
Interaction of Proteins Involved in Neuronal Proteinopathies
 ,
,
Abstract
1. Introduction
2. Pathologies Related to the Aggregation of Proteins
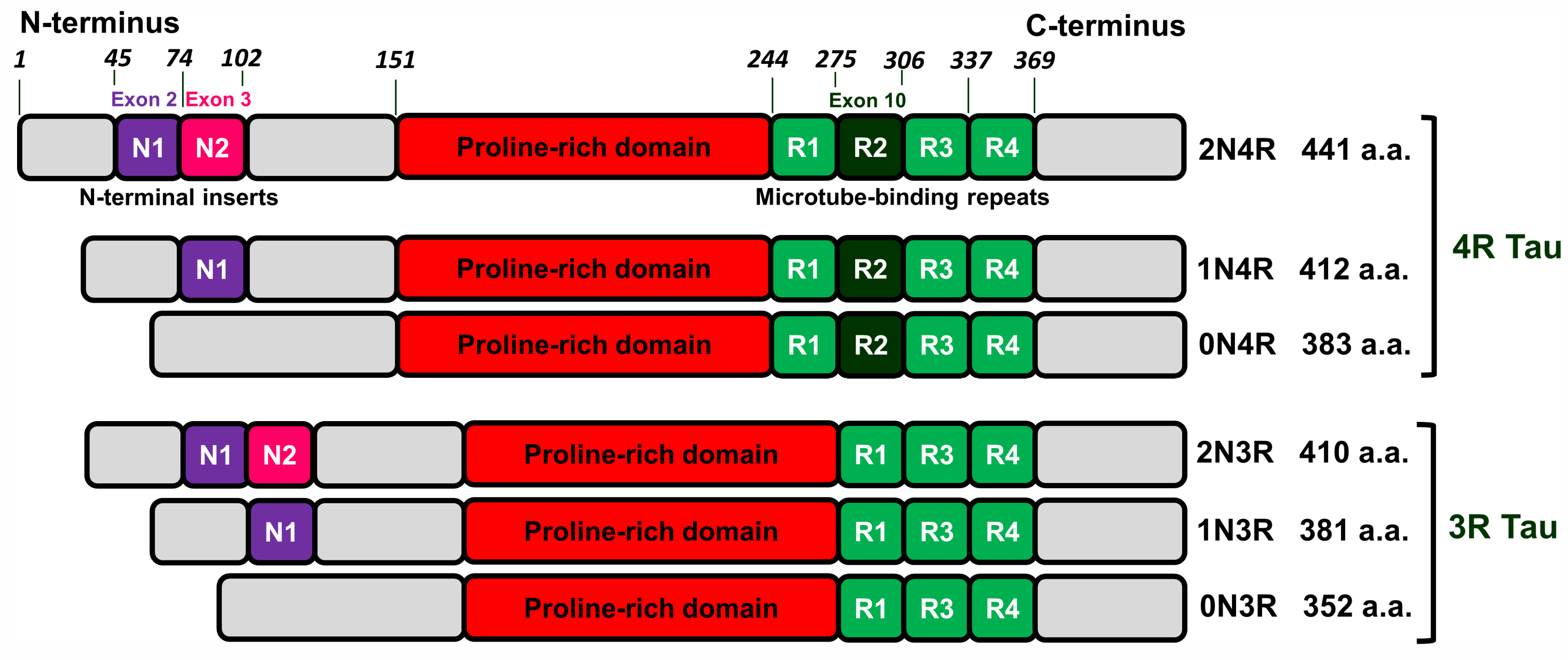
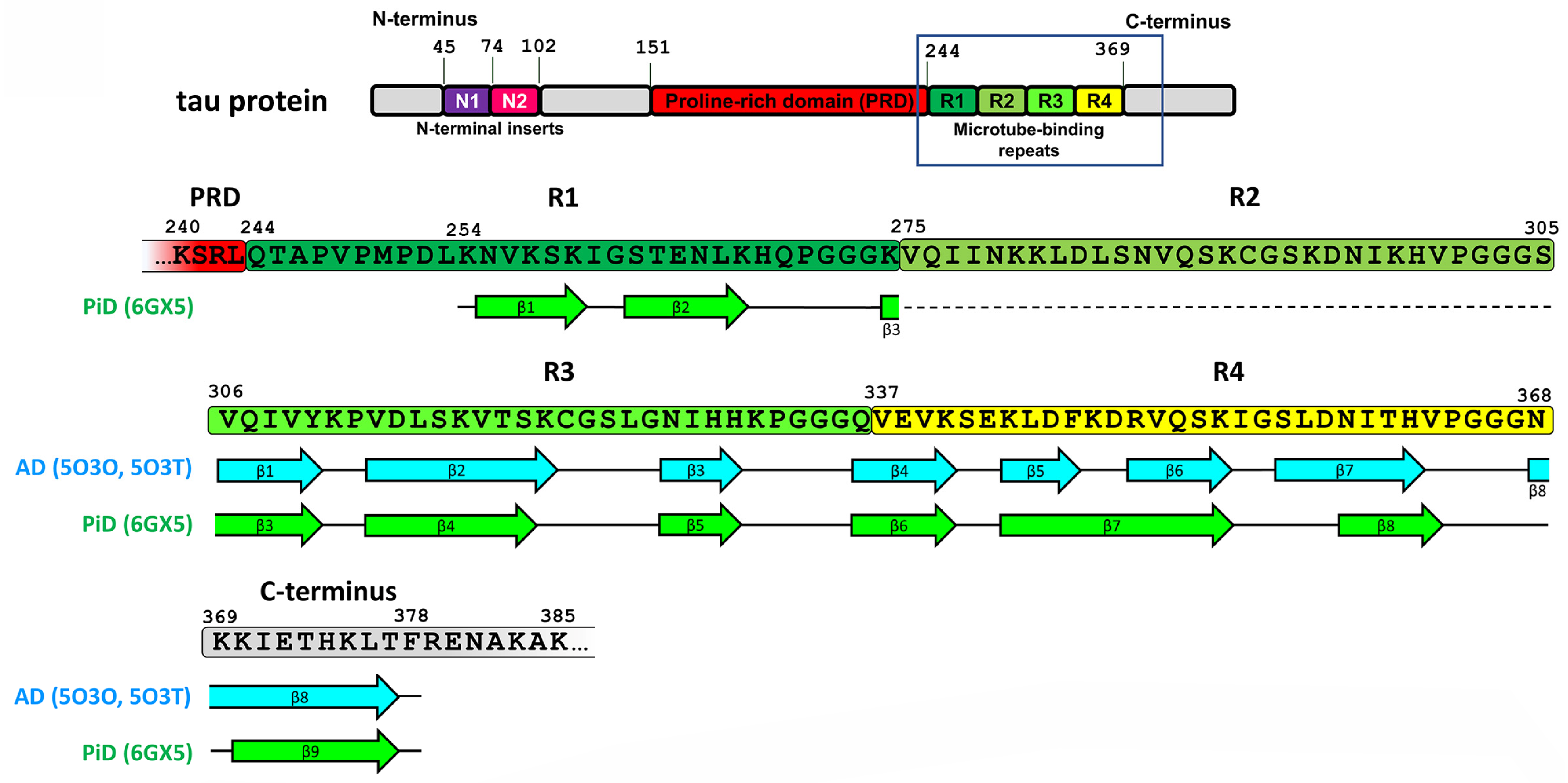
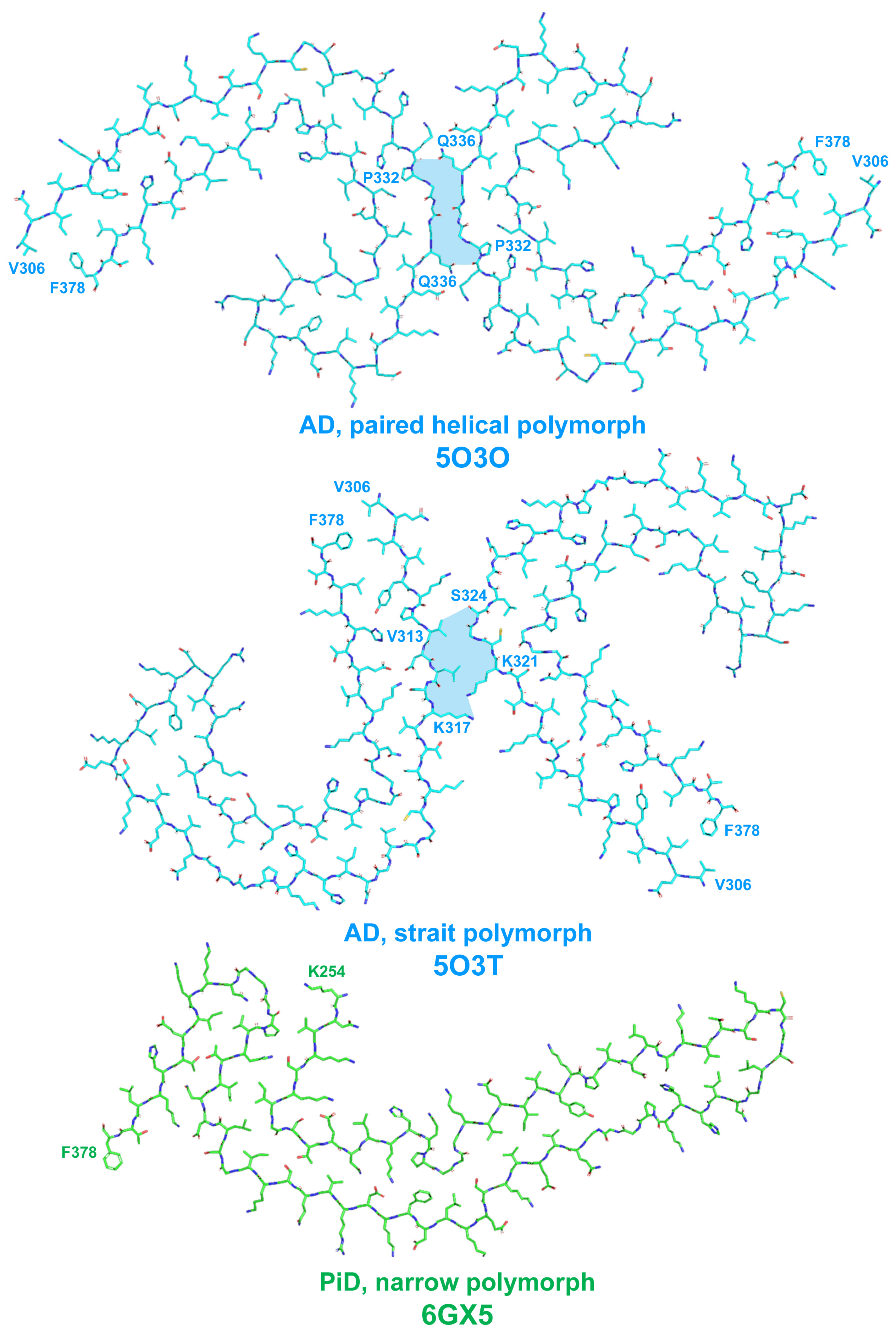
2.1. Tau Protein-Related Pathologies
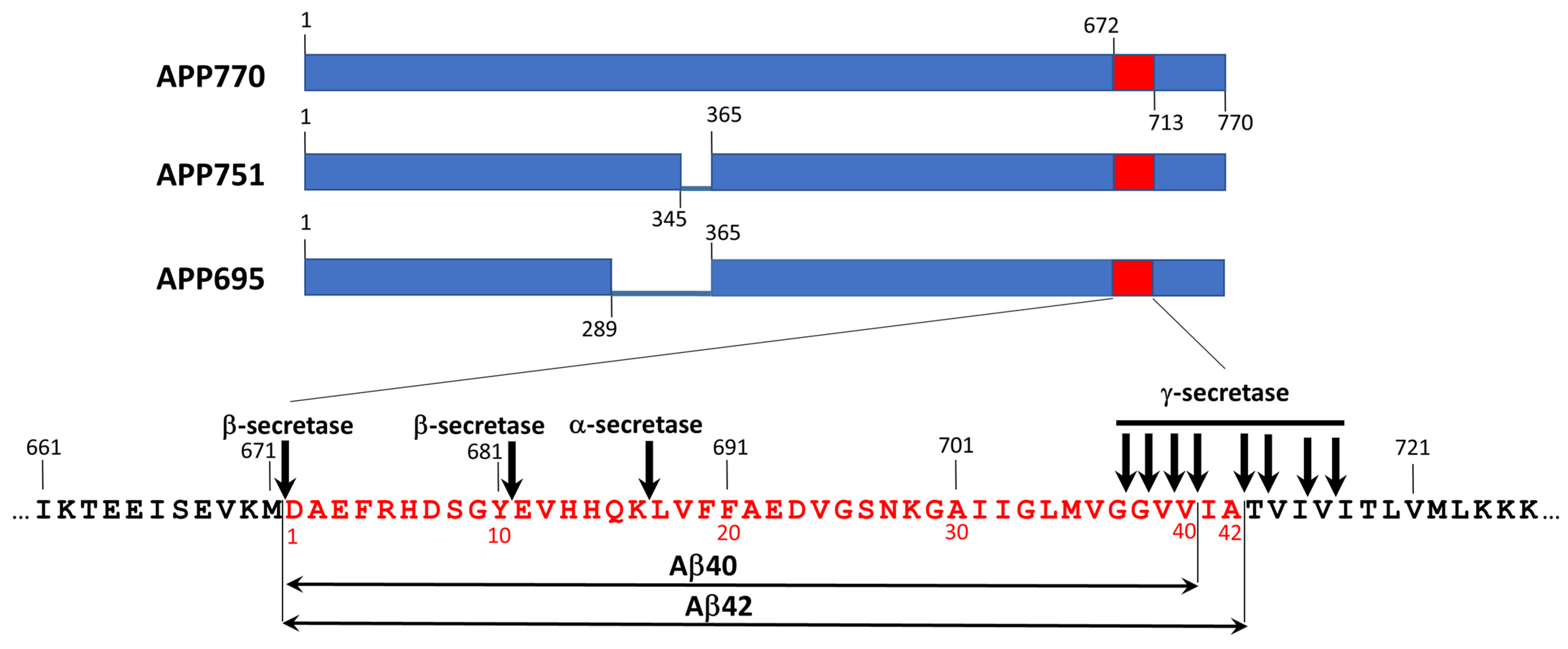
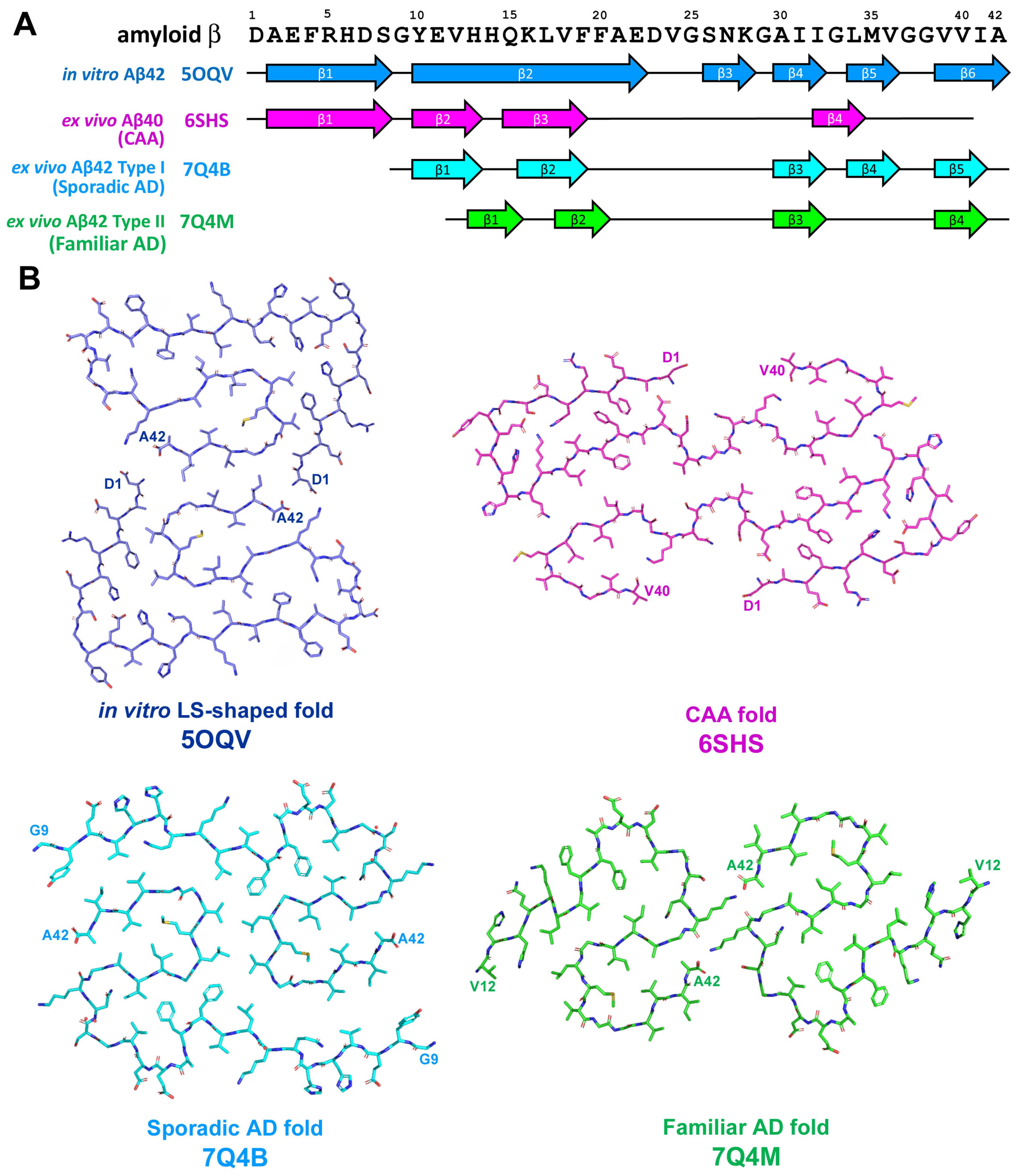
2.2. Amyloid-β-Related Pathologies
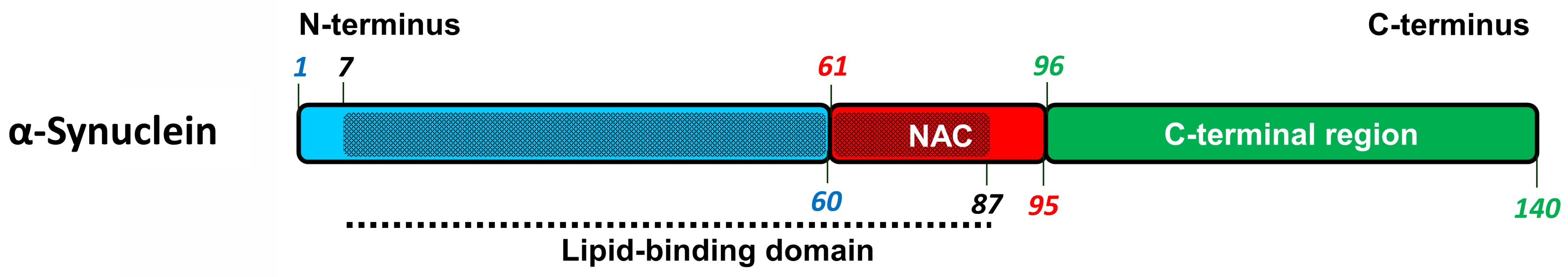
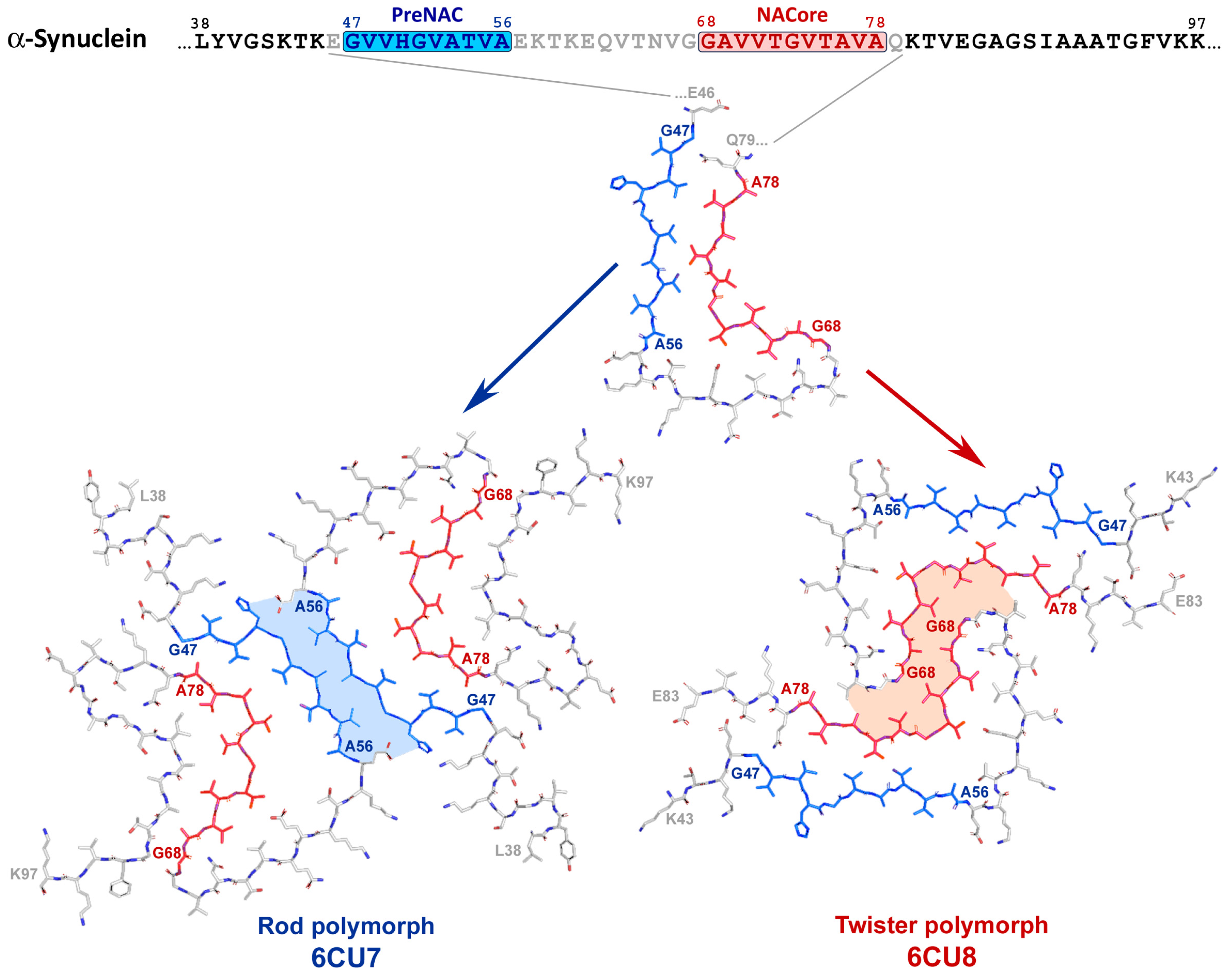
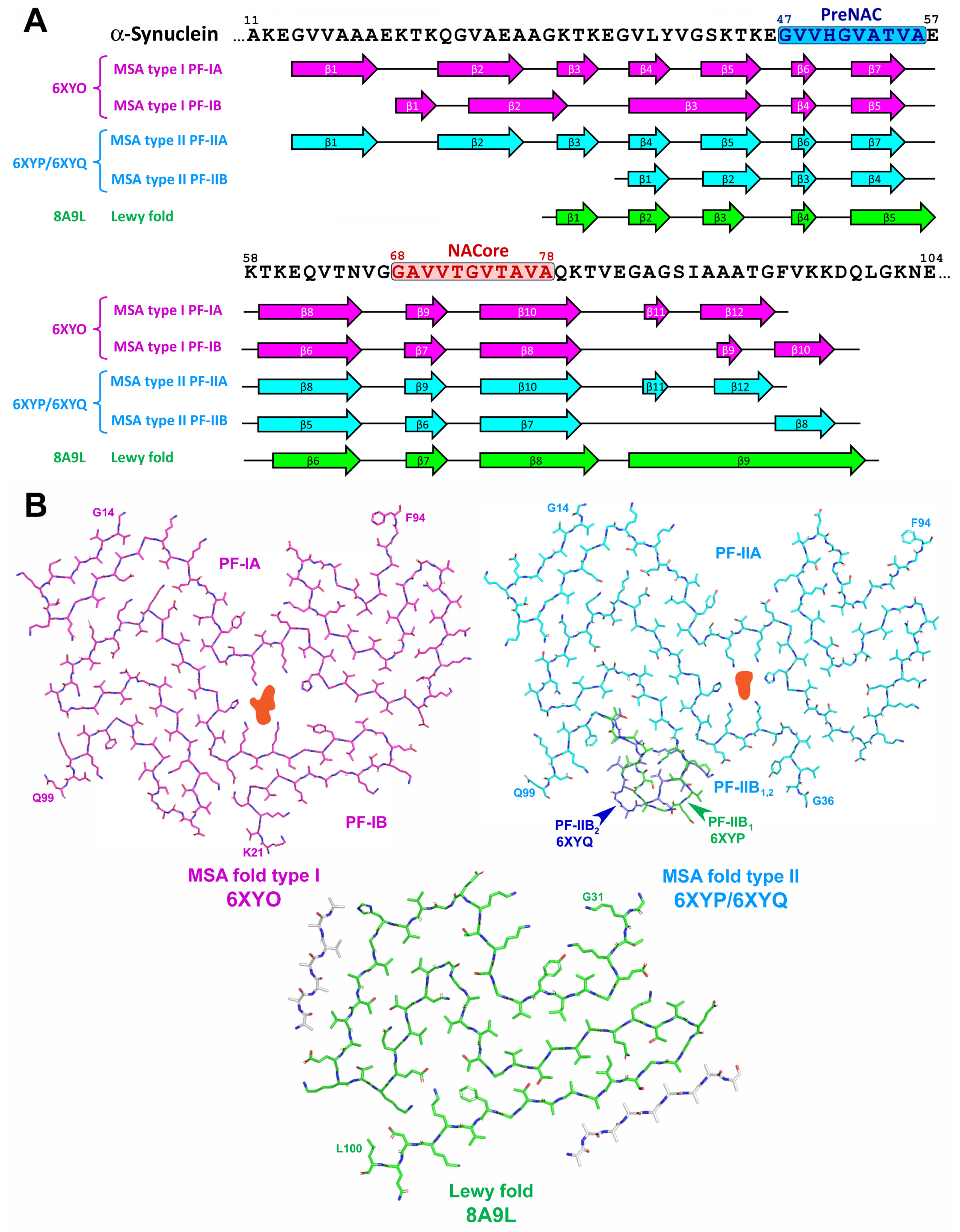
2.3. α-Synuclein-Related Pathologies
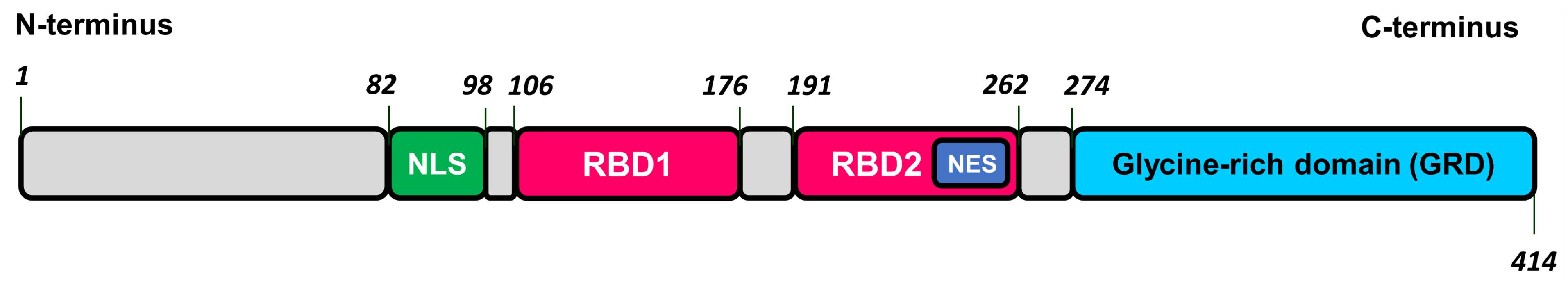
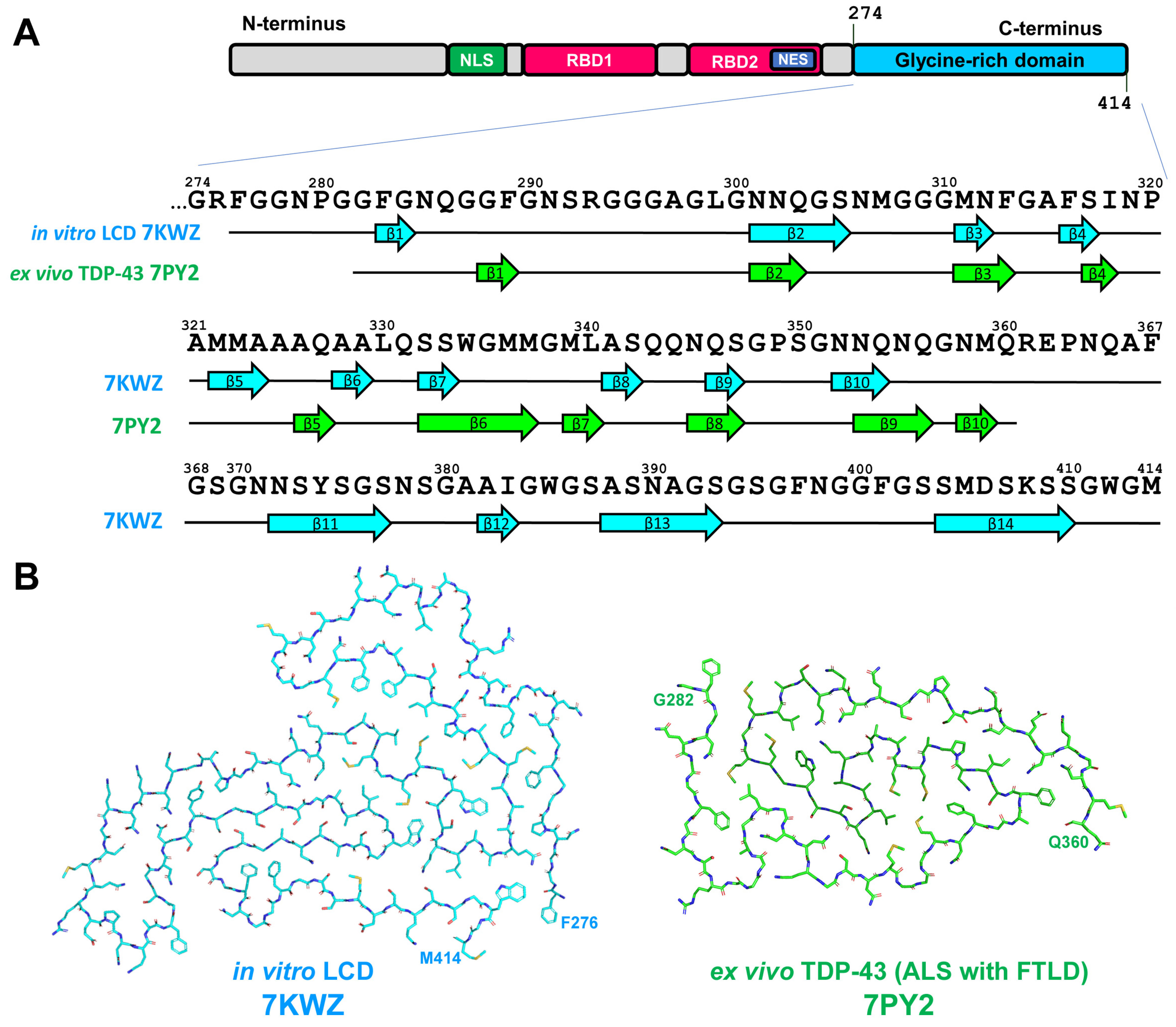
2.4. TDP-43-Related Pathologies
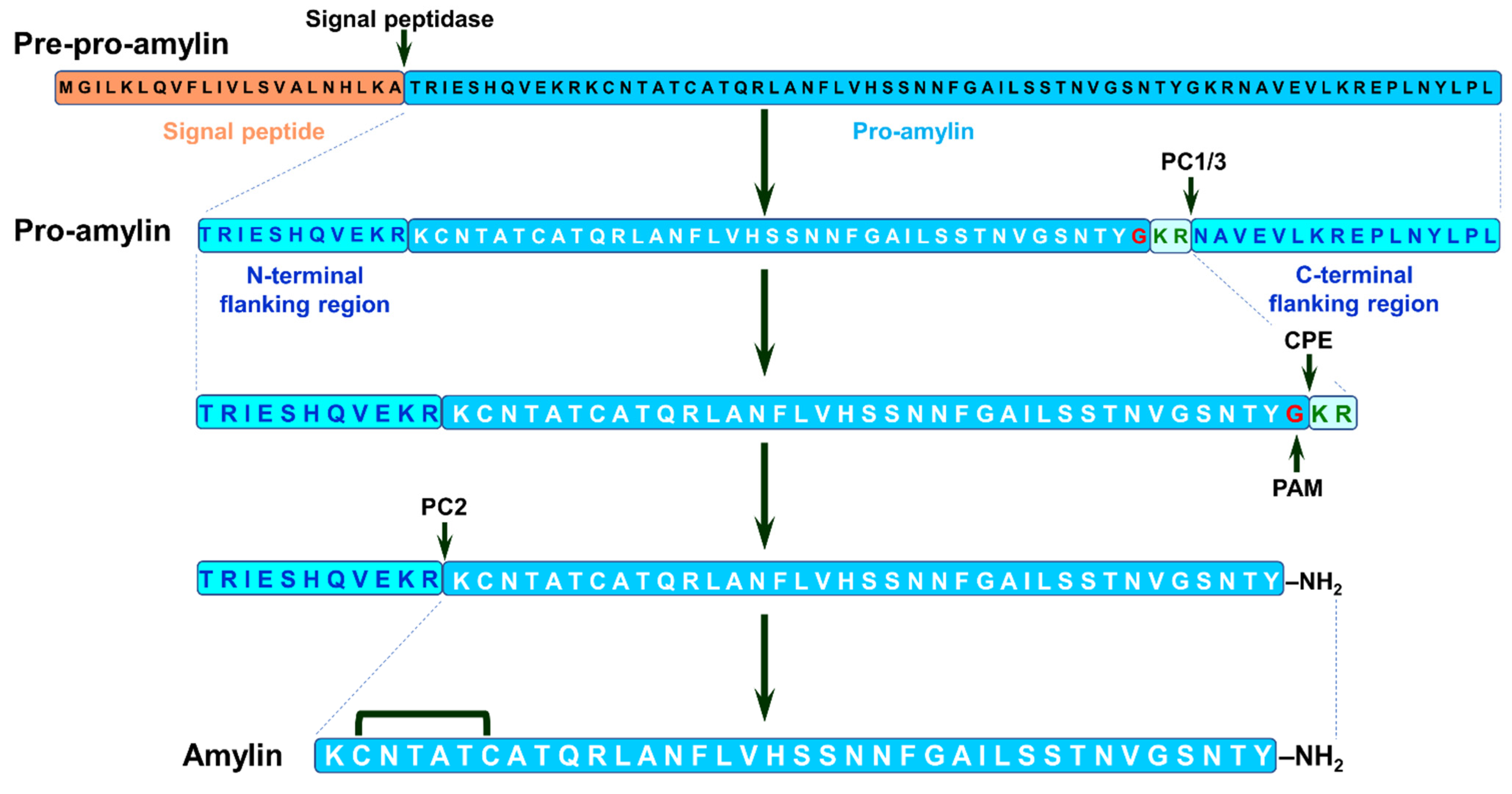
2.5. Amylin
2.5.1. Amylin, T2DM and Neurodegenerative Proteinopathies
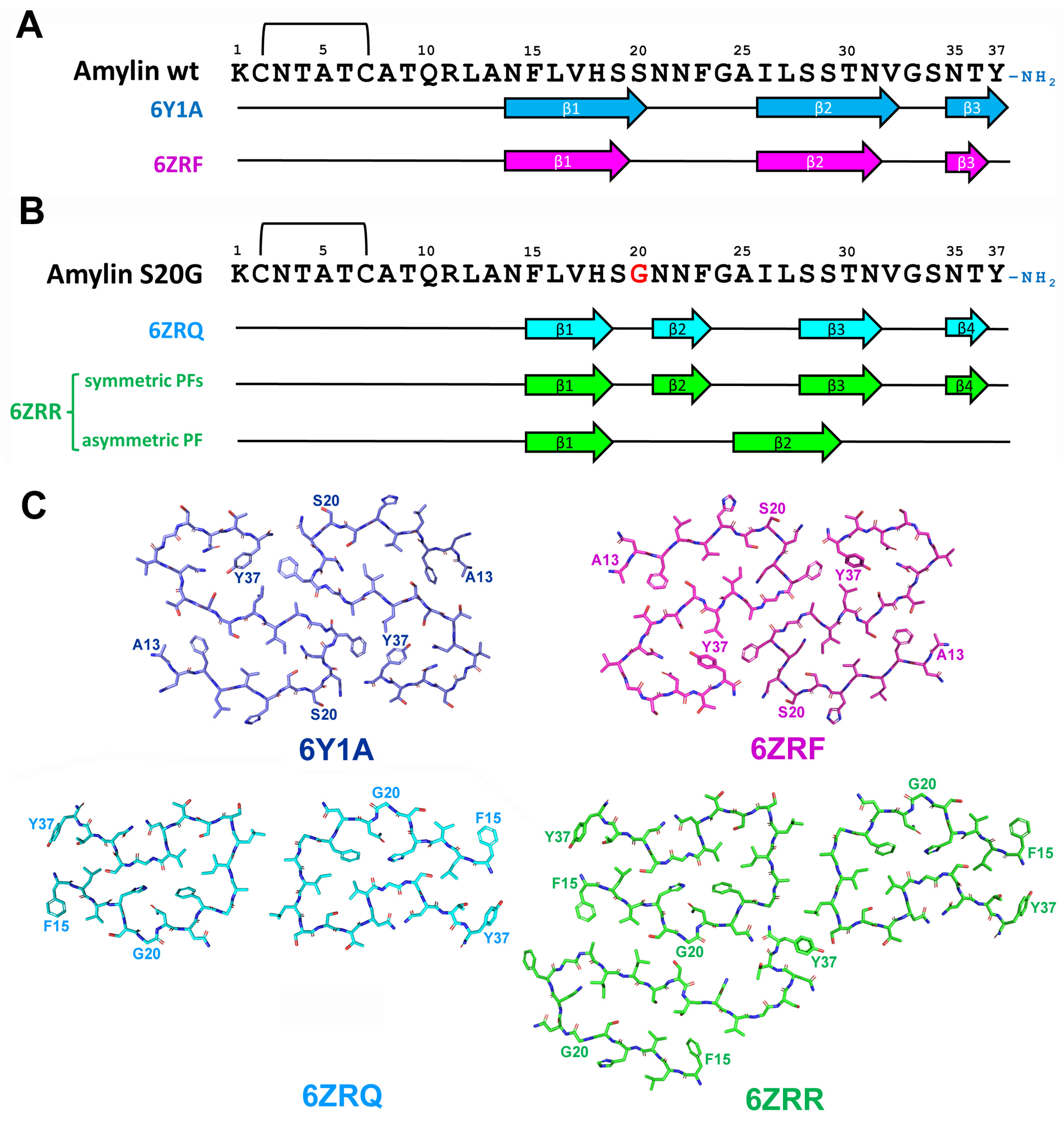
2.5.2. Structural Characterization of Amylin Amyloid Assemblies
3. Co-Pathologies: The Influence of Protein Interaction and Disease-Specific Strains of Pathological Proteins
4. Pathological Protein Interaction
4.1. Amyloid-β and Amylin Interaction
4.2. Amyloid-β and Tau Interaction
4.3. Amyloid-β and TDP-43 Interaction
4.4. Amyloid-β and α-Syn Interaction
4.5. Tau and α-Syn Interaction
4.6. Tau and TDP-43 Interaction
4.7. Tau and Amylin Interaction
4.8. α-Syn and TDP-43 Interaction
4.9. α-Syn and Amylin Interaction
4.10. TDP-43 and Amylin Interaction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| a.a. | Amino acid |
| Aβ | Amyloid-β peptide |
| AD | Alzheimer’s Disease |
| AGD | Argyrophilic grain disease |
| ALS | Amyotrophic lateral sclerosis |
| APP | Amyloid precursor protein |
| BBB | Blood–brain barrier |
| CAA | Cerebral amyloid angiopathy |
| CBD | Corticobasal degeneration |
| CPE | Carboxypeptidase E |
| CNS | Central nervous system |
| Cryo-EM | Cryo-electron microscopy |
| CSF | Cerebrospinal fluid |
| CTR | Calcitonin receptor |
| DLB | Dementia with Lewi bodies |
| EPR | Electron paramagnetic resonance spectroscopy |
| FRET | Förster resonance energy transfer |
| FTLD | Frontotemporal lobar degeneration |
| FTLD-TDP | TDP-43 positive frontotemporal lobar degeneration |
| GCI | Glial cytoplasmic inclusions |
| GRD | Glycine-rich domain |
| GSK | Glycogen synthase kinase |
| HEK293 | Human embryonic kidney 293 cells |
| IAPP | Islet amyloid polypeptide |
| IDE | Insulin-degrading enzyme |
| LB | Lewy bodies |
| LBDs | Lewy body diseases |
| LCOs | Luminescent conjugated oligothiophenes |
| LN | Lewy neurites |
| MSA | Multiply system atrophy |
| NAC | Non-amyloid component |
| NES | Nuclear export signal |
| NFTs | Neurofibrillary tangles |
| NLS | Nuclear localization signal |
| NMR | Nuclear Magnetic Resonance spectroscopy |
| NTs | Neuropil threads |
| PAGE | Polyacrylamide gel electrophoresis |
| PART | Primary age-related tauopathy |
| PAM | Peptidylglycine alpha-amidating monooxygenase |
| PD | Parkinson’s disease |
| PDD | Parkinson’s disease dementia |
| PiD | Pick’s Disease |
| PMCA | Protein Misfolding Cyclic Amplification technique |
| PK | Proteinase K |
| PrP | Prion protein |
| PSP | Progressive supranuclear palsy |
| PTMs | Post-translational modifications |
| RAMP3 | Receptor activity modifying protein 3 |
| RBD | RNA-binding domain |
| RNP | Ribonuclear protein |
| α-Syn | α-Synuclein |
| SDS | Sodium dodecyl sulphate |
| SUMO | Small Ubiquitin-like Modifier |
| T1DM | Type I diabetes mellitus |
| T2DM | Type II diabetes mellitus |
| TDP-43 | Transactive response DNA-binding protein of 43 kDa |
| TMEM106B | Transmembrane protein 106B |
References
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Toledo, J.B.; Appleby, D.H.; Xie, S.X.; Wang, L.S.; Baek, Y.; Wolk, D.A.; Lee, E.B.; Miller, B.L.; Lee, V.M.; et al. Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J. Comp. Neurol. 2013, 521, 4339–4355. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [CrossRef]
- Dickson, D.W. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 1997, 56, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; von Arnim, C.A.; Griffin, W.S.; Mrak, R.E.; Walker, L.; Attems, J.; Arzberger, T. Frontotemporal lobar degeneration FTLD-tau: Preclinical lesions, vascular, and Alzheimer-related co-pathologies. J. Neural. Transm. 2015, 122, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [CrossRef]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef]
- Walker, L.; Stefanis, L.; Attems, J. Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies-current issues and future directions. J. Neurochem. 2019, 150, 467–474. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef]
- Lee, E.B.; Porta, S.; Michael Baer, G.; Xu, Y.; Suh, E.; Kwong, L.K.; Elman, L.; Grossman, M.; Lee, V.M.; Irwin, D.J.; et al. Expansion of the classification of FTLD-TDP: Distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017, 134, 65–78. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet. J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Marin, B. ALS and frontotemporal dementia belong to a common disease spectrum. Rev. Neurol. 2017, 173, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Cao, P.; Marek, P.; Noor, H.; Patsalo, V.; Tu, L.H.; Wang, H.; Abedini, A.; Raleigh, D.P. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013, 587, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Neurodegenerative disorders associated with diabetes mellitus. J. Mol. Med. 2004, 82, 510–529. [Google Scholar] [CrossRef] [PubMed]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef]
- Kopf, D.; Frölich, L. Risk of incident Alzheimer’s disease in diabetic patients: A systematic review of prospective trials. J. Alzheimer’s Dis. 2009, 16, 677–685. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef]
- Duquette, A.; Pernègre, C.; Veilleux Carpentier, A.; Leclerc, N. Similarities and differences in the pattern of tau hyperphosphorylation in physiological and pathological conditions: Impacts on the elaboration of therapies to prevent tau pathology. Front. Neurol. 2021, 11, 607680. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 2020, 183, 1699–1713.e13. [Google Scholar] [CrossRef] [PubMed]
- Uchihara, T. Silver diagnosis in neuropathology: Principles, practice and revised interpretation. Acta Neuropathol. 2007, 113, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Tolnay, M.; Langui, D.; Goedert, M.; Spillantini, M.G. Pick’s disease: Hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol 1996, 92, 588–596. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Kulichikhin, K.Y.; Fedotov, S.A.; Rubel, M.S.; Zalutskaya, N.M.; Zobnina, A.E.; Malikova, O.A.; Neznanov, N.G.; Chernoff, Y.O.; Rubel, A.A. Development of molecular tools for diagnosis of Alzheimer’s disease that are based on detection of amyloidogenic proteins. Prion 2021, 15, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Ajit, D.; Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Chen, Y.; Ajit, A.; King, D.P.; Cohen, T.J. A unique tau conformation generated by an acetylation-mimic substitution modulates P301S-dependent tau pathology and hyperphosphorylation. J. Biol. Chem 2019, 294, 16698–16711. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 2015, 129, 221–237. [Google Scholar] [CrossRef]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef]
- Guo, J.L.; Narasimhan, S.; Changolkar, L.; He, Z.; Stieber, A.; Zhang, B.; Gathagan, R.J.; Iba, M.; McBride, J.D.; Trojanowski, J.Q.; et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016, 213, 2635–2654. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef]
- Gibbons, G.S.; Banks, R.A.; Kim, B.; Changolkar, L.; Riddle, D.M.; Leight, S.N.; Irwin, D.J.; Trojanowski, J.Q.; Lee, V.M.Y. Detection of Alzheimer disease (AD)-specific tau pathology in AD and nonAD tauopathies by immunohistochemistry with novel conformation-selective tau antibodies. J. Neuropathol. Exp. Neurol. 2018, 77, 216–228. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease-one peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Zhou, B.; Lu, J.G.; Siddu, A.; Wernig, M.; Südhof, T.C. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabn9380. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- ALZFORUM. Available online: https://www.alzforum.org/mutations/app (accessed on 6 August 2023).
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef]
- Heilbronner, G.; Eisele, Y.S.; Langer, F.; Kaeser, S.A.; Novotny, R.; Nagarathinam, A.; Aslund, A.; Hammarström, P.; Nilsson, K.P.; Jucker, M. Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013, 14, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Mahler, J.; Beschorner, N.; Kaeser, S.A.; Häsler, L.M.; Baumann, F.; Nyström, S.; Portelius, E.; Blennow, K.; Lashley, T.; et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 13018–13023. [Google Scholar] [CrossRef]
- Zhang, R.; Hu, X.; Khant, H.; Ludtke, S.J.; Chiu, W.; Schmid, M.F.; Frieden, C.; Lee, J.M. Interprotofilament interactions between Alzheimer’s Abeta1-42 peptides in amyloid fibrils revealed by cryoEM. Proc. Natl. Acad. Sci. USA 2009, 106, 4653–4658. [Google Scholar] [CrossRef]
- Schmidt, M.; Rohou, A.; Lasker, K.; Yadav, J.K.; Schiene-Fischer, C.; Fändrich, M.; Grigorieff, N. Peptide dimer structure in an Aβ(1-42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. USA 2015, 112, 11858–11863. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neurozn 2006, 52, 33–38. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef]
- Ni, X.; McGlinchey, R.P.; Jiang, J.; Lee, J.C. Structural Insights into α-Synuclein Fibril Polymorphism: Effects of Parkinson’s Disease-Related C-Terminal Truncations. J. Mol. Biol. 2019, 431, 3913–3919. [Google Scholar] [CrossRef]
- Miake, H.; Mizusawa, H.; Iwatsubo, T.; Hasegawa, M. Biochemical characterization of the core structure of alpha-synuclein filaments. J. Biol. Chem. 2002, 277, 19213–19219. [Google Scholar] [CrossRef]
- Vilar, M.; Chou, H.T.; Lührs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Papp, M.I.; Kahn, J.E.; Lantos, P.L. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J. Neurol. Sci. 1989, 94, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Campbell, B.C.; McLean, C.A.; Culvenor, J.G.; Gai, W.P.; Blumbergs, P.C.; Jäkälä, P.; Beyreuther, K.; Masters, C.L.; Li, Q.X. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 2001, 76, 87–96. [Google Scholar] [CrossRef]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Boyer, D.R.; Li, B.; Sun, C.; Fan, W.; Sawaya, M.R.; Jiang, L.; Eisenberg, D.S. Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 2019, 26, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, Y.; Schweighauser, M.; Zhang, X.; Kotecha, A.; Murzin, A.G.; Garringer, H.J.; Cullinane, P.W.; Saito, Y.; Foroud, T.; et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 2022, 610, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem 2009, 284, 20329–20339, Erratum in J. Biol. Chem. 2009, 284, 25459. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef]
- Nana, A.L.; Sidhu, M.; Gaus, S.E.; Hwang, J.L.; Li, L.; Park, Y.; Kim, E.J.; Pasquini, L.; Allen, I.E.; Rankin, K.P.; et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019, 137, 27–46. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Li, Q.; Babinchak, W.M.; Surewicz, W.K. Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nat. Commun. 2021, 12, 1620. [Google Scholar] [CrossRef]
- Arseni, D.; Hasegawa, M.; Murzin, A.G.; Kametani, F.; Arai, M.; Yoshida, M.; Ryskeldi-Falcon, B. Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 2022, 601, 139–143. [Google Scholar] [CrossRef]
- Tan, R.H.; Shepherd, C.E.; Kril, J.J.; McCann, H.; McGeachie, A.; McGinley, C.; Affleck, A.; Halliday, G.M. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol. Commun. 2013, 1, 33. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020, 139, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Del Tredici, K.; Irwin, D.J.; Grossman, M.; Robinson, J.L.; Toledo, J.B.; Fang, L.; Van Deerlin, V.M.; Ludolph, A.C.; Lee, V.M.; et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014, 127, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.X.; Cao, Q.; Sawaya, M.R.; Abskharon, R.; Ge, P.; DeTure, M.; Dickson, D.W.; Fu, J.Y.; Ogorzalek Loo, R.R.; Loo, J.A.; et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 2022, 605, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Vass, R.; Ashbridge, E.; Geser, F.; Hu, W.T.; Grossman, M.; Clay-Falcone, D.; Elman, L.; McCluskey, L.; Lee, V.M.; Van Deerlin, V.M.; et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, J.; Van Broeckhoven, C. TMEM106B a novel risk factor for frontotemporal lobar degeneration. J. Mol. Neurosci. 2011, 45, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R. Amyloid Atlas 2023. Available online: https://people.mbi.ucla.edu/sawaya/amyloidatlas/ (accessed on 6 August 2023).
- Neumann, M.; Frick, P.; Paron, F.; Kosten, J.; Buratti, E.; Mackenzie, I.R. Antibody against TDP-43 phosphorylated at serine 375 suggests conformational differences of TDP-43 aggregates among FTLD-TDP subtypes. Acta Neuropathol 2020, 140, 645–658, Erratum in Acta Neuropathol. 2021, 141, 137. [Google Scholar] [CrossRef]
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J., Jr.; Porte, D., Jr. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 1990, 39, 634–638. [Google Scholar] [CrossRef]
- Lutz, T.A. The role of amylin in the control of energy homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1475–R1484. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef]
- Denroche, H.C.; Verchere, C.B. IAPP and type 1 diabetes: Implications for immunity, metabolism and islet transplants. J. Mol. Endocrinol. 2018, 60, R57–R75. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J.; Willis, A.C.; Clark, A.; Turner, R.C.; Sim, R.B.; Reid, K.B. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. USA 1987, 84, 8628–8632. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Meyer, U. Amylin at the interface between metabolic and neurodegenerative disorders. Front. Neurosci. 2015, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.; Beare, R.; Phan, T.G.; Bruce, D.G.; Callisaya, M.L.; Srikanth, V. Alzheimer’s Disease Neuroimaging Initiative (ADNI) Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 2015, 85, 1123–1130. [Google Scholar] [CrossRef]
- Cheong, J.L.Y.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association between type 2 diabetes mellitus and Parkinson’s disease. J. Parkinsons. Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Pivovarova, O.; Höhn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Zhu, H. Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 186. [Google Scholar] [CrossRef]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.T.; Woods, C.; Zhen, J.; Antonio, T.; Carr, K.D.; Reith, M.E. Effects of diet and insulin on dopamine transporter activity and expression in rat caudate-putamen, nucleus accumbens, and midbrain. J. Neurochem. 2017, 140, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Stouffer, M.A.; Woods, C.A.; Patel, J.C.; Lee, C.R.; Witkovsky, P.; Bao, L.; Machold, R.P.; Jones, K.T.; de Vaca, S.C.; Reith, M.E.; et al. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun. 2015, 6, 8543. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Alpha-synuclein contributes to GSK-3beta-catalyzed tau phosphorylation in Parkinson’s disease models. FASEB J. 2009, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Reynolds, C.H.; Latimer, D.; Davis, D.R.; Anderton, B.H.; Gallo, J.-M.; Hanger, D.; Mulot, S.; Marquardt, B.; Stabel, S.; et al. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994, 4, 1077–1086. [Google Scholar] [CrossRef]
- Fortin, J.S.; Benoit-Biancamano, M.O. Wildlife sequences of islet amyloid polypeptide (IAPP) identify critical species variants for fibrillization. Amyloid 2015, 22, 194–202. [Google Scholar] [CrossRef]
- Nishi, M.; Chan, S.J.; Nagamatsu, S.; Bell, G.I.; Steiner, D.F. Conservation of the sequence of islet amyloid polypeptide in five mammals is consistent with its putative role as an islet hormone. Proc. Natl. Acad. Sci. USA 1989, 86, 5738–5742. [Google Scholar] [CrossRef]
- Westermark, P.; Engström, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef]
- Sakagashira, S.; Sanke, T.; Hanabusa, T.; Shimomura, H.; Ohagi, S.; Kumagaye, K.Y.; Nakajima, K.; Nanjo, K. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996, 45, 1279–1281. [Google Scholar] [CrossRef]
- Sakagashira, S.; Hiddinga, H.J.; Tateishi, K.; Sanke, T.; Hanabusa, T.; Nanjo, K.; Eberhardt, N.L. S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am. J. Pathol. 2000, 157, 2101–2109. [Google Scholar] [CrossRef]
- Ma, Z.; Westermark, G.T.; Sakagashira, S.; Sanke, T.; Gustavsson, A.; Sakamoto, H.; Engström, U.; Nanjo, K.; Westermark, P. Enhanced in vitro production of amyloid-like fibrils from mutant (S20G) islet amyloid polypeptide. Amyloid 2001, 8, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.S.; Benoit-Biancamano, M.O. Characterization of a pancreatic islet cell tumor in a polar bear (Ursus maritimus). Zoo Biol. 2014, 33, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Abedini, A.; Ridgway, Z.; Zhang, X.; Kleinberg, J.; Schmidt, A.M.; Raleigh, D.P. Evolutionary adaptation and amyloid formation: Does the reduced amyloidogenicity and cytotoxicity of ursine amylin contribute to the metabolic adaption of bears and polar bears? Isr. J. Chem. 2017, 57, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.S.; Cooper, G.J.; Goldie, K.N.; Müller, S.A.; Saafi, E.L.; Gruijters, W.T.; Misur, M.P.; Engel, A.; Aebi, U.; Kistler, J. Polymorphic fibrillar assembly of human amylin. J. Struct. Biol. 1997, 119, 17–27. [Google Scholar] [CrossRef]
- Goldsbury, C.; Goldie, K.; Pellaud, J.; Seelig, J.; Frey, P.; Müller, S.A.; Kistler, J.; Cooper, G.J.; Aebi, U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000, 130, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, R.; Iadanza, M.G.; Xu, Y.; Heath, G.R.; Foster, R.; Radford, S.E.; Ranson, N.A. Fibril structures of diabetes-related amylin variants reveal a basis for surface-templated assembly. Nat. Struct. Mol. Biol. 2020, 27, 1048–1056. [Google Scholar] [CrossRef]
- Röder, C.; Kupreichyk, T.; Gremer, L.; Schäfer, L.U.; Pothula, K.R.; Ravelli, R.B.G.; Willbold, D.; Hoyer, W.; Schröder, G.F. Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat. Struct. Mol. Biol. 2020, 27, 660–667. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 2020, 27, 653–659. [Google Scholar] [CrossRef]
- Bedrood, S.; Li, Y.; Isas, J.M.; Hegde, B.G.; Baxa, U.; Haworth, I.S.; Langen, R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012, 287, 5235–5241. [Google Scholar] [CrossRef]
- Luca, S.; Yau, W.M.; Leapman, R.; Tycko, R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry 2007, 46, 13505–13522. [Google Scholar] [CrossRef]
- Weirich, F.; Gremer, L.; Mirecka, E.A.; Schiefer, S.; Hoyer, W.; Heise, H. Structural characterization of fibrils from recombinant human islet amyloid polypeptide by solid-state NMR: The central FGAILS segment is part of the β-sheet core. PLoS ONE 2016, 11, e0161243. [Google Scholar] [CrossRef] [PubMed]
- Alexandrescu, A.T. Amide proton solvent protection in amylin fibrils probed by quenched hydrogen exchange NMR. PLoS ONE 2013, 8, e56467. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.; Sievers, S.A.; Sawaya, M.R.; Cascio, D.; Popov, D.; Riekel, C.; Eisenberg, D. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin). Protein Sci. 2008, 17, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Abskharon, R.; Saelices, L.; Nguyen, B.A.; Lu, J.; Murray, K.A.; Kandeel, F.; Eisenberg, D.S. Cryo-EM structures of hIAPP fibrils seeded by patient-extracted fibrils reveal new polymorphs and conserved fibril cores. Nat. Struct. Mol. Biol. 2021, 28, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Fîlfan, M.; Sandu, R.E.; Zăvăleanu, A.D.; GreşiŢă, A.; Glăvan, D.G.; Olaru, D.G.; Popa-Wagner, A. Autophagy in aging and disease. Rom. J. Morphol. Embryol. 2017, 58, 27–31. [Google Scholar]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7299. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards, G., III; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer disease Amyloid-β peptide and other amyloid proteins: A further aspect of the amyloid cascade hypothesis. J. Biol. Chem 2016, 291, 16485–16493. [Google Scholar] [CrossRef]
- Biessels, G.J.; Kappelle, L.J.; Utrecht Diabetic Encephalopathy Study Group. Increased risk of Alzheimer’s disease in Type II diabetes: Insulin resistance of the brain or insulin-induced amyloid pathology? Biochem. Soc. Trans. 2005, 33, 1041–1044. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.M.; Velkova, A.; Tatarek-Nossol, M.; Andreetto, E.; Kapurniotu, A. IAPP mimic blocks Abeta cytotoxic self-assembly: Cross-suppression of amyloid toxicity of Abeta and IAPP suggests a molecular link between Alzheimer’s disease and type II diabetes. Angew. Chem. Int. Ed. Engl. 2007, 46, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Andreetto, E.; Yan, L.M.; Tatarek-Nossol, M.; Velkova, A.; Frank, R.; Kapurniotu, A. Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem. Int. Ed. Engl. 2010, 49, 3081–3085. [Google Scholar] [CrossRef]
- Krotee, P.; Griner, S.L.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Shi, D.; Philipp, S.; Murray, K.; Saelices, L.; Lee, J.; et al. Common fibrillar spines of amyloid-β and human islet amyloid polypeptide revealed by microelectron diffraction and structure-based inhibitors. J. Biol. Chem. 2018, 293, 2888–2902. [Google Scholar] [CrossRef]
- Kachkin, D.V.; Lashkul, V.V.; Gorsheneva, N.A.; Fedotov, S.A.; Rubel, M.S.; Chernoff, Y.O.; Rubel, A.A. The Aβ42 peptide and IAPP physically interact in a yeast-based assay. Int. J. Mol. Sci. 2023, 24, 14122. [Google Scholar] [CrossRef]
- Wang, Y.; Westermark, G.T. The amyloid forming peptides islet amyloid polypeptide and amyloid β interact at the molecular level. Int. J. Mol. Sci. 2021, 22, 11153. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank (RCSB PDB). Available online: https://www.rcsb.org/ (accessed on 6 August 2023).
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef]
- Sepulcre, J.; Schultz, A.P.; Sabuncu, M.; Gomez-Isla, T.; Chhatwal, J.; Becker, A.; Sperling, R.; Johnson, K.A. In vivo tau, amyloid, and gray matter profiles in the aging brain. J. Neurosci. 2016, 36, 7364–7374. [Google Scholar] [CrossRef]
- Hutton, M.; Pérez-Tur, J.; Hardy, J. Genetics of Alzheimer’s disease. Essays Biochem. 1998, 33, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci 2010, 30, 11938–11950, Erratum in J. Neurosci. 2012, 32, 6052. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.P.; Arai, T.; Miklossy, J.; McGeer, P.L. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef]
- Katsumata, Y.; Fardo, D.W.; Kukull, W.A.; Nelson, P.T. Dichotomous scoring of TDP-43 proteinopathy from specific brain regions in 27 academic research centers: Associations with Alzheimer’s disease and cerebrovascular disease pathologies. Acta Neuropathol. Commun. 2018, 6, 142. [Google Scholar] [CrossRef]
- Herman, A.M.; Khandelwal, P.J.; Stanczyk, B.B.; Rebeck, G.W.; Moussa, C.E. β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011, 1386, 191–199. [Google Scholar] [CrossRef]
- Davis, S.A.; Gan, K.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 expression influences amyloidβ plaque deposition and tau aggregation. Neurobiol. Dis. 2017, 103, 154–162. [Google Scholar] [CrossRef]
- LaClair, K.D.; Donde, A.; Ling, J.P.; Jeong, Y.H.; Chhabra, R.; Martin, L.J.; Wong, P.C. Depletion of TDP-43 decreases fibril and plaque β-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta Neuropathol. 2016, 132, 859–873. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.; et al. TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef]
- Shih, Y.H.; Tu, L.H.; Chang, T.Y.; Ganesan, K.; Chang, W.W.; Chang, P.S.; Fang, Y.S.; Lin, Y.T.; Jin, L.W.; Chen, Y.R. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 5950. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Krishnamurthy, S.; Paulucci-Holthauzen, A.A.; Sengupta, U.; Lasagna-Reeves, C.A.; Ahmad, Y.; Jackson, G.R.; Kayed, R. Amyloid-β oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol. Dis. 2014, 71, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Dickson, D.W.; Murray, M.E.; Whitwell, J.L.; Knopman, D.S.; Boeve, B.F.; Jack, C.R., Jr.; Parisi, J.E.; Petersen, R.C.; Josephs, K.A. TDP-43 in Alzheimer’s disease is not associated with clinical FTLD or Parkinsonism. J. Neurol. 2014, 261, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Petrucelli, L.; Liesinger, A.M.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016, 131, 571–585. [Google Scholar] [CrossRef]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef]
- Mandal, P.K.; Pettegrew, J.W.; Masliah, E.; Hamilto, R.L.; Mandal, R. Interaction between Abeta peptide and alpha synuclein: Molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem. Res 2006, 31, 1153–1162, Erratum in Neurochem. Res. 2007, 32 2002. [Google Scholar] [CrossRef]
- Atsmon-Raz, Y.; Miller, Y. Non-Amyloid-β Component of human α-synuclein oligomers induces formation of new Aβ oligomers: Insight into the mechanisms that link Parkinson’s and Alzheimer’s diseases. ACS Chem. Neurosci. 2016, 7, 46–55. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. α-Synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef]
- Arima, K.; Mizutani, T.; Alim, M.A.; Tonozuka-Uehara, H.; Izumiyama, Y.; Hirai, S.; Uéda, K. NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: Double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol. 2000, 100, 115–121. [Google Scholar] [CrossRef]
- Duda, J.E.; Giasson, B.I.; Mabon, M.E.; Miller, D.C.; Golbe, L.I.; Lee, V.M.; Trojanowski, J.Q. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 2002, 104, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimón, J.; Lleó, A. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef] [PubMed]
- Wade-Martins, R. Genetics: The MAPT locus-a genetic paradigm in disease susceptibility. Nat. Rev. Neurol. 2012, 8, 477–478. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Gelpi, E.; Martí, M.J.; Charif, S.; Dols-Icardo, O.; Blesa, R.; Clarimón, J.; Lleó, A. MAPT H1 haplotype is associated with enhanced α-synuclein deposition in dementia with Lewy bodies. Neurobiol. Aging 2013, 34, 936–942. [Google Scholar] [CrossRef]
- Labbé, C.; Ogaki, K.; Lorenzo-Betancor, O.; Soto-Ortolaza, A.I.; Walton, R.L.; Rayaprolu, S.; Fujioka, S.; Murray, M.E.; Heckman, M.G.; Puschmann, A.; et al. Role for the microtubule-associated protein tau variant p.A152T in risk of α-synucleinopathies. Neurology 2015, 85, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Nübling, G.; Bader, B.; Levin, J.; Hildebrandt, J.; Kretzschmar, H.; Giese, A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 2012, 7, 35. [Google Scholar] [CrossRef]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef]
- Yokota, O.; Davidson, Y.; Bigio, E.H.; Ishizu, H.; Terada, S.; Arai, T.; Hasegawa, M.; Akiyama, H.; Sikkink, S.; Pickering-Brown, S.; et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol. 2010, 120, 55–66. [Google Scholar] [CrossRef]
- Koga, S.; Sanchez-Contreras, M.; Josephs, K.A.; Uitti, R.J.; Graff-Radford, N.; van Gerpen, J.A.; Cheshire, W.P.; Wszolek, Z.K.; Rademakers, R.; Dickson, D.W. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov. Disord. 2017, 32, 246–255. [Google Scholar] [CrossRef]
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.; Jope, R.S. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar] [CrossRef]
- Li, Z.G.; Zhang, W.; Sima, A.A. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007, 56, 1817–1824, Erratum in Diabetes 2007, 56, 2650. [Google Scholar] [CrossRef]
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E.; et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007, 27, 13635–13648. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; Delerue, F.; Gladbach, A.; Götz, J.; Ittner, L.M. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2009, 4, e7917. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E.L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Jiao, Z.; Sun, X.; Zhao, Y.; Ren, J.; Xu, G. Effects of streptozotocin-induced diabetes on tau phosphorylation in the rat brain. Brain Res. 2011, 1383, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Taha, C.; Klip, A. The insulin signaling pathway. J. Membr. Biol. 1999, 169, 1–12. [Google Scholar] [CrossRef]
- El Khoury, N.B.; Gratuze, M.; Papon, M.A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 2014, 8, 22. [Google Scholar] [CrossRef]
- Zhang, G.; Meng, L.; Wang, Z.; Peng, Q.; Chen, G.; Xiong, J.; Zhang, Z. Islet amyloid polypeptide cross-seeds tau and drives the neurofibrillary pathology in Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 12. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef]
- Bosco, D.; Plastino, M.; Cristiano, D.; Colica, C.; Ermio, C.; De Bartolo, M.; Mungari, P.; Fonte, G.; Consoli, D.; Consoli, A.; et al. Dementia is associated with insulin resistance in patients with Parkinson’s disease. J. Neurol. Sci. 2012, 315, 39–43. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Atsmon-Raz, Y.; Miller, Y. Molecular mechanisms of the bindings between Non-Amyloid β Component oligomers and amylin oligomers. J. Phys. Chem. B 2016, 120, 10649–10659. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Valbuena, I.; Amat-Villegas, I.; Valenti-Azcarate, R.; Carmona-Abellan, M.D.M.; Marcilla, I.; Tuñon, M.T.; Luquin, M.R. Interaction of amyloidogenic proteins in pancreatic β cells from subjects with synucleinopathies. Acta Neuropathol. 2018, 135, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Mariosa, D.; Kamel, F.; Bellocco, R.; Ye, W.; Fang, F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2015, 22, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Wang, T.; Zheng, J.; Zhou, X. Causal association of type 2 diabetes with amyotrophic lateral sclerosis: New evidence from Mendelian randomization using GWAS summary statistics. BMC Med. 2019, 17, 225. [Google Scholar] [CrossRef]
- Tsai, C.P.; Lee, J.K.; Lee, C.T. Type II diabetes mellitus and the incidence of amyotrophic lateral sclerosis. J. Neurol. 2019, 266, 2233–2243. [Google Scholar] [CrossRef]
- Kalkonde, Y.V.; Jawaid, A.; Qureshi, S.U.; Shirani, P.; Wheaton, M.; Pinto-Patarroyo, G.P.; Schulz, P.E. Medical and environmental risk factors associated with frontotemporal dementia: A case-control study in a veteran population. Alzheimer’s Dement. 2012, 8, 204–210. [Google Scholar] [CrossRef]
- Körner, S.; Kollewe, K.; Ilsemann, J.; Müller-Heine, A.; Dengler, R.; Krampfl, K.; Petri, S. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur. J. Neurol. 2013, 20, 647–654. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Pradat, P.F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.M.; et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
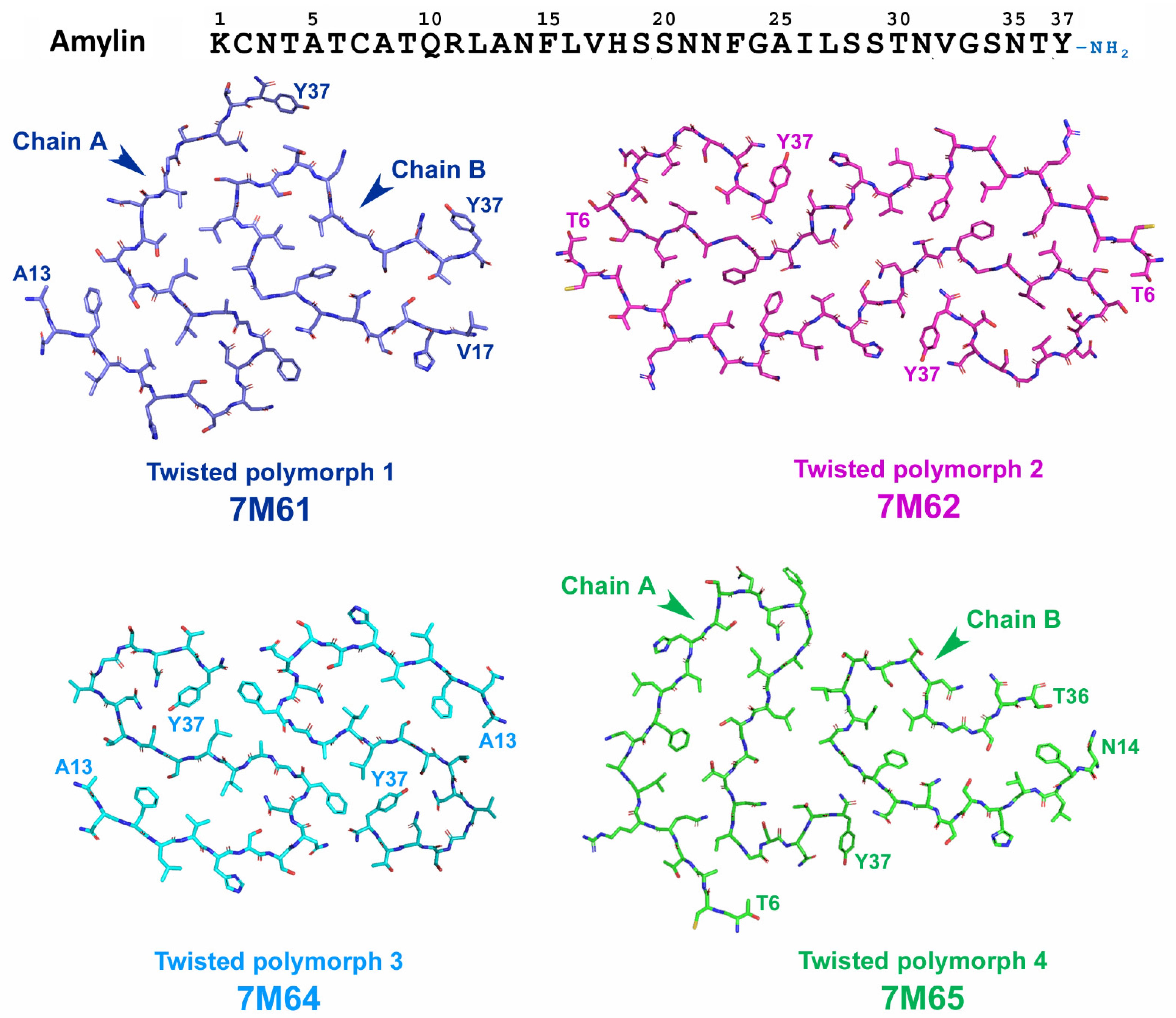{kind=link}
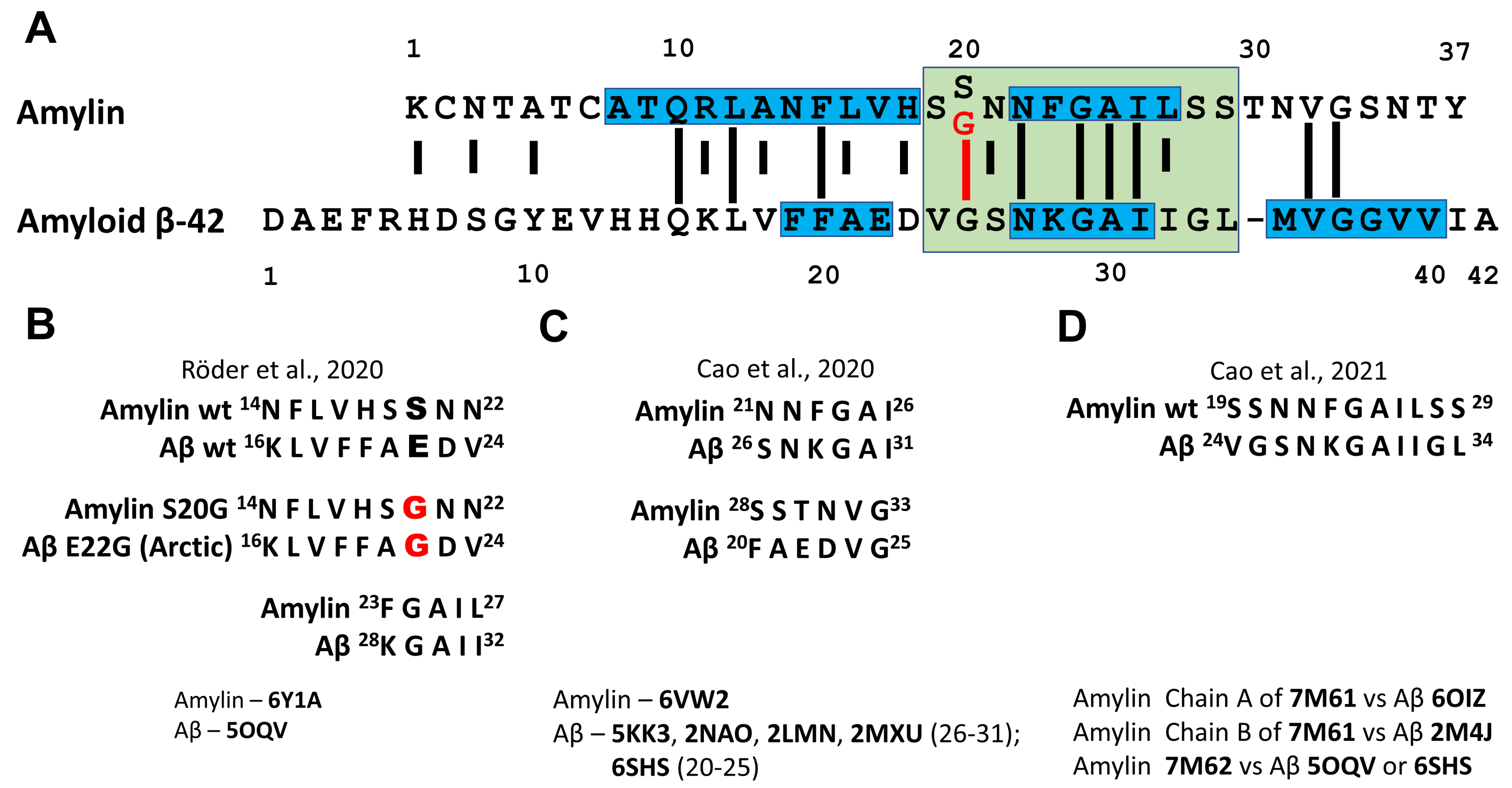{kind=link}
| Interacting Proteins | Co-Deposition | Cross-Seeding | Co-Aggregation | Interaction of Protein Monomers | ||||
|---|---|---|---|---|---|---|---|---|
| Fibrillar Seeds | Oligomeric Seeds | Mixed Fibrillar Assemblies | Chimeric Oligomer Assemblies | PTM | Templating | |||
| 1. | Aβ–amylin | + | + | + | ||||
| 2. | Aβ–tau | + | + | |||||
| 3. | Aβ–TDP-43 | + | + | |||||
| 4. | Aβ–α-Syn | + | + | + | + | + | ||
| 5. | Tau–α-Syn | + | + | + | + | + | ||
| 6. | Tau–TDP-43 | + | ||||||
| 7. | Tau–amylin | + | ||||||
| 8. | α-Syn–TPD-43 | + | ||||||
| 9. | α-Syn–amylin | + | + | + | + | + | ||
| 10. | TDP-43–amylin | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulichikhin, K.Y.; Malikova, O.A.; Zobnina, A.E.; Zalutskaya, N.M.; Rubel, A.A. Interaction of Proteins Involved in Neuronal Proteinopathies. Life 2023, 13, 1954. https://doi.org/10.3390/life13101954
Kulichikhin KY, Malikova OA, Zobnina AE, Zalutskaya NM, Rubel AA. Interaction of Proteins Involved in Neuronal Proteinopathies. Life. 2023; 13(10):1954. https://doi.org/10.3390/life13101954
Chicago/Turabian StyleKulichikhin, Konstantin Y., Oksana A. Malikova, Anastasia E. Zobnina, Natalia M. Zalutskaya, and Aleksandr A. Rubel. 2023. "Interaction of Proteins Involved in Neuronal Proteinopathies" Life 13, no. 10: 1954. https://doi.org/10.3390/life13101954
APA StyleKulichikhin, K. Y., Malikova, O. A., Zobnina, A. E., Zalutskaya, N. M., & Rubel, A. A. (2023). Interaction of Proteins Involved in Neuronal Proteinopathies. Life, 13(10), 1954. https://doi.org/10.3390/life13101954