini, D.R.; Hamid, P. Cardiovascular System Involvement in Cystic Fibrosis. Cureus 2021, 13, e16723. [Google Scholar] [CrossRef]
- Onady, G.M.; Farinet, C.L. An adult cystic fibrosis patient presenting with persistent dyspnea: Case Report. BMC Pulm. Med. 2006, 6, 9. [Google Scholar] [CrossRef]
- Sandouk, Z.; Nachawi, N.; Simon, R.; Wyckoff, J.; Putman, M.S.; Kiel, S.; Soltman, S.; Moran, A.; Moheet, A. Coronary artery disease in patients with cystic fibrosis—A case series and review of the literature. J. Clin. Transl. Endocrinol. 2022, 30, 100308. [Google Scholar] [CrossRef]
- Perrin, F.M.; Serino, W. Ischaemic heart disease—A new issue in cystic fibrosis? J. R. Soc. Med. 2010, 103 (Suppl. 1), S44–S48. [Google Scholar] [CrossRef]
- Poore, S.; Berry, B.; Eidson, D.; McKie, K.T.; Harris, R.A. Evidence of vascular endothelial dysfunction in young patients with cystic fibrosis. Chest 2013, 143, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.H.; Garrod, R.; Ho, T.B.; Knight, R.K.; Cockcroft, J.R.; Shale, D.J.; Bolton, C.E. Increased augmentation index in patients with cystic fibrosis. Eur. Respir. J. 2009, 34, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Ronan, N.J.; Elborn, J.S.; Plant, B.J. Current and emerging comorbidities in cystic fibrosis. Presse Méd. 2017, 46 Pt 2, e125–e138. [Google Scholar] [CrossRef]
- Uramoto, H.; Okada, T.; Okada, Y. Protective role of cardiac CFTR activation upon early reperfusion against myocardial infarction. Cell. Physiol. Biochem. 2012, 30, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, B.; Nash, E.F.; Tullis, E.; Pencharz, P.B.; Brotherwood, M.; Dupuis, A.; Stephenson, A. Prevalence of dyslipidemia in adults with cystic fibrosis. J. Cyst. Fibros. 2010, 9, 24–28. [Google Scholar] [CrossRef]
- Alves Cde, A.; Lima, D.S. Cystic fibrosis-related dyslipidemia. J. Bras. Pneumol. 2008, 34, 829–837. [Google Scholar] [CrossRef]
- Figueroa, V.; Milla, C.; Parks, E.J.; Schwarzenberg, S.J.; Moran, A. Abnormal lipid concentrations in cystic fibrosis. Am. J. Clin. Nutr. 2002, 75, 1005–1011. [Google Scholar] [CrossRef]
- Worgall, T.S. Lipid metabolism in cystic fibrosis. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 105–109. [Google Scholar] [CrossRef]
- Nowak, J.K.; Szczepanik, M.; Wojsyk-Banaszak, I.; Mądry, E.; Wykrętowicz, A.; Krzyżanowska-Jankowska, P.; Drzymała-Czyż, S.; Nowicka, A.; Pogorzelski, A.; Sapiejka, E.; et al. Cystic fibrosis dyslipidaemia: A cross-sectional study. J. Cyst. Fibros. 2019, 18, 566–571. [Google Scholar] [CrossRef]
- Brennan, A.L.; Beynon, J. Clinical updates in cystic fibrosis-related diabetes. Semin. Respir. Crit. Care Med. 2015, 36, 236–250. [Google Scholar]
- Tonelli, A.R. Pulmonary hypertension survival effects and treatment options in cystic fibrosis. Curr. Opin. Pulm. Med. 2013, 19, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Aldashev, A.; Welsh, D.; Peacock, A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur. Respir. J. 2007, 30, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D., Jr.; Tobias, J.D.; Mansour, H.M.; Kirkby, S.; McCoy, K.S.; Daniels, C.J.; Whitson, B.A. Pulmonary hypertension in cystic fibrosis with advanced lung disease. Am. J. Respir. Crit. Care Med. 2014, 190, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Kapnadak, S.G.; Dimango, E.; Hadjiliadis, D.; Hempstead, S.E.; Tallarico, E.; Pilewski, J.M.; Faro, A.; Albright, J.; Benden, C.; Blair, S.; et al. Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease. J. Cyst. Fibros. 2020, 19, 344–354. [Google Scholar] [CrossRef]
- Spicuzza, L.; Sciuto, C.; Leonardi, S.; La Rosa, M. Early occurrence of obstructive sleep apnea in infants and children with cystic fibrosis. Arch. Pediatr. Adolesc. Med. 2012, 166, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Dancey, D.R.; Tullis, E.D.; Heslegrave, R.; Thornley, K.; Hanly, P.J. Sleep quality and daytime function in adults with cystic fibrosis and severe lung disease. Eur. Respir. J. 2002, 19, 504–510. [Google Scholar] [CrossRef]
- Toledano, M.B.; Mukherjee, S.K.; Howell, J.; Westaby, D.; Khan, S.A.; Bilton, D.; Simmonds, N.J. The emerging burden of liver disease in cystic fibrosis patients: A UK nationwide study. PLoS ONE 2019, 14, e0212779. [Google Scholar] [CrossRef]
- Sasame, A.; Stokes, D.; Bourke, B.; Connolly, L.; Fitzpatrick, E.; Rowland, M. The impact of liver disease on mortality in cystic fibrosis—A systematic review. J. Cyst. Fibros. 2022, 21, 202–211. [Google Scholar] [CrossRef]
- Issa, Z.; Gohy, S.; Zech, F.; Baldin, P.; Delire, B.; Dahlqvist, G. Prevalence and Characteristics of Cystic Fibrosis Liver Disease: A Study highlighting the lack of historical diagnosis. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101977. [Google Scholar] [CrossRef]
- Mendizabal, M.; Reddy, K.R.; Cassuto, J.; Olthoff, K.M.; Faust, T.W.; Makar, G.A.; Rand, E.B.; Shaked, A.; Abt, P.L. Liver transplantation in patients with cystic fibrosis: Analysis of United Network for Organ Sharing data. Liver Transpl. 2011, 17, 243–250. [Google Scholar] [CrossRef]
- Nichols, D.P.; Donaldson, S.; Frederick, C.A.; Freedman, S.D.; Gedfond, D. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J. Cyst. Fibros. 2021, 20, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Aris, R.M.; Renner, J.B.; Winders, A.D.; Buell, H.E.; Riggs, D.B.; Lester, G.E.; Onties, D.A. Increased rate of fractures and severe kyphosis: Sequela of living to adulthood with cystic fibrosis. Ann. Intern. Med. 1998, 128, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, J.; Delion, M.; Gangloff, S.; Braux, J.; Velard, F. Bone Disease in cystic fibrosis: New pathogenic insights opening novel therapies. Osteoporos. Int. 2016, 27, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Putman, M.S.; Baker, J.F.; Uluer, A.; Herlyn, K.; Lapey, A.; Sicilian, L.; Tillotson, A.P.; Gordon, C.M.; Merkel, P.A.; Finkelstein, J.S. Trends in bone mineral density in young adults with cystic fibrosis over a 15 year period. J. Cyst. Fibros. 2015, 14, 526–532. [Google Scholar] [CrossRef]
- Marquette, M.; Haworth, C.S. Bone health and disease in cystic fibrosis. Paediatr. Respir. Rev. 2016, 20, 2–5. [Google Scholar] [CrossRef]
- Shead, E.F.; Haworth, C.S.; Gunn, E.; Bilton, D.; Scott, M.A.; Compston, J.E. Osteoclastogenesis during infective exacerbations in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 306–311. [Google Scholar] [CrossRef]
- Jeffery, T.C.; Chang, A.B.; Conwell, L.S. Bisphosphonates for osteoporosis in people with cystic fibrosis. Cochrane Database Syst. Rev. 2023, 1, CD002010. [Google Scholar]
- Aris, R.M.; Merkel, P.A.; Bachrach, L.K.; Borowitz, D.S.; Boyle, M.P.; Elkin, S.L.; Guise, T.A.; Hardin, D.S.; Haworth, C.S.; Holick, M.F.; et al. Guide to bone health and disease in cystic fibrosis. J. Clin. Endocrinol. Metab. 2005, 90, 1888–1896. [Google Scholar] [CrossRef]
- Robertson, C.M.; Hawkins, M.M. Childhood cancer and cystic fibrosis. J. Natl. Cancer Inst. 1995, 87, 1486–1487. [Google Scholar] [CrossRef]
- Maisonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer Risk in Cystic Fibrosis: A 20-Year Nationwide Study from the United States. JNCI J. Natl. Cancer Inst. 2013, 105, 122–129. [Google Scholar] [CrossRef]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Song, C.; Li, J.; Sun, Q. CFTR Functions as a Tumor Suppressor and Is Regulated by DNA Methylation in Colorectal Cancer. Cancer Manag. Res. 2020, 12, 4261–4270. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wei, J.; Na, R.; Resurreccion, W.K.; Zheng, S.L.; Hulick, P.J.; Helfand, B.T.; Talamonti, M.S.; Xu, J. Cystic fibrosis F508del carriers and cancer risk: Results from the UK Biobank. Int. J. Cancer 2021, 148, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Appelt, D.; Fuchs, T.; Steinkamp, G.; Ellemunter, H. Malignancies in patients with cystic fibrosis: A case series. J. Med. Case Rep. 2022, 16, 27. [Google Scholar] [CrossRef]
- Hadjiliadis, D.; Khoruts, A.; Zauber, A.G.; Hempstead, S.E.; Maisonneuve, P.; Lowenfels, A.B.; Cystic Fibrosis Colorectal Cancer Screening Task Force. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology 2018, 154, 736–745.e14. [Google Scholar] [CrossRef]
- Niccum, D.E.; Billings, J.L.; Dunitz, J.M.; Khoruts, A. Colonoscopic screening shows increased early incidence and progression of adenomas in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 548–553. [Google Scholar] [CrossRef]
- Fink, A.K.; Yanik, E.L.; Marshall, B.C.; Wilschanski, M.; Lynch, C.F.; Austin, A.A.; Copeland, G.; Safaeian, M.; Engels, E.A. Cancer risk among lung transplant recipients with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 91–97. [Google Scholar] [CrossRef]
- Allen, U.D.; Preiksaitis, J.K.; AST Infectious Diseases Community of Practice. Post-transplant lymphoproliferative disorders, Epstein-Barr virus infection, and disease in solid organ transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13652. [Google Scholar] [CrossRef]
- Lowery, E.M.; Adams, W.; Grim, S.A.; Clark, N.M.; Edwards, L.; Layden, J.E. Increased risk of PTLD in lung transplant recipients with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 727–734. [Google Scholar] [CrossRef]


{kind=link}
| BMD T/Z Score | Interpretation | Management | Follow-Up DXA Screening |
|---|---|---|---|
| ≥1.0 | Normal | ● Monitor ● Maintain nutritional health | Repeat screening every 5 years |
| Between −1.0 and −2.0 | Osteopenia | ● Address calcium, vitamin D insufficiency ● Minimize corticosteroid use ● Improve glycemic control in CFRD ● Optimize respiratory health to reduce Pex ● Consider bisphosphonate therapy in patients with history of fragility fractures, BMD loss of > 3–5%, and/or are awaiting transplant | Repeat every 2 to 4 years |
| ≤−2.0 | Osteoporosis | ● Start treatment (e.g., bisphosphonate, denosumab) | Annually |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ticona, J.H.; Lapinel, N.; Wang, J. Future Comorbidities in an Aging Cystic Fibrosis Population. Life 2023, 13, 1305. https://doi.org/10.3390/life13061305
Ticona JH, Lapinel N, Wang J. Future Comorbidities in an Aging Cystic Fibrosis Population. Life. 2023; 13(6):1305. https://doi.org/10.3390/life13061305
Chicago/Turabian StyleTicona, Javier Humberto, Nicole Lapinel, and Janice Wang. 2023. "Future Comorbidities in an Aging Cystic Fibrosis Population" Life 13, no. 6: 1305. https://doi.org/10.3390/life13061305
APA StyleTicona, J. H., Lapinel, N., & Wang, J. (2023). Future Comorbidities in an Aging Cystic Fibrosis Population. Life, 13(6), 1305. https://doi.org/10.3390/life13061305