
Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Patients
- Worsening respiratory symptoms;
- Absolute decline in the forced vital capacity (FVC) > 5% predicted or absolute decline in the diffusive capacity of the lungs (DLco) > 10% predicted within 1 year of follow-up;
- Radiological evidence of disease progression.
2.3. Data Collection
2.4. Statistical Analysis
3. Results
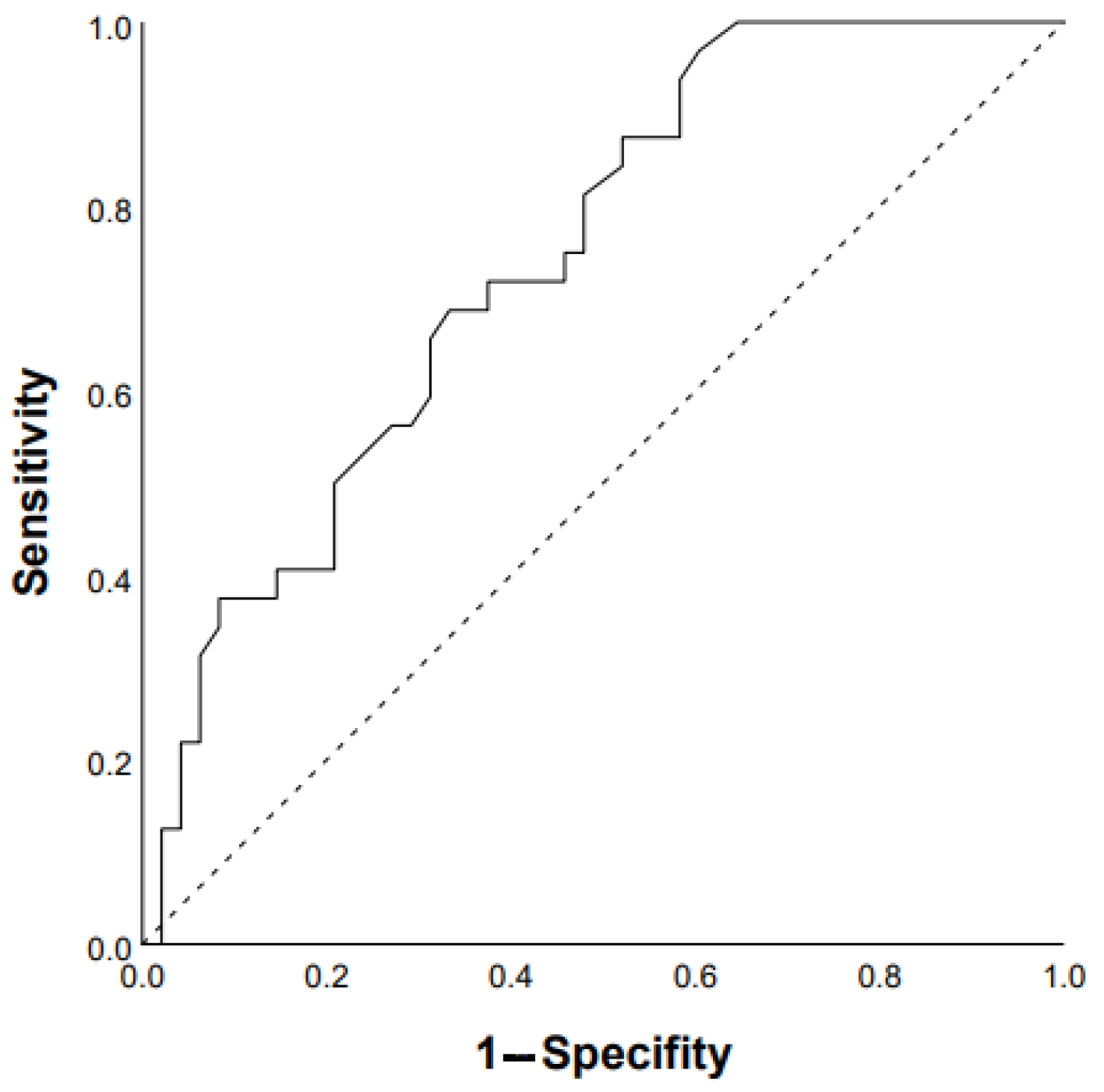
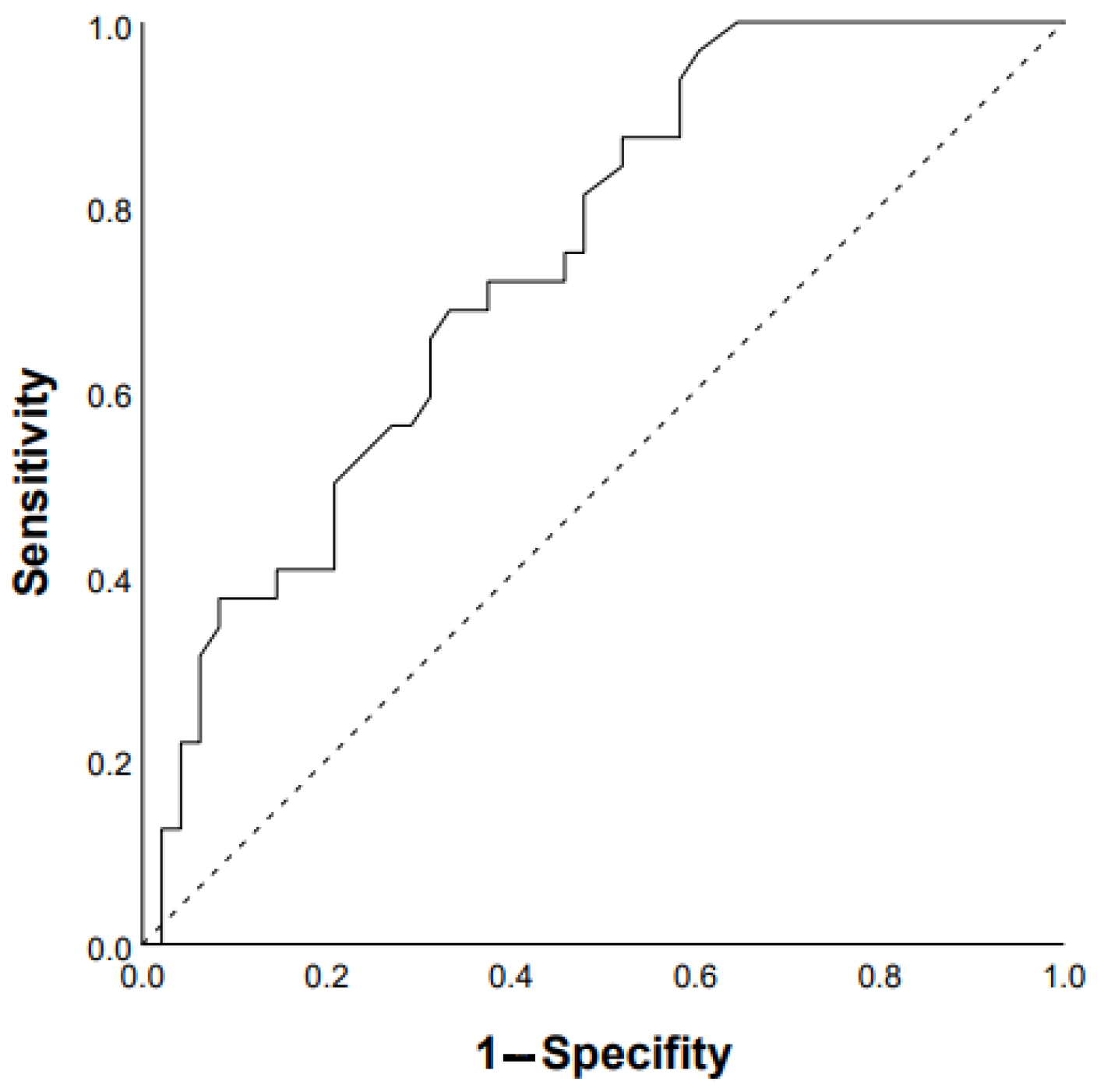
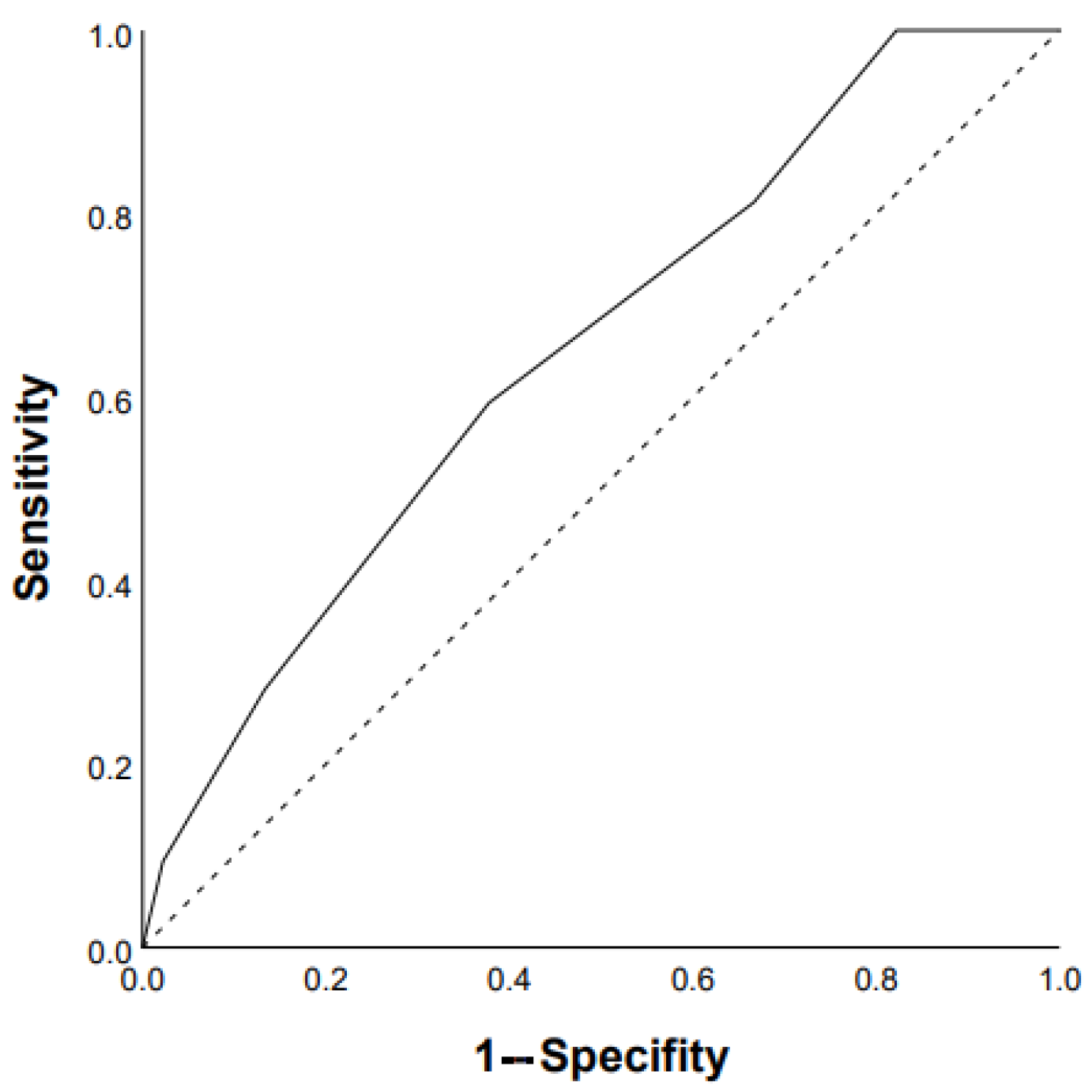
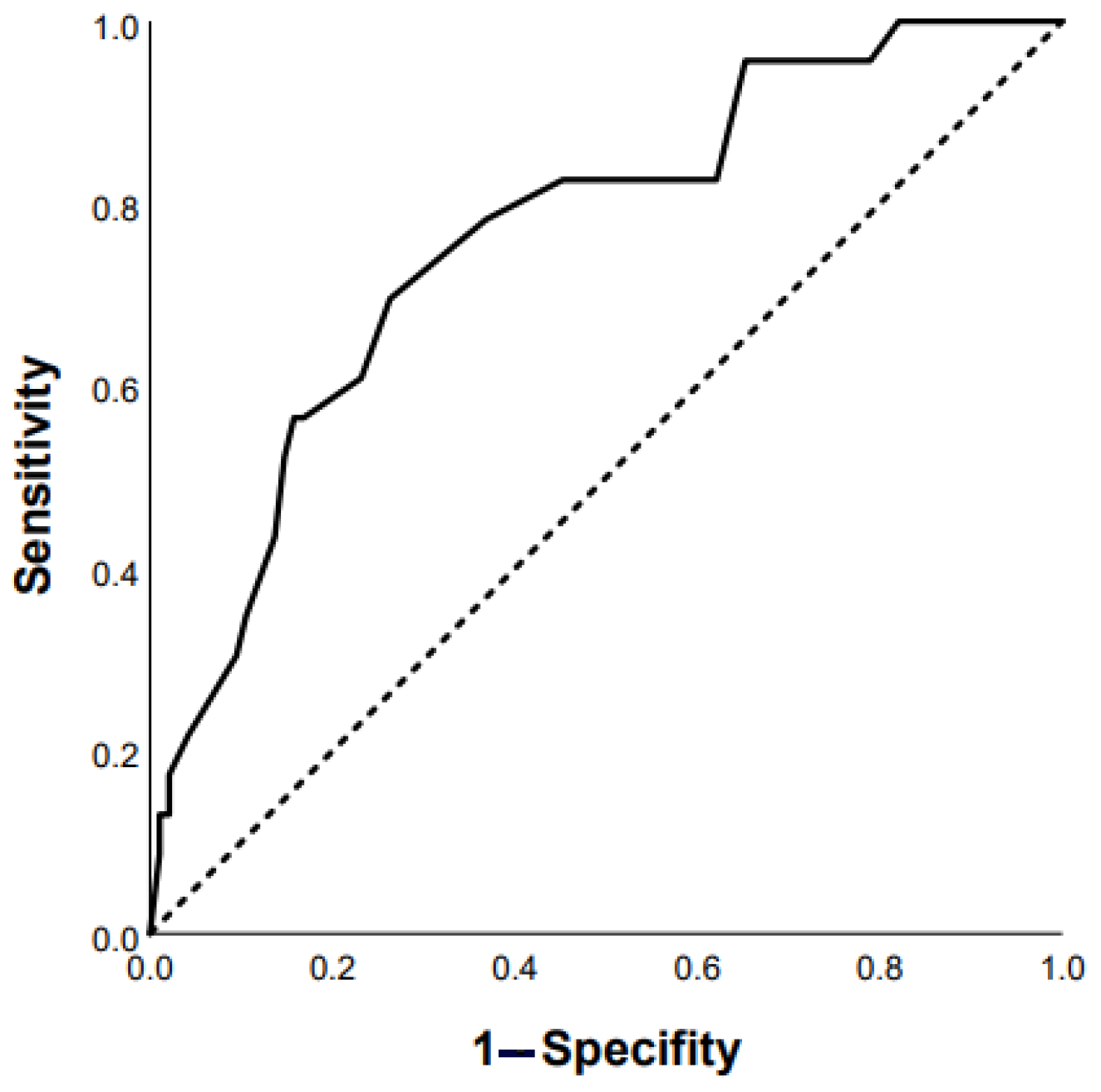
3.1. Predictors of Progression
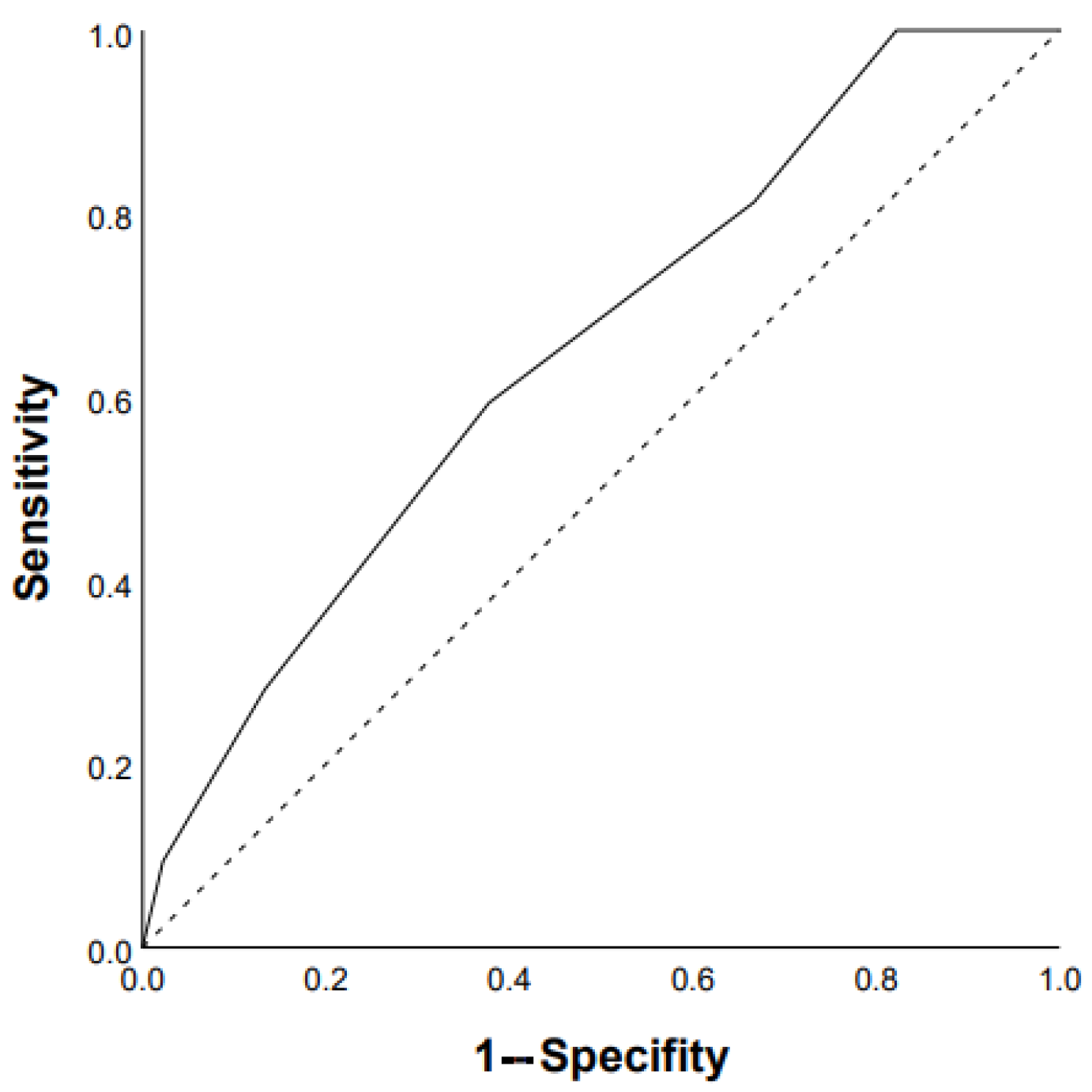
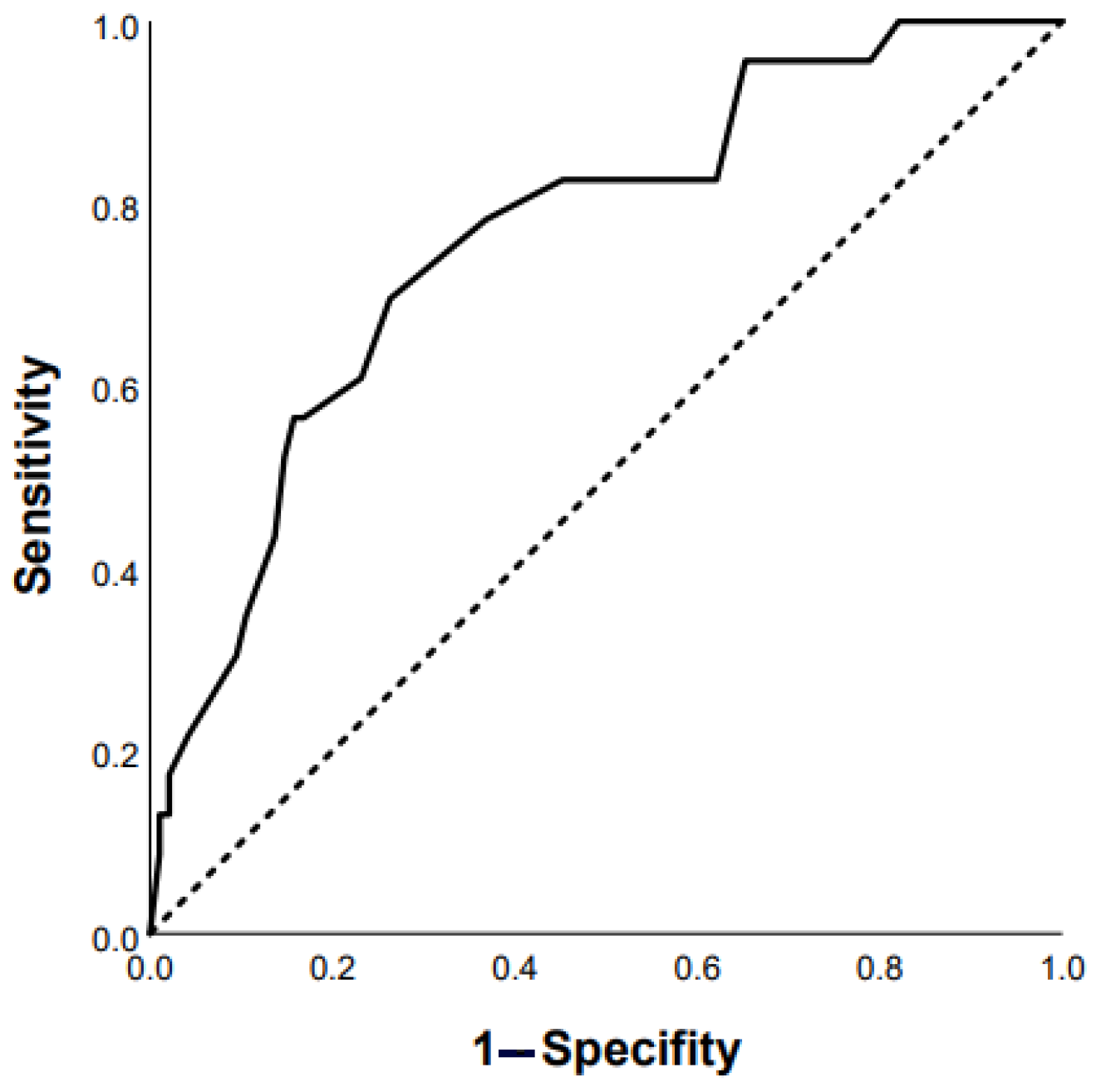
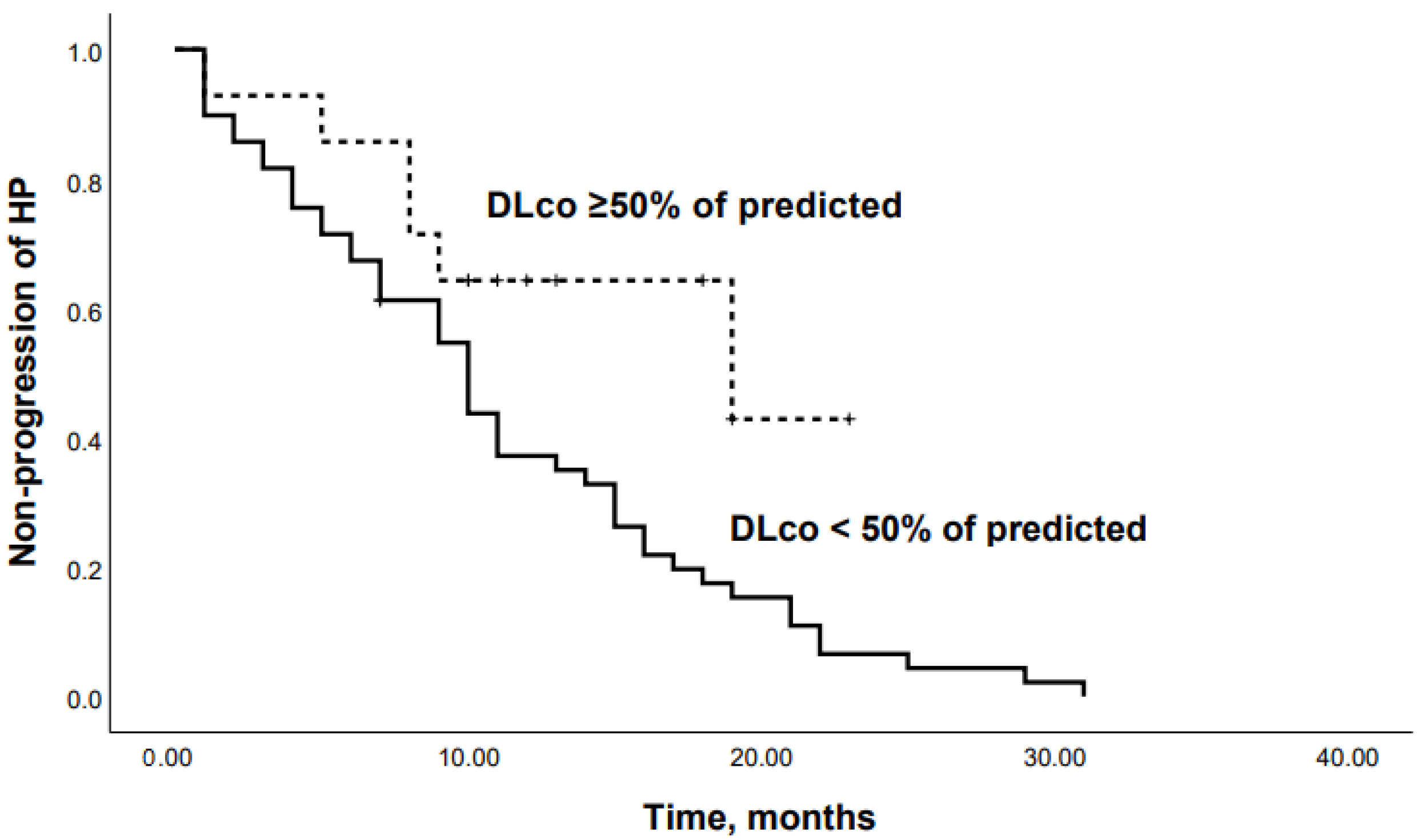
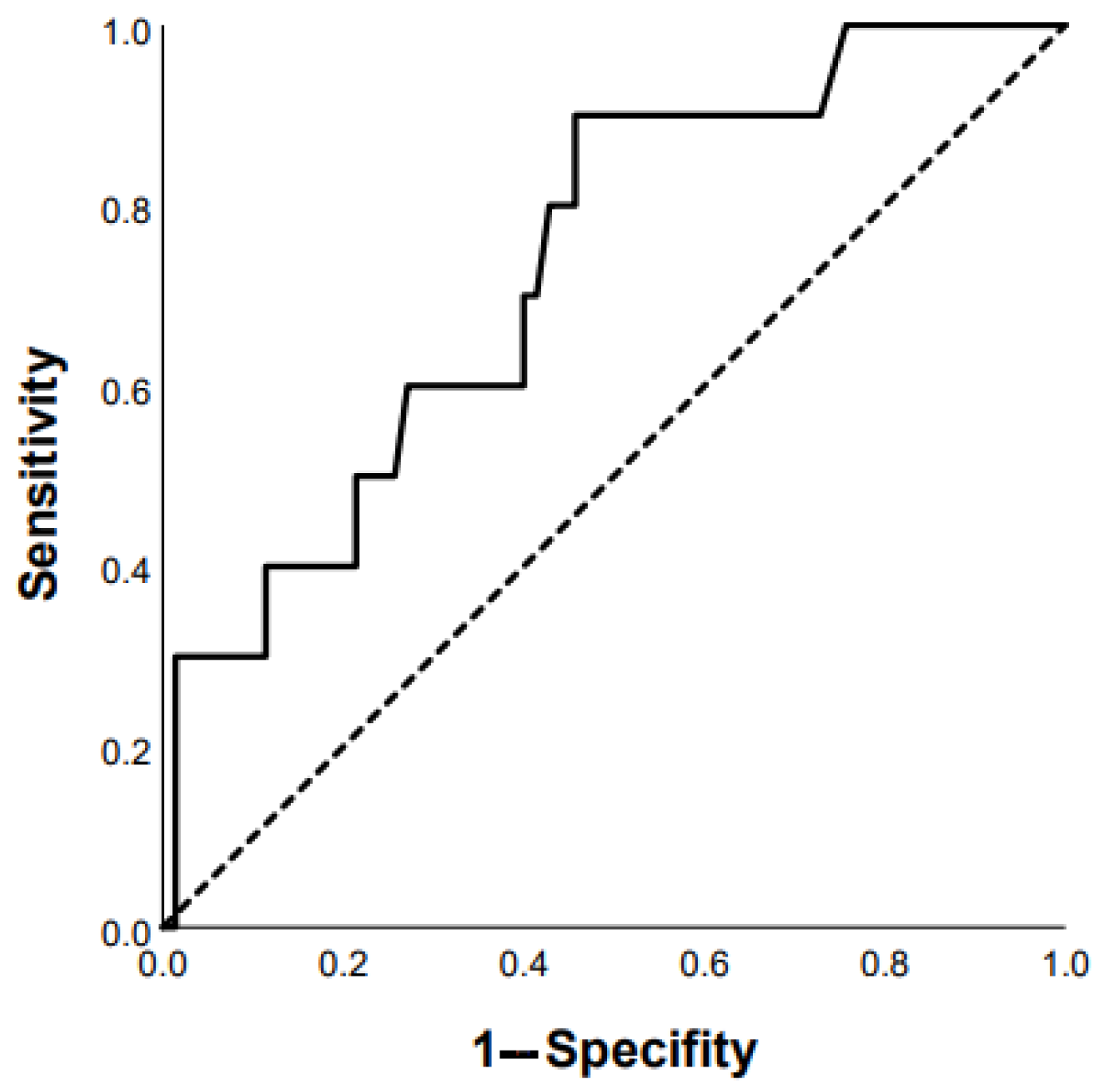
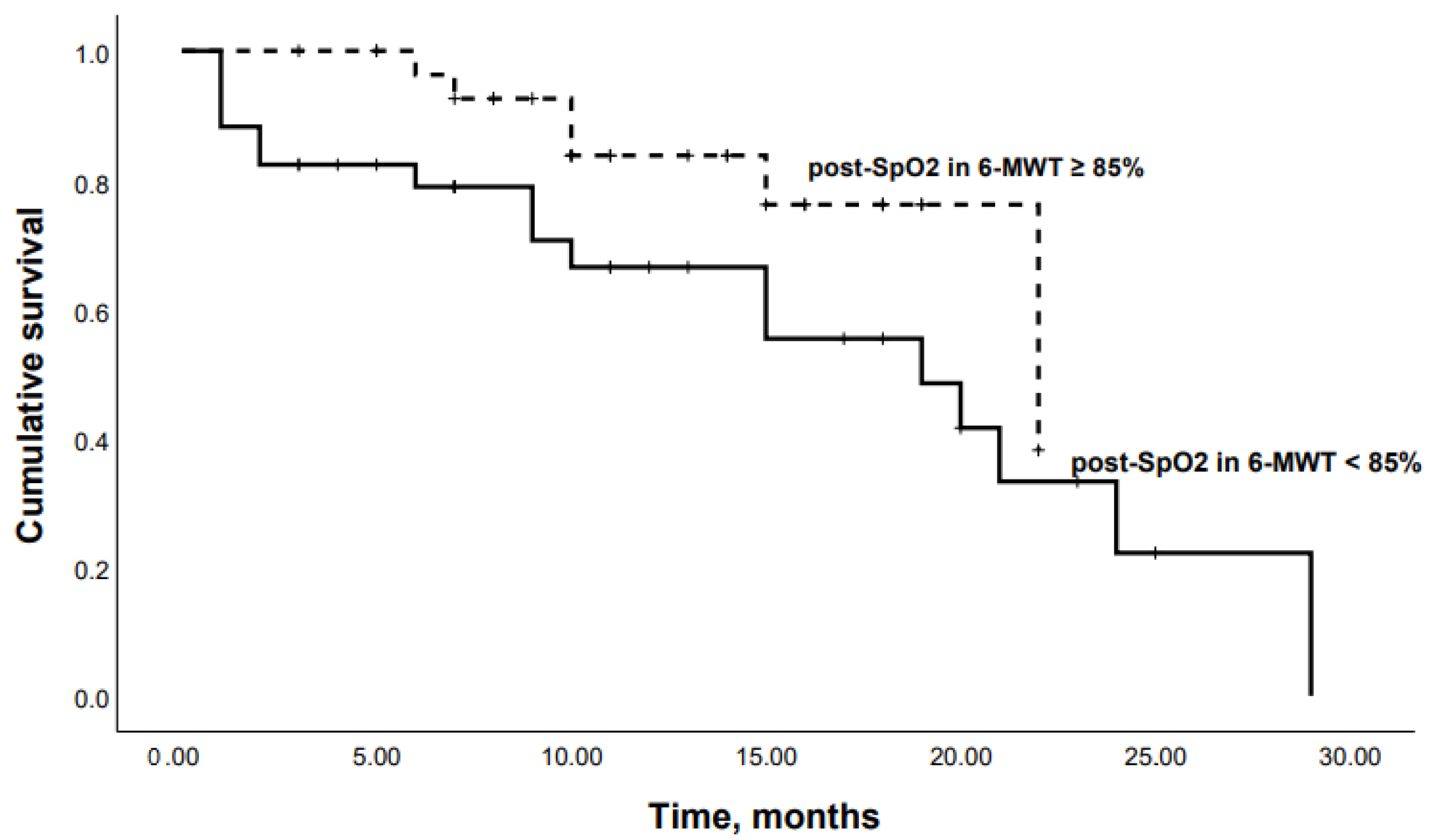
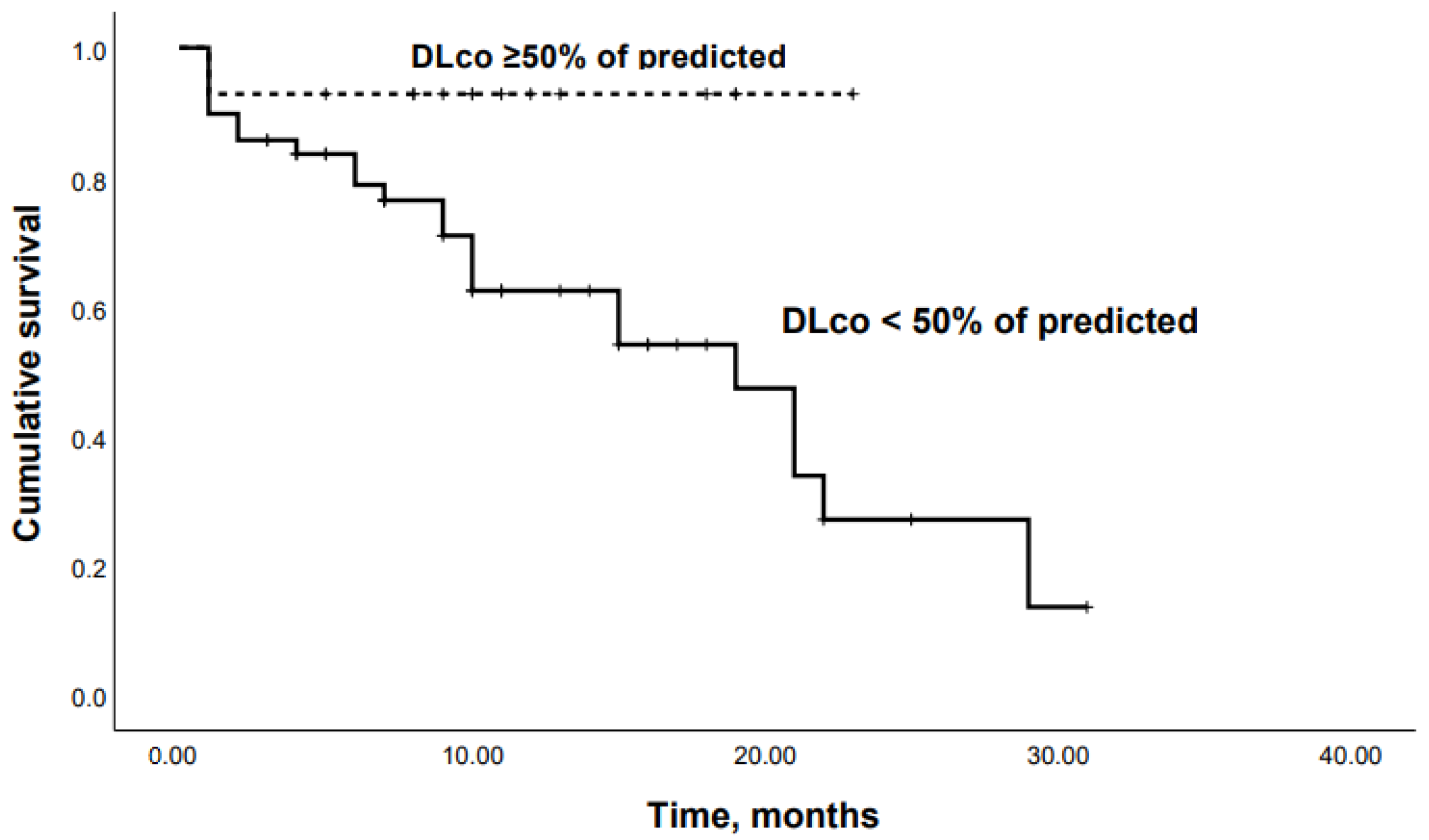
3.2. Predictors of Mortality
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomeer, M.J.; Costabel, U.; Rizzato, G.; Poletti, V.; Demedts, M. Comparison of registries of interstitial lung diseases in three European countries. Eur. Respir. J. 2001, 18, 114–118. [Google Scholar]
- Rittig, A.H.; Hilberg, O.; Ibsen, R.; Løkke, A. Incidence, comorbidity and survival rate of hypersensitivity pneumonitis: A national population-based study. ERJ Open Res. 2019, 5, 00259–2018. [Google Scholar] [CrossRef]
- Solaymani-Dodaran, M.; West, J.; Smith, C.; Hubbard, R. Extrinsic allergic alveolitis: Incidence and mortality in the general population. QJM 2007, 100, 233–237. [Google Scholar] [CrossRef]
- Ojanguren, I.; Morell, F.; Ramón, M.A.; Villar, A.; Romero, C.; Cruz, M.J.; Muñoz, X. Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy 2019, 74, 944–952. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Sprunger, D.B.; Ratanawatkul, P.; Maier, L.A.; Huie, T.J.; Swigris, J.J.; Solomon, J.J.; Mohning, M.P.; Keith, R.C.; Brown, K.K. Increasing hypersensitivity pneumonitis–related mortality in the United States from 1988 to 2016. Am. J. Respir. Crit. Care Med. 2019, 199, 1284–1287. [Google Scholar]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, 36–69. [Google Scholar]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar]
- Fernández Pérez, E.R.; Swigris, J.J.; Forssén, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Travis, W.D.; Lynch, D.A.; Brown, K.K.; Johannson, K.A.; Selman, M.; Ryu, J.H.; Wells, A.U.; Huang, Y.T.; Pereiraet, C.A.C.; et al. Executive summary diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 2021, 160, 595–615. [Google Scholar] [CrossRef]
- Pérez-Padilla, R.; Salas, J.; Chapela, R.; Sánchez, M.; Carrillo, G.; Pérez, R.; Sansores, R.; Gaxiola, M.; Selman, M. Mortality in Mexican patients with chronic pigeon breeder’s lung compared with those with usual interstitial pneumonia. Am. Rev. Respir. Dis. 1993, 148, 49–53. [Google Scholar]
- Sahin, H.; Brown, K.K.; Curran-Everett, D.; Hale, V.; Cool, C.D.; Vourlekis, J.S.; Lynch, D.A. Chronic hypersensitivity pneumonitis: CT features comparison with pathologic evidence of fibrosis and survival. Radiology 2007, 244, 591–598. [Google Scholar]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. INBUILD Trial Investigators. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, 18–47. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya, A.; Abhijit, R.D.; Roy Chowdhury, S.; Chaudhury, K. Risk factors associated with mortality in hypersensitivity pneumonitis: A meta-analysis. Expert. Rev. Respir. Med. 2022, 16, 801–811. [Google Scholar] [CrossRef]
- Chikina, S.Y.; Chernyak, A.V.; Merzhoeva, Z.M.; Tyurin, I.E.; Avdeev, S.N. Idiopathic pulmonary fibrosis Registry in Russia. Pulmonologiya 2020, 30, 173–183. (In Russian) [Google Scholar]
- Salisbury, M.L.; Gu, T.; Murray, S.; Gross, B.H.; Chughtai, A.; Sayyouh, M.; Kazerooni, E.A.; Myers, J.L.; Lagstein, A.; Konopka, K.E.; et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated with Distinct Survival Time and Pulmonary Function Trajectory. Chest 2019, 155, 699–711. [Google Scholar]
- Hanak, V.; Golbin, J.M.; Hartman, T.E.; Ryu, J.H. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest 2008, 134, 133–138. [Google Scholar] [CrossRef]
- Brown, K.K.; Martinez, F.J.; Walsh, S.L.F.; Thannickal, V.J.; Prasse, A.; Schlenker-Herceg, R.; Goeldner, R.G.; Clerisme-Beaty, E.; Tetzlaff, K.; Cottin, V.; et al. The natural history of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2020, 55, 2000085. [Google Scholar] [CrossRef]
- Alberti, M.L.; Malet Ruiz, J.M.; Fernández, M.E.; Fassola, L.; Caro, F.; Roldán, I.B.; Paulin, F. Comparative survival analysis between idiopathic pulmonary fibrosis and chronic hypersensitivity pneumonitis. Pulmonology 2020, 26, 3–9. [Google Scholar] [CrossRef]
- Lewandowska, K.B.; Barańska, I.; Sobiecka, M.; Radwan-Rohrenschef, P.; Dybowska, M.; Franczuk, M.; Roży, A.; Skoczylas, A.; Bestry, I.; Kuś, J.; et al. Factors Predictive for Immunomodulatory Therapy Response and Survival in Patients with Hypersensitivity Pneumonitis-Retrospective Cohort Analysis. Diagnostics 2022, 12, 2767. [Google Scholar] [CrossRef]
- Mooney, J.J.; Elicker, B.M.; Urbania, T.H.; Agarwal, M.R.; Ryerson, C.J.; Nguyen, M.L.T.; Woodruff, P.G.; Jones, K.D.; Collard, H.R.; King, T.E., Jr.; et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest 2013, 144, 586–592. [Google Scholar] [CrossRef]
- Leuschner, G.; Klotsche, J.; Kreuter, M.; Prasse, A.; Wirtz, H.; Pittrow, D.; Frankenberger, M.; Behr, J.; Kneidinger, N.; INSIGHTS-IPF Registry Group. Idiopathic Pulmonary Fibrosis in Elderly Patients: Analysis of the INSIGHTS-IPF Observational Study. Front. Med. 2020, 7, 601279. [Google Scholar]
- Clarson, L.E.; Bajpai, R.; Whittle, R.; Belcher, J.; Sultan, A.A.; Kwok, C.S.; Welsh, V.; Mamas, M.; Mallen, C.D. Interstitial lung disease is a risk factor for ischaemic heart disease and myocardial infarction. Heart 2020, 106, 916–922. [Google Scholar] [CrossRef]
- Hyldgaard, C.; Hilberg, O.; Bendstrup, E. How does comorbidity influence survival in idiopathic pulmonary fibrosis? Respir. Med. 2014, 108, 647–653. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, J.W.; Kyung, S.Y.; Lee, S.P.; Chung, M.P.; Kim, Y.H.; Lee, J.H.; Kim, Y.C.; Ryu, J.S.; Lee, H.L.; et al. Clinical characteristics of idiopathic pulmonary fibrosis patients with diabetes mellitus: The national survey in Korea from 2003 to 2007. J. Korean Med. Sci. 2012, 27, 756–760. [Google Scholar] [CrossRef]
- Zhan, X.; Du, Y.; Luo, J.; Que, Y.; Hu, C.; Xu, L.; Wang, Z.; Wu, Y.; Jin, M.; Zheng, C.; et al. Features of transbronchial lung cryobiopsy-diagnosed fibrotic hypersensitivity pneumonitis. Clin. Respir. J. 2022, 17, 50–58. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Vittinghoff, E.; Ley, B.; Lee, J.S.; Mooney, J.J.; Jones, K.D.; Elicker, B.M.; Wolters, P.J.; Koth, L.L.; King, T.E., Jr.; et al. Predicting survival across chronic interstitial lung disease: The ILD-GAP model. Chest 2014, 145, 723–728. [Google Scholar] [CrossRef]
- Lama, V.N.; Flaherty, K.R.; Toews, G.B.; Colby, T.V.; Travis, W.D.; Long, Q.; Murray, S.; Kazerooni, E.A.; Gross, B.H.; Lynch, J.P.; et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2003, 168, 1084–1090. [Google Scholar] [CrossRef]
- Amano, M.; Izumi, C.; Baba, M.; Abe, R.; Matsutani, H.; Inao, T.; Miyake, M.; Nishimoto, Y.; Tamura, T.; Noma, S.; et al. Progression of right ventricular dysfunction and predictors of mortality in patients with idiopathic interstitial pneumonias. J. Cardiol. 2020, 75, 242–249. [Google Scholar] [CrossRef]
- Rivera-Lebron, B.N.; Forfia, P.R.; Kreider, M.; Lee, J.C.; Holmes, J.H.; Kawut, S.M. Echocardiographic and hemodynamic predictors of mortality in idiopathic pulmonary fibrosis. Chest 2013, 144, 564–570. [Google Scholar] [CrossRef]
- Zhu, W.W.; Li, H.; Li, Y.D.; Sun, L.; Kong, L.; Ye, X.; Cai, Q.; Lv, X.Z. Risk assessment in interstitial lung disease: The incremental prognostic value of cardiopulmonary ultrasound. BMC Pulm. Med. 2021, 21, 237. [Google Scholar] [CrossRef]
- Cai, M.; Zhu, M.; Ban, C.; Su, J.; Ye, Q.; Liu, Y.; Zhao, W.; Wang, C.; Dai, H. Clinical features and outcomes of 210 patients with idiopathic pulmonary fibrosis. Chin. Med. J. 2014, 127, 1868–1873. [Google Scholar] [CrossRef]
- Wang, P.; Jones, K.D.; Urisman, A.; Elicker, B.M.; Urbania, T.; Johannson, K.A.; Assayag, D.; Lee, J.; Wolters, P.J.; Collard, H.R.; et al. Pathologic Findings and Prognosis in a Large Prospective Cohort of Chronic Hypersensitivity Pneumonitis. Chest 2017, 152, 502–509. [Google Scholar] [CrossRef]
- Adegunsoye, A.; Oldham, J.M.; Chung, J.H.; Montner, S.M.; Lee, C.; Witt, L.J.; Stahlbaum, D.; Bermea, R.S.; Chen, L.W.; Hsu, S.; et al. Phenotypic Clusters Predict Outcomes in a Longitudinal Interstitial Lung Disease Cohort. Chest 2018, 153, 349–360. [Google Scholar] [CrossRef]
- Morisset, J.; Johannson, K.A.; Vittinghoff, E.; Aravena, C.; Elicker, B.M.; Jones, K.D.; Fell, C.D.; Manganas, H.; Dubé, B.P.; Wolters, P.J.; et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017, 151, 619–625. [Google Scholar]
- Fiddler, C.A.; Simler, N.; Thillai, M.; Parfrey, H. Use of mycophenolate mofetil and azathioprine for the treatment of chronic hypersensitivity pneumonitis-A single-centre experience. Clin. Respir. J. 2019, 13, 791–794. [Google Scholar] [CrossRef]
- Sterclova, M.; Smetakova, M.; Stehlik, L.; Skibova, J.; Vasakova, M. Bronchoalveolar lavage cell profiles and proteins concentrations can be used to phenotype extrinsic allergic alveolitis patients. Multidiscip. Respir. Med. 2019, 14, 13. [Google Scholar] [CrossRef]
- Furusawa, H.; Peljto, A.L.; Walts, A.D.; Cardwell, J.; Molyneaux, P.L.; Lee, J.S.; Fernández Pérez, E.R.; Wolters, P.J.; Yang, I.V.; Schwartz, D.A. Common idiopathic pulmonary fibrosis risk variants are associated with hypersensitivity pneumonitis. Thorax 2022, 77, 508–510. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Age, years | 65 (58–71) |
| Gender (M/F), n (%) | 133 (43.8%)/159 (52.3%) |
| BMI, kg/m2 | 28.0 (24.9–31.6) |
| Smoking history, pack/years | 20 (13–32) |
| Time from symptom onset to diagnosis, months | 11 (3–33) |
| Fibrotic phenotype, n (%) Nonfibrotic phenotype, n (%) | 279 (91.8%) 13 (4.3%) |
| Known history of exposure, n (%) | 46 (15.1%) |
| GAP score, points | 3 (2–4) |
| GAP, stages (%) I II III | 52.6% 42.1% 5.3% |
| Cough, n (%) | 223 (73.4%) |
| Dyspnea, n (%) | 263 (86.5%) |
| Dyspnea mMRC score, points | 2 (2–3) |
| General weakness, n (%) | 190 (62.5%) |
| Chest pain, n (%) | 33 (10.9%) |
| Respiratory rate, min−1 | 20 (18–22) |
| Heart rate, min−1 | 85 (77–94) |
| SpO2, % | 94 (90–96) |
| Cyanosis, n (%) | 93 (17.2%) |
| Peripheral edema, n (%) | 32 (5.9%) |
| Finger clubbing, n (%) | 117 (21.6%) |
| Comorbidities: Arterial hypertension, n (%) Cardiovascular diseases, n (%) Pulmonary hypertension, n (%) GERD, n (%) Diabetes mellitus, n (%) COPD, n (%) Chronic kidney diseases, n (%) Liver diseases, n (%) Thrombosis, n (%) Oncological diseases, n (%) | 166 (54.6%) 108 (35.5%) 55 (18.1%) 54 (17.8%) 44 (14.5%) 8 (2.6%) 14 (4.6%) 10 (3.3%) 8 (2.6%) 8 (2.6%) |
| Charlson Comorbidity Index, points | 3 (2–4) |
| Hemoglobin, g/L | 142 (130–156) |
| Erythrocytes, 1012/L | 4.74 (4.33–5.2) |
| Platelets, 109/L | 233 (193–286) |
| Leukocytes, 109/L | 9.4 (7.4–12.1) |
| Lymphocytes, 109/L | 2.3 (1.7–3.1) |
| Monocytes, 109/L | 1.68 (1.13–2.4) |
| Neutrophils, 109/L | 6.0 (4.5–8.3) |
| Neutrophils to Lymphocytes ratio | 2.7 (1.8–3.8) |
| ESR, mm/h | 16 (8–31) |
| CRP, mg/L | 5.0 (2.8–12.9) |
| FVC, L | 2.2 (1.7–2.7) |
| FVC, % pred | 67.0 (57.0–78.0) |
| FEV1, L | 1.8 (1.3–2.3) |
| FEV1, % pred | 67.0 (57.0–78.0) |
| FEV1/FVC, % | 87.3 (81.3–92.4) |
| TLC, L | 4.0 (3.2–4.8) |
| TLC, % pred | 64.3 (54.5–74.5) |
| DLco, % pred | 51.2 (41.6–59.8) |
| RA area, cm2 | 17.1 (13.3–19.8) |
| SPAP, mm Hg | 36 (29–43) |
| TAPSE, mm | 23 (18–27) |
| Distance 6-MWT, m | 450 (390–500) |
| Pre-SpO2 in 6-MWT | 95 (92–96) |
| Post-SpO2 6-MWT | 86 (81–90) |
| PaO2, mmHg | 59.8 (51.8–70.1) |
| PaCO2, mmHg | 33.5 (29.2–39.7) |
| Treatment | |
| Long-term oxygen therapy, n (%) | 61 (20.1%) |
| Antifibrotic therapy, n (%) | 38 (11.5%) |
| SCS, n (%) | 125 (41.1%) |
| Immunosuppressants, n (%) | 32 (4.6%) |
| Follow-up | |
| Progression | 92 (30.3%) |
| Mortality | 44 (14.5%) |
| Characteristic | Nonprogressive Phenotype (n = 200) | Progressive Phenotype (n = 92) | p Value |
|---|---|---|---|
| Age, years | 65.0 (58.0–70.5) | 64.5 (59.3–71.8) | 0.32 |
| Gender (M/F), n (%) | 83 (41.5%)/117 (58.5%) | 50 (54.3%)/42 (45.7%) | 0.04 |
| BMI, kg/m2 | 28.2 (24.8–31.8) | 27.8 (25.3–31.1) | 0.47 |
| Smoking history, pack/years | 20 (9–30) | 30 (19–40) | 0.003 |
| Time from symptom onset to diagnosis, months | 11 (3–32) | 12 (5–35) | 0.41 |
| Fibrotic phenotype, n (%) | 187 (93.5%) | 92 (100%) | |
| Known history of exposure, n (%) | 32 (16.0%) | 14 (15.2%) | 0.89 |
| GAP score, points | 3 (2–4) | 4 (3–5) | 0.02 |
| GAP, stages (%) I II III | 61.4% 36.4% 2.3% | 40.6% 50.0% 9.4% | |
| Dyspnea mMRC, points | 2 (2–3) | 2 (2–3) | 0.94 |
| Cough, n (%) | 149 (74.5%) | 74 (80.4%) | 0.27 |
| General weakness, n (%) | 135 (67.5%) | 55 (59.8%) | 0.19 |
| Chest pain, n (%) | 21 (10.5%) | 12 (13%) | 0.52 |
| Respiratory rate, min−1 | 20 (18–22) | 21 (18–22) | 0.87 |
| Heart rate, min−1 | 84 (77–92) | 88 (75–100) | 0.15 |
| Cyanosis, n (%) | 65 (23.5%) | 29 (32.5%) | 0.61 |
| Peripheral edema, n (%) | 20 (10%) | 8 (8.7%) | 0.06 |
| Finger clubbing, n (%) | 73 (36.5%) | 44 (47.8%) | 0.09 |
| Comorbidities: Arterial hypertension, n (%) Cardiovascular diseases, n (%) Pulmonary hypertension, n (%) GERD, n (%) Diabetes mellitus, n (%) COPD, n (%) Chronic kidney diseases, n (%) Liver diseases, n (%) Thrombosis, n (%) Oncological diseases, n (%) | 105 (53%) 70 (35%) 32 (16%) 38 (19%) 22 (11%) 6 (3%) 8 (4%) 7 (3.5%) 5 (2.5%) 2 (1%) | 61 (66.3%) 38 (41.3%) 23 (25%) 16 (17.4%) 22 (23.9%) 2 (2.2%) 6 (6.5%) 3 (3.3%) 3 (3.3%) 6 (6.5%) | 0.03 0.3 0.07 0.74 0.004 0.69 0.35 0.92 0.71 0.01 |
| Charlson comorbidity index, points | 3 (2–4) | 3 (3–5) | 0.04 |
| Hemoglobin, g/L | 142 (129–156) | 142 (131–153) | 0.74 |
| Erythrocytes, 1012/L | 4.7 (4.3–5.2) | 4.8 (4.4–5.2) | 0.49 |
| Platelets, 109/L | 242 (200–285) | 217 (177–288) | 0.06 |
| Leukocytes, 109/L | 9.6 (7.7–12.5) | 9.1 (7.2–11.7) | 0.25 |
| Lymphocytes, 109/L | 2.3 (1.6–3.1) | 2.3 (1.8–3.2) | 0.21 |
| Neutrophils to lymphocytes ratio | 2.8 (1.8–4.0) | 2.3 (1.7–3.4) | 0.12 |
| Monocytes, 109/L | 1.5 (0.9–2.4) | 1.9 (1.2–2.5) | 0.27 |
| Neutrophils, 109/L | 6.4 (4.7–8.5) | 5.9 (3.6–7.7) | 0.14 |
| ESR, mm/h | 14 (8–25) | 24 (9–37) | 0.04 |
| CRP, mg/L | 5.3 (2.9–14.5) | 5 (2.5–12.0) | 0.68 |
| FVC, L | 2.2 (1.7–2.7) | 2.1 (1.7–2.6) | 0.57 |
| FVC, % pred | 67 (57–82) | 66.0 (57.0–74.5) | 0.20 |
| TLC, L | 4.0 (3.2–4.9) | 3.7 (2.9–4.6) | 0.43 |
| TLC, % pred | 64.7 (60.0–84.5) | 62.1 (54.9–71.4) | 0.96 |
| DLco, % pred | 55.0 (45.0–65.0) | 44.5 (35.1–53.2) | 0.0001 |
| SPAP, mmHg | 35 (29–43) | 38 (29–43) | 0.46 |
| TAPSE, mm | 27.5 (19.8–43.0) | 21 (18–24) | 0.16 |
| RA area, cm2 | 16.8 (13.5–19.2) | 19 (13.0–24.9) | 0.37 |
| Distance 6-MWT, m | 460 (360–550) | 450 (390–493) | 0.74 |
| Pre-SpO2 6-MWT | 95 (93–96) | 95 (92–96) | 0.48 |
| Post-SpO2 6-MWT | 88 (84–90) | 84 (79–88) | 0.001 |
| PaO2, mmHg | 59.8 (52.9–72.1) | 59.0 (51.1–67.2) | 0.48 |
| PaCO2, mmHg | 34.8 (28.4–40.3) | 33 (29–38) | 0.63 |
| Treatment | |||
| Long-term oxygen therapy, n (%) | 38 (19.0%) | 70 (76.1%) | 0.0001 |
| Antifibrotics, n (%) | 18 (17.1%) | 20 (24.4%) | 0.01 |
| SCS, n (%) | 81 (77.1%) | 44 (60.3%) | 0.24 |
| Immunosuppressants, n (%) | 14 (13.3%) | 18 (24.7%) | 0.03 |
| At the end of follow-up | |||
| Dyspnea mMRC, points | 3 (2–4) | 3 (3–4) | 0.01 |
| FEV1, L | 2.1 (1.6–2.5) | 1.6 (1.2–2.2) | 0.005 |
| FEV1, % pred | 65.0 (55.0–78.5) | 54.2 (45.0–63.0) | 0.0001 |
| Delta FEV1, % | 4.0 (1.0–6.0) | 11.0 (10.0–13.0) | 0.0001 |
| DLco, % pred | 50.5 (45.6–57.8) | 32 (22.0–40.5) | 0.0001 |
| Delta DLco,% | 5.0 (1.3–10.3) | 14.5 (11.0–16.4) | 0.0001 |
| Distance 6-MWT, m | 430 (385–515) | 315 (204–366) | 0.001 |
| Delta distance 6-MWT, m | 50 (33–75) | 132 (103 -178) | 0.0001 |
| Pre-SpO2 6-MWT | 94 (92–98) | 92 (89–95) | 0.009 |
| Post-SpO2 6-MWT | 87 (80–90) | 80 (73–87) | 0.02 |
| Mortality | 0 | 44 (47.8%) | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trushenko, N.V.; Suvorova, O.A.; Pershina, E.S.; Nekludova, G.V.; Chikina, S.Y.; Levina, I.A.; Chernyaev, A.L.; Samsonova, M.V.; Tyurin, I.E.; Mustafina, M.K.; et al. Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases. Life 2023, 13, 467. https://doi.org/10.3390/life13020467
Trushenko NV, Suvorova OA, Pershina ES, Nekludova GV, Chikina SY, Levina IA, Chernyaev AL, Samsonova MV, Tyurin IE, Mustafina MK, et al. Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases. Life. 2023; 13(2):467. https://doi.org/10.3390/life13020467
Chicago/Turabian StyleTrushenko, Natalia V., Olga A. Suvorova, Ekaterina S. Pershina, Galina V. Nekludova, Svetlana Yu. Chikina, Iuliia A. Levina, Andrey L. Chernyaev, Maria V. Samsonova, Igor E. Tyurin, Malika Kh. Mustafina, and et al. 2023. "Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases" Life 13, no. 2: 467. https://doi.org/10.3390/life13020467
APA StyleTrushenko, N. V., Suvorova, O. A., Pershina, E. S., Nekludova, G. V., Chikina, S. Y., Levina, I. A., Chernyaev, A. L., Samsonova, M. V., Tyurin, I. E., Mustafina, M. K., Yaroshetskiy, A. I., Nadtochiy, N. B., Merzhoeva, Z. M., Proshkina, A. A., & Avdeev, S. N. (2023). Predictors of Progression and Mortality in Patients with Chronic Hypersensitivity Pneumonitis: Retrospective Analysis of Registry of Fibrosing Interstitial Lung Diseases. Life, 13(2), 467. https://doi.org/10.3390/life13020467