
Regulation of Physiological Barrier Function by the Commensal Microbiota


Abstract
1. Introduction
2. The Gut Microbiota and the Intestinal Epithelium
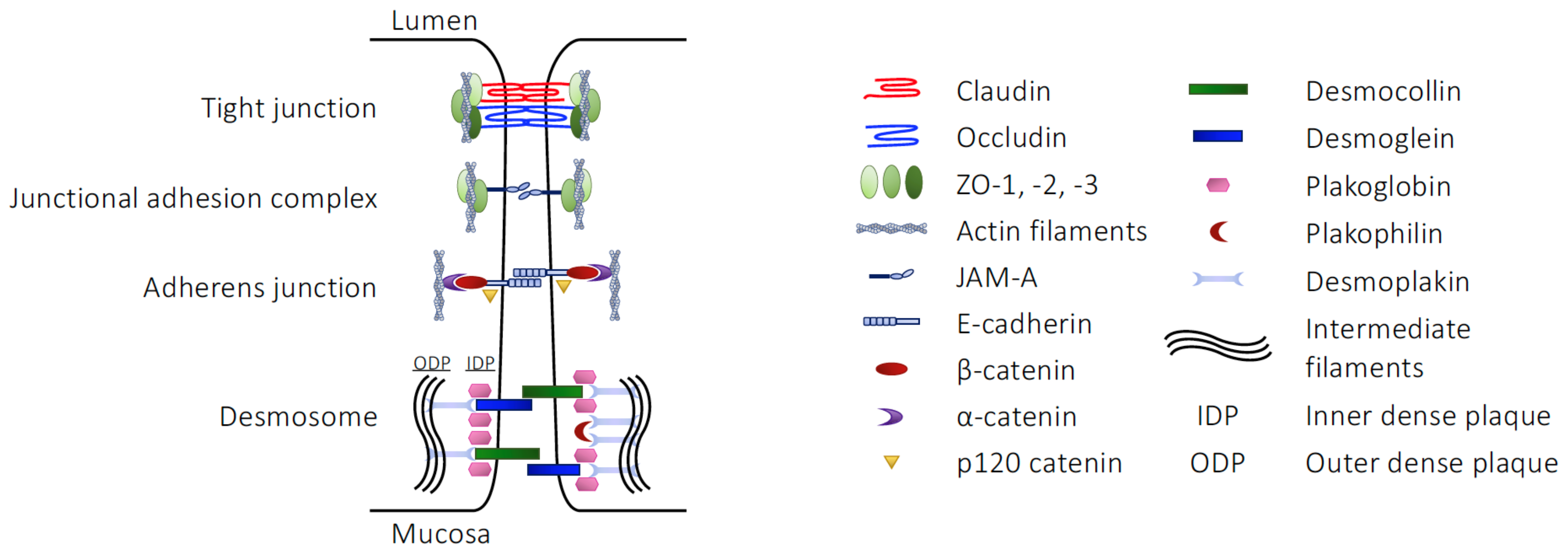
2.1. The Structural Barriers of the Gut
2.2. Microbial Influences on the Intestinal Epithelial Barrier Structures
3. The Skin Microbiome as a Regulator of the Epidermal Barrier
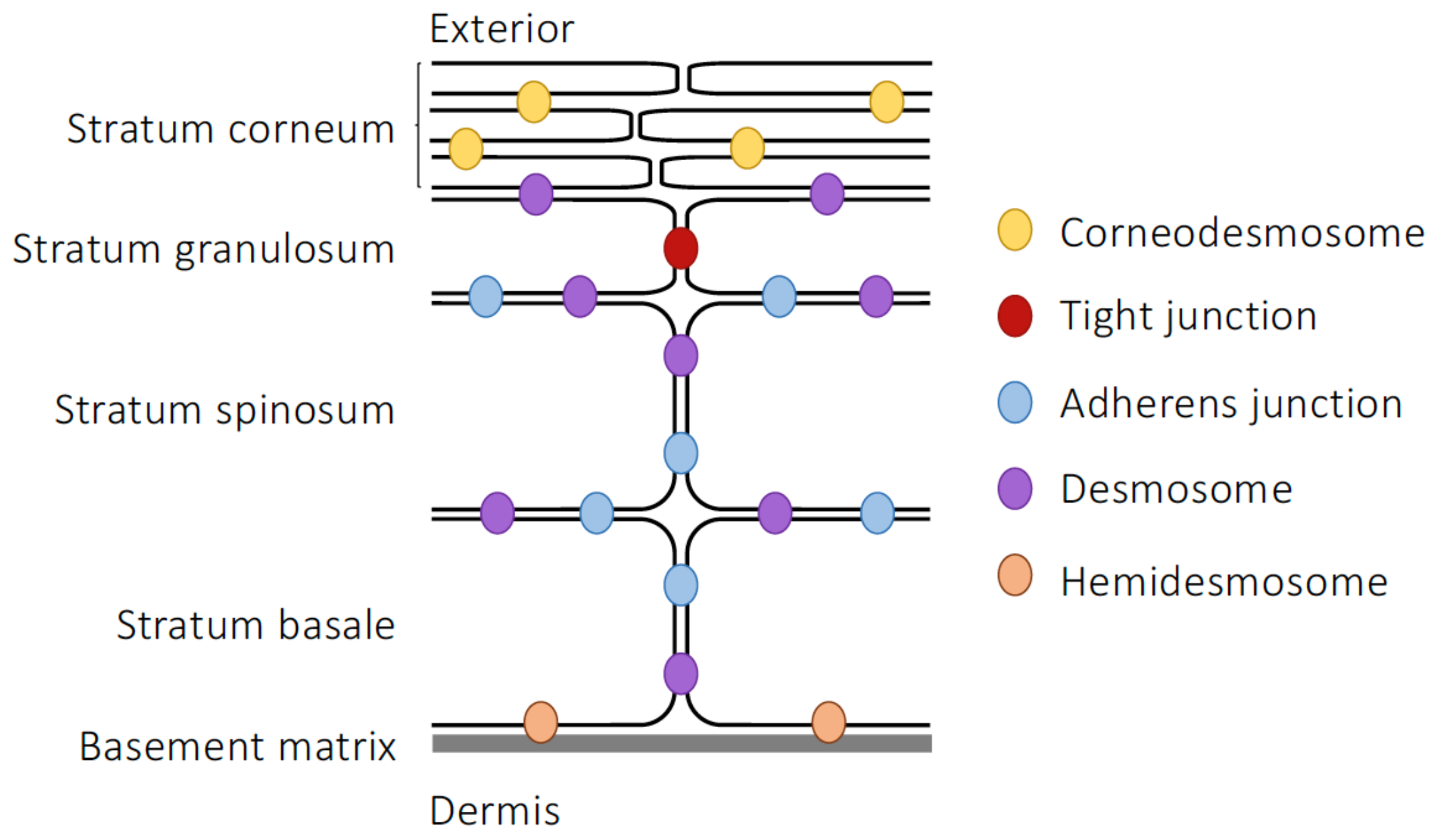
3.1. Epidermal Barrier Structure
3.2. Microbial Influences on Skin Integrity
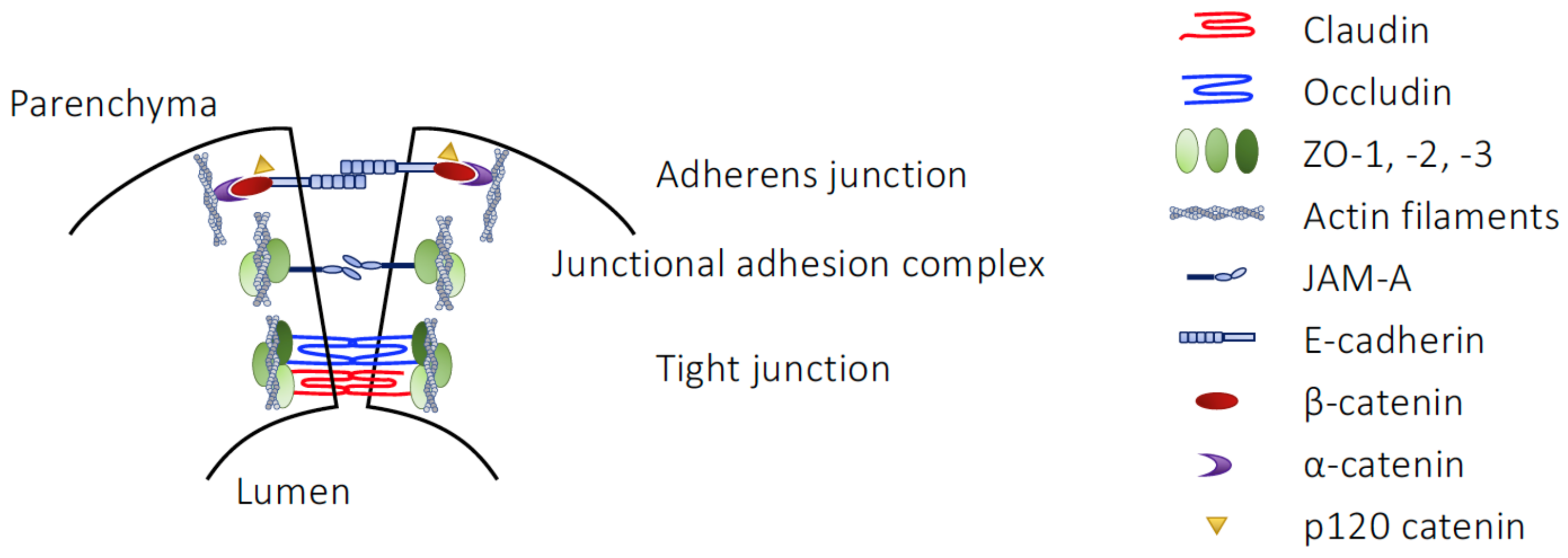
4. The Blood–Brain Barrier and the Gut–Brain Axis
4.1. Structural Elements of the BBB
4.2. Regulation of BBB Integrity by Microbial Metabolites
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martino, C.; Dilmore, A.; Burcham, Z.; Metcalf, J.; Jeste, D.; Knight, R. Microbiota Succession throughout Life from the Cradle to the Grave. Nat. Rev. Microbiol. 2022, 20, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Thriene, K.; Michels, K.B. Human Gut Microbiota Plasticity throughout the Life Course. Int. J. Environ. Res. Public Health 2023, 20, 1463. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Helander, H.F.; Fändriks, L. Surface Area of the Digestive Tract—Revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]
- Gillois, K.; Lévêque, M.; Théodorou, V.; Robert, H.; Mercier-Bonin, M. Mucus: An Underestimated Gut Target for Environmental Pollutants and Food Additives. Microorganisms 2018, 6, 53. [Google Scholar] [CrossRef]
- Birchenough, G.M.H.; Nyström, E.E.L.; Johansson, M.E.V.; Hansson, G.C. A Sentinel Goblet Cell Guards the Colonic Crypt by Triggering Nlrp6-Dependent Muc2 Secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef]
- Bansil, R.; Turner, B.S. The Biology of Mucus: Composition, Synthesis and Organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef]
- König, J.; Wells, J.; Cani, P.D.; García-Ródenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.-J. Human Intestinal Barrier Function in Health and Disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef]
- Dupont, A.; Heinbockel, L.; Brandenburg, K.; Hornef, M.W. Antimicrobial Peptides and the Enteric Mucus Layer Act in Concert to Protect the Intestinal Mucosa. Gut Microbes 2014, 5, 761–765. [Google Scholar] [CrossRef]
- Strugnell, R.A.; Wijburg, O.L.C. The Role of Secretory Antibodies in Infection Immunity. Nat. Rev. Microbiol. 2010, 8, 656–667. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage Adhering to Mucus Provide a Non-Host-Derived Immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef]
- Ermund, A.; Schütte, A.; Johansson, M.E.V.; Gustafsson, J.K.; Hansson, G.C. Studies of Mucus in Mouse Stomach, Small Intestine, and Colon. I. Gastrointestinal Mucus Layers Have Different Properties Depending on Location as Well as over the Peyer’s Patches. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G341–G347. [Google Scholar] [CrossRef]
- Matsuo, K.; Ota, H.; Akamatsu, T.; Sugiyama, A.; Katsuyama, T. Histochemistry of the Surface Mucous Gel Layer of the Human Colon. Gut 1997, 40, 782–789. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and Function of the Tight Junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel Integral Membrane Proteins Localizing at Tight Junctions with No Sequence Similarity to Occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A Novel Integral Membrane Protein Localizing at Tight Junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Umeda, K.; Tsukita, S.; Furuse, M.; Tsukita, S. Requirement of ZO-1 for the Formation of Belt-like Adherens Junctions during Epithelial Cell Polarization. J. Cell Biol. 2007, 176, 779–786. [Google Scholar] [CrossRef]
- Gumbiner, B. Structure, Biochemistry, and Assembly of Epithelial Tight Junctions. Am. J. Physiol. 1987, 253, C749–C758. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Khatib, K.; Guo, S.; Ye, D.; Youssef, M.; Ma, T. Occludin Regulates Macromolecule Flux across the Intestinal Epithelial Tight Junction Barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1054–G1064. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Günzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin Forms a Barrier to Macromolecules in Tricellular Tight Junctions without Affecting Ion Permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ding, L.; Lu, Q.; Chen, Y.-H. Claudins in Intestines: Distribution and Functional Significance in Health and Diseases. Tissue Barriers 2013, 1, e24978. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef]
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 Forms Paracellular Chloride Channel in the Kidney and Requires Claudin-8 for Tight Junction Localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef]
- Markov, A.G.; Veshnyakova, A.; Fromm, M.; Amasheh, M.; Amasheh, S. Segmental Expression of Claudin Proteins Correlates with Tight Junction Barrier Properties in Rat Intestine. J. Comp. Physiol. B 2010, 180, 591–598. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsui, C.; Furuse, K.; Mimori-Kiyosue, Y.; Furuse, M.; Tsukita, S. Dynamic Behavior of Paired Claudin Strands within Apposing Plasma Membranes. Proc. Natl. Acad. Sci. USA 2003, 100, 3971–3976. [Google Scholar] [CrossRef]
- Matter, K.; Balda, M.S. Signalling to and from Tight Junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–237. [Google Scholar] [CrossRef]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A Coordinately Regulate Tight Junction Formation and Epithelial Polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef]
- Liu, Y.; Nusrat, A.; Schnell, F.J.; Reaves, T.A.; Walsh, S.; Pochet, M.; Parkos, C.A. Human Junction Adhesion Molecule Regulates Tight Junction Resealing in Epithelia. J. Cell Sci. 2000, 113, 2363–2374. [Google Scholar] [CrossRef]
- Mandell, K.J.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Junctional Adhesion Molecule 1 Regulates Epithelial Cell Morphology through Effects on Β1 Integrins and Rap1 Activity. J. Biol. Chem. 2005, 280, 11665–11674. [Google Scholar] [CrossRef]
- Hartmann, C.; Schwietzer, Y.A.; Otani, T.; Furuse, M.; Ebnet, K. Physiological Functions of Junctional Adhesion Molecules (JAMs) in Tight Junctions. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183299. [Google Scholar] [CrossRef]
- Perez-Moreno, M.; Fuchs, E. Catenins: Keeping Cells from Getting Their Signals Crossed. Dev. Cell 2006, 11, 601–612. [Google Scholar] [CrossRef]
- Meng, W.; Takeichi, M. Adherens Junction: Molecular Architecture and Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef]
- Schnoor, M. E-Cadherin Is Important for the Maintenance of Intestinal Epithelial Homeostasis Under Basal and Inflammatory Conditions. Dig. Dis. Sci. 2015, 60, 816–818. [Google Scholar] [CrossRef]
- Zhang, Y.; Sivasankar, S.; Nelson, W.J.; Chu, S. Resolving Cadherin Interactions and Binding Cooperativity at the Single-Molecule Level. Proc. Natl. Acad. Sci. USA 2009, 106, 109–114. [Google Scholar] [CrossRef]
- Madara, J.L. Intestinal Absorptive Cell Tight Junctions Are Linked to Cytoskeleton. Am. J. Physiol. 1987, 253, C171–C175. [Google Scholar] [CrossRef]
- Schneider, M.R.; Kolligs, F.T. E-Cadherin’s Role in Development, Tissue Homeostasis and Disease: Insights from Mouse Models: Tissue-Specific Inactivation of the Adhesion Protein E-Cadherin in Mice Reveals Its Functions in Health and Disease. Bioessays 2015, 37, 294–304. [Google Scholar] [CrossRef]
- Garcia, M.A.; Nelson, W.J.; Chavez, N. Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a029181. [Google Scholar] [CrossRef]
- Lowndes, M.; Rakshit, S.; Shafraz, O.; Borghi, N.; Harmon, R.M.; Green, K.J.; Sivasankar, S.; Nelson, W.J. Different Roles of Cadherins in the Assembly and Structural Integrity of the Desmosome Complex. J. Cell Sci. 2014, 127, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Getsios, S.; Green, K.J. Desmosome Regulation and Signaling in Disease. Cell Tissue Res. 2015, 360, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Delva, E.; Tucker, D.K.; Kowalczyk, A.P. The Desmosome. Cold Spring Harb. Perspect. Biol. 2009, 1, a002543. [Google Scholar] [CrossRef] [PubMed]
- Abrams, G.D.; Bauer, H.; Sprinz, H. Influence of the Normal Flora on Mucosal Morphology and Cellular Renewal in the Ileum. A Comparison of Germ-Free and Conventional Mice. Lab. Investig. 1963, 12, 355–364. [Google Scholar] [PubMed]
- Johansson, M.E.V.; Jakobsson, H.E.; Holmén-Larsson, J.; Schütte, A.; Ermund, A.; Rodríguez-Piñeiro, A.M.; Arike, L.; Wising, C.; Svensson, F.; Bäckhed, F.; et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe 2015, 18, 582–592. [Google Scholar] [CrossRef]
- Pullan, R.D.; Thomas, G.A.; Rhodes, M.; Newcombe, R.G.; Williams, G.T.; Allen, A.; Rhodes, J. Thickness of Adherent Mucus Gel on Colonic Mucosa in Humans and Its Relevance to Colitis. Gut 1994, 35, 353–359. [Google Scholar] [CrossRef]
- Nakamori, S.; Ota, D.M.; Cleary, K.R.; Shirotani, K.; Irimura, T. MUC1 Mucin Expression as a Marker of Progression and Metastasis of Human Colorectal Carcinoma. Gastroenterology 1994, 106, 353–361. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus Barrier, Mucins and Gut Microbiota: The Expected Slimy Partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Kozakova, H.; Schwarzer, M.; Tuckova, L.; Srutkova, D.; Czarnowska, E.; Rosiak, I.; Hudcovic, T.; Schabussova, I.; Hermanova, P.; Zakostelska, Z.; et al. Colonization of Germ-Free Mice with a Mixture of Three Lactobacillus Strains Enhances the Integrity of Gut Mucosa and Ameliorates Allergic Sensitization. Cell. Mol. Immunol. 2016, 13, 251–262. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, W.; Zuo, L.; Dong, J.; Zhu, W.; Li, Y.; Gu, L.; Gong, J.; Li, Q.; Li, N.; et al. Intestinal Dysbacteriosis Contributes to Decreased Intestinal Mucosal Barrier Function and Increased Bacterial Translocation. Lett. Appl. Microbiol. 2014, 58, 384–392. [Google Scholar] [CrossRef]
- Hayes, C.L.; Dong, J.; Galipeau, H.J.; Jury, J.; McCarville, J.; Huang, X.; Wang, X.-Y.; Naidoo, A.; Anbazhagan, A.N.; Libertucci, J.; et al. Commensal Microbiota Induces Colonic Barrier Structure and Functions That Contribute to Homeostasis. Sci. Rep. 2018, 8, 14184. [Google Scholar] [CrossRef]
- Nakata, K.; Sugi, Y.; Narabayashi, H.; Kobayakawa, T.; Nakanishi, Y.; Tsuda, M.; Hosono, A.; Kaminogawa, S.; Hanazawa, S.; Takahashi, K. Commensal Microbiota-Induced MicroRNA Modulates Intestinal Epithelial Permeability through the Small GTPase ARF4. J. Biol. Chem. 2017, 292, 15426–15433. [Google Scholar] [CrossRef]
- Ran, Y.; Fukui, H.; Xu, X.; Wang, X.; Ebisutani, N.; Tanaka, Y.; Maeda, A.; Makizaki, Y.; Ohno, H.; Kondo, T.; et al. Alteration of Colonic Mucosal Permeability during Antibiotic-Induced Dysbiosis. Int. J. Mol. Sci. 2020, 21, 6108. [Google Scholar] [CrossRef]
- Nevado, R.; Forcén, R.; Layunta, E.; Murillo, M.D.; Grasa, L. Neomycin and Bacitracin Reduce the Intestinal Permeability in Mice and Increase the Expression of Some Tight-Junction Proteins. Rev. Esp. Enferm. Dig. 2015, 107, 672–676. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Nighot, P.; Nighot, M.; Haque, M.; Rawat, M.; Ma, T.Y. Lactobacillus Acidophilus Induces a Strain-Specific and Toll-Like Receptor 2-Dependent Enhancement of Intestinal Epithelial Tight Junction Barrier and Protection Against Intestinal Inflammation. Am. J. Pathol. 2021, 191, 872–884. [Google Scholar] [CrossRef]
- Anderson, R.C.; Cookson, A.L.; McNabb, W.C.; Park, Z.; McCann, M.J.; Kelly, W.J.; Roy, N.C. Lactobacillus Plantarum MB452 Enhances the Function of the Intestinal Barrier by Increasing the Expression Levels of Genes Involved in Tight Junction Formation. BMC Microbiol. 2010, 10, 316. [Google Scholar] [CrossRef]
- Ukena, S.N.; Singh, A.; Dringenberg, U.; Engelhardt, R.; Seidler, U.; Hansen, W.; Bleich, A.; Bruder, D.; Franzke, A.; Rogler, G.; et al. Probiotic Escherichia Coli Nissle 1917 Inhibits Leaky Gut by Enhancing Mucosal Integrity. PLoS ONE 2007, 2, e1308. [Google Scholar] [CrossRef]
- Sato, K.; Yokoji, M.; Yamada, M.; Nakajima, T.; Yamazaki, K. An Orally Administered Oral Pathobiont and Commensal Have Comparable and Innocuous Systemic Effects in Germ-Free Mice. J. Periodontal Res. 2018, 53, 950–960. [Google Scholar] [CrossRef]
- Gou, H.-Z.; Zhang, Y.-L.; Ren, L.-F.; Li, Z.-J.; Zhang, L. How Do Intestinal Probiotics Restore the Intestinal Barrier? Front. Microbiol. 2022, 13, 929346. [Google Scholar] [CrossRef]
- Karczewski, J.; Troost, F.J.; Konings, I.; Dekker, J.; Kleerebezem, M.; Brummer, R.-J.M.; Wells, J.M. Regulation of Human Epithelial Tight Junction Proteins by Lactobacillus Plantarum in Vivo and Protective Effects on the Epithelial Barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G851–G859. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like Receptor 2 Enhances ZO-1-Associated Intestinal Epithelial Barrier Integrity via Protein Kinase C. Gastroenterology 2004, 127, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Paveljšek, D.; Ivičak-Kocjan, K.; Treven, P.; Benčina, M.; Jerala, R.; Rogelj, I. Distinctive Probiotic Features Share Common TLR2-Dependent Signalling in Intestinal Epithelial Cells. Cell. Microbiol. 2021, 23, e13264. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.A.; Mariño, E. An Insight Into the Intestinal Web of Mucosal Immunity, Microbiota, and Diet in Inflammation. Front. Immunol. 2018, 9, 2617. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium Perfringens Enterotoxin Fragment Removes Specific Claudins from Tight Junction Strands: Evidence for Direct Involvement of Claudins in Tight Junction Barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar] [CrossRef]
- Kondoh, M.; Masuyama, A.; Takahashi, A.; Asano, N.; Mizuguchi, H.; Koizumi, N.; Fujii, M.; Hayakawa, T.; Horiguchi, Y.; Watanbe, Y. A Novel Strategy for the Enhancement of Drug Absorption Using a Claudin Modulator. Mol. Pharmacol. 2005, 67, 749–756. [Google Scholar] [CrossRef]
- Simonovic, I.; Rosenberg, J.; Koutsouris, A.; Hecht, G. Enteropathogenic Escherichia Coli Dephosphorylates and Dissociates Occludin from Intestinal Epithelial Tight Junctions. Cell. Microbiol. 2000, 2, 305–315. [Google Scholar] [CrossRef]
- Shifflett, D.E.; Clayburgh, D.R.; Koutsouris, A.; Turner, J.R.; Hecht, G.A. Enteropathogenic E. Coli Disrupts Tight Junction Barrier Function and Structure in Vivo. Lab. Investig. 2005, 85, 1308–1324. [Google Scholar] [CrossRef]
- Fasano, A.; Nataro, J.P. Intestinal Epithelial Tight Junctions as Targets for Enteric Bacteria-Derived Toxins. Adv. Drug Deliv. Rev. 2004, 56, 795–807. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-Activated Receptors: Contribution to Physiology and Disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef]
- Pontarollo, G.; Mann, A.; Brandão, I.; Malinarich, F.; Schöpf, M.; Reinhardt, C. Protease-Activated Receptor Signaling in Intestinal Permeability Regulation. FEBS J. 2020, 287, 645–658. [Google Scholar] [CrossRef]
- Reinhardt, C.; Bergentall, M.; Greiner, T.U.; Schaffner, F.; Ostergren-Lundén, G.; Petersen, L.C.; Ruf, W.; Bäckhed, F. Tissue Factor and PAR1 Promote Microbiota-Induced Intestinal Vascular Remodelling. Nature 2012, 483, 627–631. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, Y.; Heo, G.; Jeong, S.; Park, S.; Yoo, J.-W.; Jung, Y.; Im, E. Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy. Cells 2022, 11, 878. [Google Scholar] [CrossRef]
- Jacob, C.; Yang, P.-C.; Darmoul, D.; Amadesi, S.; Saito, T.; Cottrell, G.S.; Coelho, A.-M.; Singh, P.; Grady, E.F.; Perdue, M.; et al. Mast Cell Tryptase Controls Paracellular Permeability of the Intestine. Role of Protease-Activated Receptor 2 and Beta-Arrestins. J. Biol. Chem. 2005, 280, 31936–31948. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic Bacterial Metabolites Regulate Gastrointestinal Barrier Function via the Xenobiotic Sensor PXR and Toll-like Receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Grosheva, I.; Zheng, D.; Levy, M.; Polansky, O.; Lichtenstein, A.; Golani, O.; Dori-Bachash, M.; Moresi, C.; Shapiro, H.; Del Mare-Roumani, S.; et al. High-Throughput Screen Identifies Host and Microbiota Regulators of Intestinal Barrier Function. Gastroenterology 2020, 159, 1807–1823. [Google Scholar] [CrossRef]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.-J. Review Article: The Role of Butyrate on Colonic Function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef]
- Roediger, W.E. Role of Anaerobic Bacteria in the Metabolic Welfare of the Colonic Mucosa in Man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef]
- Della Ragione, F.; Criniti, V.; Della Pietra, V.; Borriello, A.; Oliva, A.; Indaco, S.; Yamamoto, T.; Zappia, V. Genes Modulated by Histone Acetylation as New Effectors of Butyrate Activity. FEBS Lett. 2001, 499, 199–204. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef]
- Harding, S.D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.H.; Davenport, A.P.; Pawson, A.J.; Spedding, M.; Davies, J.A. NC-IUPHAR The IUPHAR/BPS Guide to PHARMACOLOGY in 2022: Curating Pharmacology for COVID-19, Malaria and Antibacterials. Nucleic Acids Res. 2022, 50, D1282–D1294. [Google Scholar] [CrossRef]
- Ohata, A.; Usami, M.; Miyoshi, M. Short-Chain Fatty Acids Alter Tight Junction Permeability in Intestinal Monolayer Cells via Lipoxygenase Activation. Nutrition 2005, 21, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Suzuki, Y.; Saito, Y. Butyrate Reduces Colonic Paracellular Permeability by Enhancing PPARgamma Activation. Biochem. Biophys. Res. Commun. 2002, 293, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, M.; Usami, M.; Ohata, A. Short-Chain Fatty Acids and Trichostatin A Alter Tight Junction Permeability in Human Umbilical Vein Endothelial Cells. Nutrition 2008, 24, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological Concentrations of Short-Chain Fatty Acids Immediately Suppress Colonic Epithelial Permeability. Br. J. Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor–Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef]
- Wang, H.-B.; Wang, P.-Y.; Wang, X.; Wan, Y.-L.; Liu, Y.-C. Butyrate Enhances Intestinal Epithelial Barrier Function via Up-Regulation of Tight Junction Protein Claudin-1 Transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-Dependent Regulation of Claudin-1 Is Central to Intestinal Epithelial Tight Junction Integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef]
- Ma, X.; Fan, P.X.; Li, L.S.; Qiao, S.Y.; Zhang, G.L.; Li, D.F. Butyrate Promotes the Recovering of Intestinal Wound Healing through Its Positive Effect on the Tight Junctions. J. Anim. Sci. 2012, 90 (Suppl. 4), 266–268. [Google Scholar] [CrossRef]
- Saleri, R.; Borghetti, P.; Ravanetti, F.; Cavalli, V.; Ferrari, L.; De Angelis, E.; Andrani, M.; Martelli, P. Effects of Different Short-Chain Fatty Acids (SCFA) on Gene Expression of Proteins Involved in Barrier Function in IPEC-J2. Porc. Health Manag. 2022, 8, 21. [Google Scholar] [CrossRef]
- Kondrashina, A.; Brodkorb, A.; Giblin, L. Sodium Butyrate Converts Caco-2 Monolayers into a Leaky but Healthy Intestinal Barrier Resembling That of a Newborn Infant. Food Funct. 2021, 12, 5066–5076. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Feng, W.; Wu, Y.; Chen, G.; Fu, S.; Li, B.; Huang, B.; Wang, D.; Wang, W.; Liu, J. Sodium Butyrate Attenuates Diarrhea in Weaned Piglets and Promotes Tight Junction Protein Expression in Colon in a GPR109A-Dependent Manner. Cell. Physiol. Biochem. 2018, 47, 1617–1629. [Google Scholar] [CrossRef]
- Landman, C.; Grill, J.-P.; Mallet, J.-M.; Marteau, P.; Humbert, L.; Le Balc’h, E.; Maubert, M.-A.; Perez, K.; Chaara, W.; Brot, L.; et al. Inter-Kingdom Effect on Epithelial Cells of the N-Acyl Homoserine Lactone 3-Oxo-C12:2, a Major Quorum-Sensing Molecule from Gut Microbiota. PLoS ONE 2018, 13, e0202587. [Google Scholar] [CrossRef]
- Aguanno, D.; Coquant, G.; Postal, B.G.; Osinski, C.; Wieckowski, M.; Stockholm, D.; Grill, J.-P.; Carrière, V.; Seksik, P.; Thenet, S. The Intestinal Quorum Sensing 3-Oxo-C12:2 Acyl Homoserine Lactone Limits Cytokine-Induced Tight Junction Disruption. Tissue Barriers 2020, 8, 1832877. [Google Scholar] [CrossRef]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.-E. Colonic Bacterial Metabolites and Human Health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef]
- Zheng, X.; Xie, G.; Zhao, A.; Zhao, L.; Yao, C.; Chiu, N.H.L.; Zhou, Z.; Bao, Y.; Jia, W.; Nicholson, J.K.; et al. The Footprints of Gut Microbial-Mammalian Co-Metabolism. J. Proteome Res. 2011, 10, 5512–5522. [Google Scholar] [CrossRef]
- Gallo, R.L. Human Skin Is the Largest Epithelial Surface for Interaction with Microbes. J. Investig. Dermatol. 2017, 137, 1213–1214. [Google Scholar] [CrossRef]
- Bay, L.; Barnes, C.J.; Fritz, B.G.; Thorsen, J.; Restrup, M.E.M.; Rasmussen, L.; Sørensen, J.K.; Hesselvig, A.B.; Odgaard, A.; Hansen, A.J.; et al. Universal Dermal Microbiome in Human Skin. mBio 2020, 11, e02945-19. [Google Scholar] [CrossRef]
- Bay, L.; Ring, H.C. Human Skin Microbiota in Health and Disease: The Cutaneous Communities’ Interplay in Equilibrium and Dysbiosis. Apmis 2021, 130, 706–718. [Google Scholar] [CrossRef]
- Leshem, A.; Liwinski, T.; Elinav, E. Immune-Microbiota Interplay and Colonization Resistance in Infection. Mol. Cell 2020, 78, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Asmuth, J.; Baker, S.E.; Langhofer, M.; Roth, S.I.; Hopkinson, S.B. Hemidesmosomes: Extracellular Matrix/Intermediate Filament Connectors. Exp. Cell Res. 1994, 213, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Marekov, L.N. The Proteins Elafin, Filaggrin, Keratin Intermediate Filaments, Loricrin, and Small Proline-Rich Proteins 1 and 2 Are Isodipeptide Cross-Linked Components of the Human Epidermal Cornified Cell Envelope. J. Biol. Chem. 1995, 270, 17702–17711. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.; Lee, M.J.; Norton, D.; MacLeod, A.S. The Skin and Intestinal Microbiota and Their Specific Innate Immune Systems. Front. Immunol. 2019, 10, 2950. [Google Scholar] [CrossRef]
- Egelrud, T. Desquamation in the Stratum Corneum. Acta Derm. Venereol. Suppl. 2000, 208, 44–45. [Google Scholar] [CrossRef]
- Lin, T.-K.; Crumrine, D.; Ackerman, L.D.; Santiago, J.-L.; Roelandt, T.; Uchida, Y.; Hupe, M.; Fabriàs, G.; Abad, J.L.; Rice, R.H.; et al. Cellular Changes That Accompany Shedding of Human Corneocytes. J. Investig. Dermatol. 2012, 132, 2430–2439. [Google Scholar] [CrossRef]
- Abhishek, S.; Palamadai Krishnan, S. Epidermal Differentiation Complex: A Review on Its Epigenetic Regulation and Potential Drug Targets. Cell J. 2016, 18, 1–6. [Google Scholar] [CrossRef]
- Hashimoto, K. Intercellular Spaces of the Human Epidermis as Demonstrated with Lanthanum. J. Investig. Dermatol. 1971, 57, 17–31. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-Based Tight Junctions Are Crucial for the Mammalian Epidermal Barrier: A Lesson from Claudin-1-Deficient Mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Kirschner, N.; Houdek, P.; Fromm, M.; Moll, I.; Brandner, J.M. Tight Junctions Form a Barrier in Human Epidermis. Eur. J. Cell Biol. 2010, 89, 839–842. [Google Scholar] [CrossRef]
- Brandner, J.M.; Kief, S.; Grund, C.; Rendl, M.; Houdek, P.; Kuhn, C.; Tschachler, E.; Franke, W.W.; Moll, I. Organization and Formation of the Tight Junction System in Human Epidermis and Cultured Keratinocytes. Eur. J. Cell Biol. 2002, 81, 253–263. [Google Scholar] [CrossRef]
- Zorn-Kruppa, M.; Vidal-y-Sy, S.; Houdek, P.; Wladykowski, E.; Grzybowski, S.; Gruber, R.; Gorzelanny, C.; Harcup, J.; Schneider, S.W.; Majumdar, A.; et al. Tight Junction Barriers in Human Hair Follicles—Role of Claudin-1. Sci. Rep. 2018, 8, 12800. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Kishibe, M.; Murakami, M.; Honma, M.; Takahashi, H.; Iizuka, H. Lamellar Granule Secretion Starts before the Establishment of Tight Junction Barrier for Paracellular Tracers in Mammalian Epidermis. PLoS ONE 2012, 7, e31641. [Google Scholar] [CrossRef]
- Brandner, J.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.; De Benedetto, A. Epidermal Tight Junctions in Health and Disease. Tissue Barriers 2014, 3, e974451. [Google Scholar] [CrossRef]
- Haftek, M.; Oji, V.; Feldmeyer, L.; Hohl, D.; Hadj-Rabia, S.; Abdayem, R. The Fate of Epidermal Tight Junctions in the Stratum Corneum: Their Involvement in the Regulation of Desquamation and Phenotypic Expression of Certain Skin Conditions. Int. J. Mol. Sci. 2022, 23, 7486. [Google Scholar] [CrossRef]
- Kaiser, H.W.; Ness, W.; Jungblut, I.; Briggaman, R.A.; Kreysel, H.W.; O’Keefe, E.J. Adherens Junctions: Demonstration in Human Epidermis. J. Investig. Dermatol. 1993, 100, 180–185. [Google Scholar] [CrossRef]
- Ishiko, A.; Matsunaga, Y.; Masunaga, T.; Aiso, S.; Nishikawa, T.; Shimizu, H. Immunomolecular Mapping of Adherens Junction and Desmosomal Components in Normal Human Epidermis. Exp. Dermatol. 2003, 12, 747–754. [Google Scholar] [CrossRef]
- Tunggal, J.A.; Helfrich, I.; Schmitz, A.; Schwarz, H.; Günzel, D.; Fromm, M.; Kemler, R.; Krieg, T.; Niessen, C.M. E-Cadherin Is Essential for in Vivo Epidermal Barrier Function by Regulating Tight Junctions. EMBO J. 2005, 24, 1146–1156. [Google Scholar] [CrossRef]
- Schmitt, T.; Pircher, J.; Steinert, L.; Meier, K.; Ghoreschi, K.; Vielmuth, F.; Kugelmann, D.; Waschke, J. Dsg1 and Dsg3 Composition of Desmosomes Across Human Epidermis and Alterations in Pemphigus Vulgaris Patient Skin. Front. Immunol. 2022, 13, 884241. [Google Scholar] [CrossRef]
- Hegazy, M.; Perl, A.L.; Svoboda, S.A.; Green, K.J. Desmosomal Cadherins in Health and Disease. Annu. Rev. Pathol. 2022, 17, 47–72. [Google Scholar] [CrossRef]
- Harrison, O.J.; Brasch, J.; Lasso, G.; Katsamba, P.S.; Ahlsen, G.; Honig, B.; Shapiro, L. Structural Basis of Adhesive Binding by Desmocollins and Desmogleins. Proc. Natl. Acad. Sci. USA 2016, 113, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.J.; Walsh, A. Desmosomes, Corneosomes and Desquamation. An Ultrastructural Study of Adult Pig Epidermis. Arch. Dermatol. Res. 1990, 282, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Jonca, N.; Leclerc, E.A.; Caubet, C.; Simon, M.; Guerrin, M.; Serre, G. Corneodesmosomes and Corneodesmosin: From the Stratum Corneum Cohesion to the Pathophysiology of Genodermatoses. Eur. J. Dermatol. 2011, 21 (Suppl. 2), 35–42. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Igawa, S. The Biology and Regulation of Corneodesmosomes. Cell Tissue Res. 2015, 360, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Yokouchi, M.; Kubo, A. Maintenance of Tight Junction Barrier Integrity in Cell Turnover and Skin Diseases. Exp. Dermatol. 2018, 27, 876–883. [Google Scholar] [CrossRef]
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin Microbiota-Host Interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef]
- Darlenski, R.; Kozyrskyj, A.L.; Fluhr, J.W.; Caraballo, L. Association between Barrier Impairment and Skin Microbiota in Atopic Dermatitis from a Global Perspective: Unmet Needs and Open Questions. J. Allergy Clin. Immunol. 2021, 148, 1387–1393. [Google Scholar] [CrossRef]
- Uberoi, A.; Bartow-McKenney, C.; Zheng, Q.; Flowers, L.; Campbell, A.; Knight, S.A.B.; Chan, N.; Wei, M.; Lovins, V.; Bugayev, J.; et al. Commensal Microbiota Regulates Skin Barrier Function and Repair via Signaling through the Aryl Hydrocarbon Receptor. Cell Host Microbe 2021, 29, 1235–1248. [Google Scholar] [CrossRef]
- Lee, E.; Min, K.; Ahn, H.; Jeon, B.; Park, S.; Yun, C.; Jeon, H.; Yeon, J.; Kim, H.; Park, H. Potential Therapeutic Skin Microbiomes Suppressing Staphylococcus Aureus-Derived Immune Responses and Upregulating Skin Barrier Function-Related Genes via the AhR Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 9551. [Google Scholar] [CrossRef]
- Loub, W.D.; Wattenberg, L.W.; Davis, D.W. Aryl Hydrocarbon Hydroxylase Induction in Rat Tissues by Naturally Occurring Indoles of Cruciferous Plants. J. Natl. Cancer Inst. 1975, 54, 985–988. [Google Scholar]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Salgaonkar, N.; Kadamkode, V.; Kumaran, S.; Mallemala, P.; Christy, E.; Appavoo, S.; Majumdar, A.; Mitra, R.; Dasgupta, A. Glycerol Fermentation by Skin Bacteria Generates Lactic Acid and Upregulates the Expression Levels of Genes Associated with the Skin Barrier Function. Exp. Dermatol. 2022, 31, 1364–1372. [Google Scholar] [CrossRef]
- Wang, Y.; Kuo, S.; Shu, M.; Yu, J.; Huang, S.; Dai, A.; Two, A.; Gallo, R.L.; Huang, C.-M. Staphylococcus Epidermidis in the Human Skin Microbiome Mediates Fermentation to Inhibit the Growth of Propionibacterium Acnes: Implications of Probiotics in Acne Vulgaris. Appl. Microbiol. Biotechnol. 2014, 98, 411–424. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, H.Y.; Kim, A.R.; Yun, C.-H.; Han, S.H. Propionate Ameliorates Staphylococcus Aureus Skin Infection by Attenuating Bacterial Growth. Front. Microbiol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Sanford, J.A.; Zhang, L.-J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.-M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by Microbial Short-Chain Fatty Acids Breaks Immune Tolerance of the Epidermis to TLR Ligands. Sci. Immunol. 2016, 1, eaah4609. [Google Scholar] [CrossRef]
- Trompette, A.; Pernot, J.; Perdijk, O.; Alqahtani, R.A.A.; Domingo, J.S.; Camacho-Muñoz, D.; Wong, N.C.; Kendall, A.C.; Wiederkehr, A.; Nicod, L.P.; et al. Gut-Derived Short-Chain Fatty Acids Modulate Skin Barrier Integrity by Promoting Keratinocyte Metabolism and Differentiation. Mucosal Immunol. 2022, 15, 908–926. [Google Scholar] [CrossRef]
- Aragón-González, A.; Shaw, P.J.; Ferraiuolo, L. Blood-Brain Barrier Disruption and Its Involvement in Neurodevelopmental and Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 15271. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Franke, W.W.; Cowin, P.; Grund, C.; Kuhn, C.; Kapprell, H.-P. The Endothelial Junction. In Endothelial Cell Biology in Health and Disease; Simionescu, N., Simionescu, M., Eds.; Springer: Boston, MA, USA, 1988; pp. 147–166. ISBN 978-1-4613-0937-6. [Google Scholar]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood-Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane Proteins of the Tight Junctions at the Blood-Brain Barrier: Structural and Functional Aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef]
- Saunders, N.R.; Habgood, M.D.; Møllgård, K.; Dziegielewska, K.M. The Biological Significance of Brain Barrier Mechanisms: Help or Hindrance in Drug Delivery to the Central Nervous System? F1000Res 2016, 5, 313. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Birkelund, S.; Burkhart, A.; Stensballe, A.; Moos, T. Synthesis and Deposition of Basement Membrane Proteins by Primary Brain Capillary Endothelial Cells in a Murine Model of the Blood–Brain Barrier. J. Neurochem. 2017, 140, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Sorokin, L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin. Immunopathol. 2009, 31, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Milner, R. Integrin-Matrix Interactions in the Cerebral Microvasculature. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.J.; McClenahan, F.K.; Leiton, C.V.; Aranmolate, A.; Shan, X.; Colognato, H. The Extracellular Matrix Protein Laminin A2 Regulates the Maturation and Function of the Blood-Brain Barrier. J. Neurosci. 2014, 34, 15260–15280. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, Z.-L.; Norris, E.H.; Strickland, S. Astrocytic Laminin Regulates Pericyte Differentiation and Maintains Blood Brain Barrier Integrity. Nat. Commun. 2014, 5, 3413. [Google Scholar] [CrossRef]
- Yang, T.; Guo, R.; Zhang, F. Brain Perivascular Macrophages: Recent Advances and Implications in Health and Diseases. CNS Neurosci. Ther. 2019, 25, 1318–1328. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, but Not Cells, Drain from the Brain Parenchyma along Basement Membranes of Capillaries and Arteries: Significance for Cerebral Amyloid Angiopathy and Neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef]
- Willis, C.L.; Garwood, C.J.; Ray, D.E. A Size Selective Vascular Barrier in the Rat Area Postrema Formed by Perivascular Macrophages and the Extracellular Matrix. Neuroscience 2007, 150, 498–509. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Yilmaz-Ozcan, S.; Yemisci, M.; Schallek, J.; Kılıç, K.; Can, A.; Di Polo, A.; Dalkara, T. Capillary Pericytes Express α-Smooth Muscle Actin, Which Requires Prevention of Filamentous-Actin Depolymerization for Detection. eLife 2018, 7, e34861. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary Pericytes Regulate Cerebral Blood Flow in Health and Disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Crivellato, E. The Role of Pericytes in Angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of Pericytes Leads to Endothelial Hyperplasia and Abnormal Vascular Morphogenesis. J. Cell Biol. 2001, 153, 543–553. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Janzer, R.C.; Raff, M.C. Astrocytes Induce Blood-Brain Barrier Properties in Endothelial Cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Yamamura, T.; Arima, K.; Konno, H. Human Astrocytes Express Aquaporin-1 and Aquaporin-4 in Vitro and in Vivo. Neuropathology 2007, 27, 245–256. [Google Scholar] [CrossRef]
- Asgari, M.; De Zélicourt, D.; Kurtcuoglu, V. How Astrocyte Networks May Contribute to Cerebral Metabolite Clearance. Sci. Rep. 2015, 5, 15024. [Google Scholar] [CrossRef]
- McConnell, H.L.; Mishra, A. Cells of the Blood-Brain Barrier: An Overview of the Neurovascular Unit in Health and Disease. Methods Mol. Biol. 2022, 2492, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Dziegielewska, K.M.; Møllgård, K.; Habgood, M.D. Markers for Blood-Brain Barrier Integrity: How Appropriate Is Evans Blue in the Twenty-First Century and What Are the Alternatives? Front. Neurosci. 2015, 9, 385. [Google Scholar] [CrossRef] [PubMed]
- Eckman, P.L.; King, W.M.; Brunson, J.G. Studies on the Blood Brain Barrier. I. Effects Produced by a Single Injection of Gramnegative Endotoxin on the Permeability of the Cerebral Vessels. Am. J. Pathol. 1958, 34, 631–643. [Google Scholar] [PubMed]
- Banks, W.A.; Kastin, A.J.; Brennan, J.M.; Vallance, K.L. Adsorptive Endocytosis of HIV-1gp120 by Blood-Brain Barrier Is Enhanced by Lipopolysaccharide. Exp. Neurol. 1999, 156, 165–171. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Moor, A.C.; Blom-Roosemalen, M.C.; De Boer, A.G.; Breimer, D.D.; Van Berkel, T.J.; Kuiper, J. Lymphocyte Adhesion to Brain Capillary Endothelial Cells in Vitro. J. Neuroimmunol. 1994, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Bauer, B.; Fricker, G.; Miller, D.S. Rapid Modulation of P-Glycoprotein-Mediated Transport at the Blood-Brain Barrier by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. Mol. Pharmacol. 2006, 69, 462–470. [Google Scholar] [CrossRef]
- Salkeni, M.A.; Lynch, J.L.; Otamis-Price, T.; Banks, W.A. Lipopolysaccharide Impairs Blood-Brain Barrier P-Glycoprotein Function in Mice through Prostaglandin- and Nitric Oxide-Independent Pathways. J. Neuroimmune Pharmacol. 2009, 4, 276–282. [Google Scholar] [CrossRef]
- Singh, A.K.; Jiang, Y.; Gupta, S. Effects of Bacterial Toxins on Endothelial Tight Junction in Vitro: A Mechanism-Based Investigation. Toxicol. Mech. Methods 2007, 17, 331–347. [Google Scholar] [CrossRef]
- Choi, J.J.; Choi, Y.J.; Chen, L.; Zhang, B.; Eum, S.Y.; Abreu, M.T.; Toborek, M. Lipopolysaccharide Potentiates Polychlorinated Biphenyl-Induced Disruption of the Blood-Brain Barrier via TLR4/IRF-3 Signaling. Toxicology 2012, 302, 212–220. [Google Scholar] [CrossRef]
- Zhou, T.; Zhao, L.; Zhan, R.; He, Q.; Tong, Y.; Tian, X.; Wang, H.; Zhang, T.; Fu, Y.; Sun, Y.; et al. Blood-Brain Barrier Dysfunction in Mice Induced by Lipopolysaccharide Is Attenuated by Dapsone. Biochem. Biophys. Res. Commun. 2014, 453, 419–424. [Google Scholar] [CrossRef]
- Hoyles, L.; Snelling, T.; Umlai, U.-K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome-Host Systems Interactions: Protective Effects of Propionate upon the Blood-Brain Barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef]
- Qin, L.; Huang, W.; Mo, X.; Chen, Y.; Wu, X. LPS Induces Occludin Dysregulation in Cerebral Microvascular Endothelial Cells via MAPK Signaling and Augmenting MMP-2 Levels. Oxid. Med. Cell. Longev. 2015, 2015, 120641. [Google Scholar] [CrossRef]
- Zhao, Z.; Hu, J.; Gao, X.; Liang, H.; Liu, Z. Activation of AMPK Attenuates Lipopolysaccharide-Impaired Integrity and Function of Blood-Brain Barrier in Human Brain Microvascular Endothelial Cells. Exp. Mol. Pathol. 2014, 97, 386–392. [Google Scholar] [CrossRef]
- Gasparotto, J.; Ribeiro, C.T.; Bortolin, R.C.; Somensi, N.; Fernandes, H.S.; Teixeira, A.A.; Guasselli, M.O.R.; Agani, C.A.J.O.; Souza, N.C.; Grings, M.; et al. Anti-RAGE Antibody Selectively Blocks Acute Systemic Inflammatory Responses to LPS in Serum, Liver, CSF and Striatum. Brain Behav. Immun. 2017, 62, 124–136. [Google Scholar] [CrossRef]
- Maggioli, E.; McArthur, S.; Mauro, C.; Kieswich, J.; Kusters, D.H.M.; Reutelingsperger, C.P.M.; Yaqoob, M.; Solito, E. Estrogen Protects the Blood-Brain Barrier from Inflammation-Induced Disruption and Increased Lymphocyte Trafficking. Brain Behav. Immun. 2016, 51, 212–222. [Google Scholar] [CrossRef]
- Asarian, L.; Langhans, W. A New Look on Brain Mechanisms of Acute Illness Anorexia. Physiol. Behav. 2010, 100, 464–471. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The Gut Microbiota Influences Blood-Brain Barrier Permeability in Mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Fröhlich, E.E.; Farzi, A.; Mayerhofer, R.; Reichmann, F.; Jačan, A.; Wagner, B.; Zinser, E.; Bordag, N.; Magnes, C.; Fröhlich, E.; et al. Cognitive Impairment by Antibiotic-Induced Gut Dysbiosis: Analysis of Gut Microbiota-Brain Communication. Brain Behav. Immun. 2016, 56, 140–155. [Google Scholar] [CrossRef]
- Sun, N.; Hu, H.; Wang, F.; Li, L.; Zhu, W.; Shen, Y.; Xiu, J.; Xu, Q. Antibiotic-Induced Microbiome Depletion in Adult Mice Disrupts Blood-Brain Barrier and Facilitates Brain Infiltration of Monocytes after Bone-Marrow Transplantation. Brain Behav. Immun. 2021, 92, 102–114. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Wang, F.; Ding, G.; Chen, W.; Fang, R.; Yao, Y.; Pang, M.; Lu, Z.-Q.; Liu, J. Sodium Butyrate Exerts Neuroprotective Effects by Restoring the Blood-Brain Barrier in Traumatic Brain Injury Mice. Brain Res. 2016, 1642, 70–78. [Google Scholar] [CrossRef]
- Wang, Z.; Leng, Y.; Tsai, L.-K.; Leeds, P.; Chuang, D.-M. Valproic Acid Attenuates Blood-Brain Barrier Disruption in a Rat Model of Transient Focal Cerebral Ischemia: The Roles of HDAC and MMP-9 Inhibition. J. Cereb. Blood Flow Metab. 2011, 31, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Knox, E.G.; Aburto, M.R.; Tessier, C.; Nagpal, J.; Clarke, G.; O’Driscoll, C.M.; Cryan, J.F. Microbial-Derived Metabolites Induce Actin Cytoskeletal Rearrangement and Protect Blood-Brain Barrier Function. iScience 2022, 25, 105648. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Adu, J.; Davey, A.J.; Abbott, N.J.; Bradbury, M.W. The Effect of Bile Salts on the Permeability and Ultrastructure of the Perfused, Energy-Depleted, Rat Blood-Brain Barrier. J. Cereb. Blood Flow Metab. 1991, 11, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.; McMillin, M.; Galindo, C.; Frampton, G.; Pae, H.Y.; DeMorrow, S. Bile Acids Permeabilize the Blood Brain Barrier after Bile Duct Ligation in Rats via Rac1-Dependent Mechanisms. Dig. Liver Dis. 2014, 46, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Palmela, I.; Correia, L.; Silva, R.F.M.; Sasaki, H.; Kim, K.S.; Brites, D.; Brito, M.A. Hydrophilic Bile Acids Protect Human Blood-Brain Barrier Endothelial Cells from Disruption by Unconjugated Bilirubin: An in Vitro Study. Front. Neurosci. 2015, 9, 80. [Google Scholar] [CrossRef]
- Thomas, M.S.; Fernandez, M.L. Trimethylamine N-Oxide (TMAO), Diet and Cardiovascular Disease. Curr. Atheroscler. Rep. 2021, 23, 12. [Google Scholar] [CrossRef]
- Bordoni, L.; Samulak, J.J.; Sawicka, A.K.; Pelikant-Malecka, I.; Radulska, A.; Lewicki, L.; Kalinowski, L.; Gabbianelli, R.; Olek, R.A. Trimethylamine N-Oxide and the Reverse Cholesterol Transport in Cardiovascular Disease: A Cross-Sectional Study. Sci. Rep. 2020, 10, 18675. [Google Scholar] [CrossRef]
- Skagen, K.; Trøseid, M.; Ueland, T.; Holm, S.; Abbas, A.; Gregersen, I.; Kummen, M.; Bjerkeli, V.; Reier-Nilsen, F.; Russell, D.; et al. The Carnitine-Butyrobetaine-Trimethylamine-N-Oxide Pathway and Its Association with Cardiovascular Mortality in Patients with Carotid Atherosclerosis. Atherosclerosis 2016, 247, 64–69. [Google Scholar] [CrossRef]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine Intake and High Trimethylamine N-Oxide Plasma Levels Correlate with Low Aortic Lesions in ApoE(-/-) Transgenic Mice Expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef]
- Huc, T.; Drapala, A.; Gawrys, M.; Konop, M.; Bielinska, K.; Zaorska, E.; Samborowska, E.; Wyczalkowska-Tomasik, A.; Pączek, L.; Dadlez, M.; et al. Chronic, Low-Dose TMAO Treatment Reduces Diastolic Dysfunction and Heart Fibrosis in Hypertensive Rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1805–H1820. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Xin, F.-Z.; Zhou, D.; Xue, Y.-Q.; Liu, X.-L.; Yang, R.-X.; Pan, Q.; Fan, J.-G. Trimethylamine N-Oxide Attenuates High-Fat High-Cholesterol Diet-Induced Steatohepatitis by Reducing Hepatic Cholesterol Overload in Rats. World J. Gastroenterol. 2019, 25, 2450–2462. [Google Scholar] [CrossRef]
- Dumas, M.E.; Rothwell, A.R.; Hoyles, L.; Aranias, T.; Chilloux, J.; Calderari, S.; Noll, E.M.; Péan, N.; Boulangé, C.L.; Blancher, C.; et al. Microbial-Host Co-Metabolites Are Prodromal Markers Predicting Phenotypic Heterogeneity in Behavior, Obesity, and Impaired Glucose Tolerance. Cell Rep. 2017, 20, 136–148. [Google Scholar] [CrossRef]
- Hoyles, L.; Pontifex, M.G.; Rodriguez-Ramiro, I.; Anis-Alavi, M.A.; Jelane, K.S.; Snelling, T.; Solito, E.; Fonseca, S.; Carvalho, A.L.; Carding, S.R.; et al. Regulation of Blood-Brain Barrier Integrity by Microbiome-Associated Methylamines and Cognition by Trimethylamine N-Oxide. Microbiome 2021, 9, 235. [Google Scholar] [CrossRef]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an Essential Endogenous Regulator of Blood-Brain Barrier Integrity, and Its Pathological and Therapeutic Implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of Phenol- and p-Cresol-Producing Intestinal Bacteria by Using Media Supplemented with Tyrosine and Its Metabolites. FEMS Microbiol. Ecol. 2018, 94, fiy125. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Lesaffer, G. P-Cresol: A Toxin Revealing Many Neglected but Relevant Aspects of Uraemic Toxicity. Nephrol. Dial. Transplant. 1999, 14, 2813–2815. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. P-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef]
- Shah, S.N.; Knausenberger, T.B.-A.; Connell, E.; Gall, G.L.; Hardy, T.A.J.; Randall, D.W.; McCafferty, K.; Yaqoob, M.M.; Solito, E.; Müller, M.; et al. Cerebrovascular Damage Caused by the Gut Microbe-Derived Uraemic Toxin p-Cresol Sulfate Is Prevented by Blockade of the Epidermal Growth Factor Receptor. bioRxiv 2022. [Google Scholar] [CrossRef]
- Stachulski, A.V.; Knausenberger, T.B.-A.; Shah, S.N.; Hoyles, L.; McArthur, S. A Host-Gut Microbial Amino Acid Co-Metabolite, p-Cresol Glucuronide, Promotes Blood-Brain Barrier Integrity in Vivo. Tissue Barriers 2022, 11, 2073175. [Google Scholar] [CrossRef]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic Distribution of Three Pathways for Propionate Production within the Human Gut Microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Component | Junction Type | Junctional Role |
|---|---|---|
| Claudins | TJ | Homotypic/heterotypic cell–cell interactions |
| Occludin | TJ | Homotypic cell–cell interactions |
| Tricellulin | TJ | Homotypic cell–cell interactions at tripartite or greater junctions |
| ZO-1, -2, -3 | TJ | Link between claudins, occludin or JAM-A and the actin cytoskeleton |
| JAM-A | JAM | Cell–cell communication, stabilisation of the TJ environment |
| E-cadherin | AJ | Homotypic cell–cell interactions |
| β-catenin | AJ | Act as a multipartite complex linking E-cadherin with the actin cytoskeleton |
| α-catenin | AJ | |
| p120-catenin | AJ | |
| Desmocollin | Desmosome | Cadherin family proteins, bringing neighbouring cell membranes into apposition |
| Desmoglein | Desmosome | |
| Plakoglobin | Desmosome | Catenin family proteins, linking desmocollin and desmoglein to desmoplakin |
| Plakophilin | Desmosome | |
| Desmoplakin | Desmosome | Link other desmosomal proteins to keratin intermediate fibres of the cytoskeleton |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McArthur, S. Regulation of Physiological Barrier Function by the Commensal Microbiota. Life 2023, 13, 396. https://doi.org/10.3390/life13020396
McArthur S. Regulation of Physiological Barrier Function by the Commensal Microbiota. Life. 2023; 13(2):396. https://doi.org/10.3390/life13020396
Chicago/Turabian StyleMcArthur, Simon. 2023. "Regulation of Physiological Barrier Function by the Commensal Microbiota" Life 13, no. 2: 396. https://doi.org/10.3390/life13020396
APA StyleMcArthur, S. (2023). Regulation of Physiological Barrier Function by the Commensal Microbiota. Life, 13(2), 396. https://doi.org/10.3390/life13020396