
The Bootstrap Model of Prebiotic Networks of Proteins and Nucleic Acids


Abstract
:1. Introduction
2. The Background and the Model
2.1. The Premises and Assumptions
- A nonequilibrium driver. Because life is now, and always must have been, out of equilibrium, we are at liberty to suppose some persistent nonequilibrium drive was present. There are many potential sources. Here, we assume the availability of amino acids and nucleic acids, and that both are persistently being polymerized. At first, these would produce only short-chain random sequences of xNAs or peptides. Such plausible syntheses have previously been demonstrated [16,17,18,19];
- A propagation principle. Today, life as a whole sustains, and never dies out, due to the survival-of-the-fittest propagation principle. Moreover, it is resourceful, creative and innovative, due to its ability to search and choose by mutation and selection. Without it, there is no biology. It results because changes in biomolecules lead to changes in cell growth rates, which lead to changes in cell populations. Here, we assume a simple physical precursor dynamic. We assume peptides and xNAs are encapsulated and polymerize inside colloids or vesicles, causing such protocells to grow and to divide by known surface-to-volume effects [20,21,22,23]. We assume, as others have done, that the amino acid and nucleic acid monomers from the surroundings can pass freely into the protocells, but that the chains inside are too long to pass back out [24,25,26];
- Funneling in the molecule space. We believe life originated more as a disorder-to-order process, and less as specific sequence actions or specific binding actions or specific recognition between polymers (such as the genetic code). Rather, we believe such specificity must have emerged from the propagation mechanism (see above) acting on random molecules.
2.2. The Growth and Split Mechanism
2.3. Intermolecular Interactions Drive Network Formation
Protein Copiers as Peptides
- Contemporary organisms are known to have some nonribosomal peptide syntheses which are facilitated by other protein structures (i.e., nonribosomal peptide synthetase) [40]. As far as we know, there are no known equivalents for xNAs being duplicated by solely other xNAs;
- At the current level of coarseness of the present modeling, we simply approximate ribosomes as being catalytic elongators. Therefore, the network structure would not differ much from the observed results.
2.4. Computing the Growth Dynamics
2.5. Mutations Drive the Network to Discover New Functional Relations, Affecting the Protocell’s Growth Rate
2.6. Polymer Aggregation Decrease Proto-Cellular Growth Rate
2.7. Mutations Can Be Advantageous or Noise
2.8. Mutations of the Individual Cells Propagate through the Population
2.9. Computer Simulations of the Model
3. Results and Discussion
3.1. When a Network Discovers Complete Copier Subgraphs, Its Protocell Grows Faster
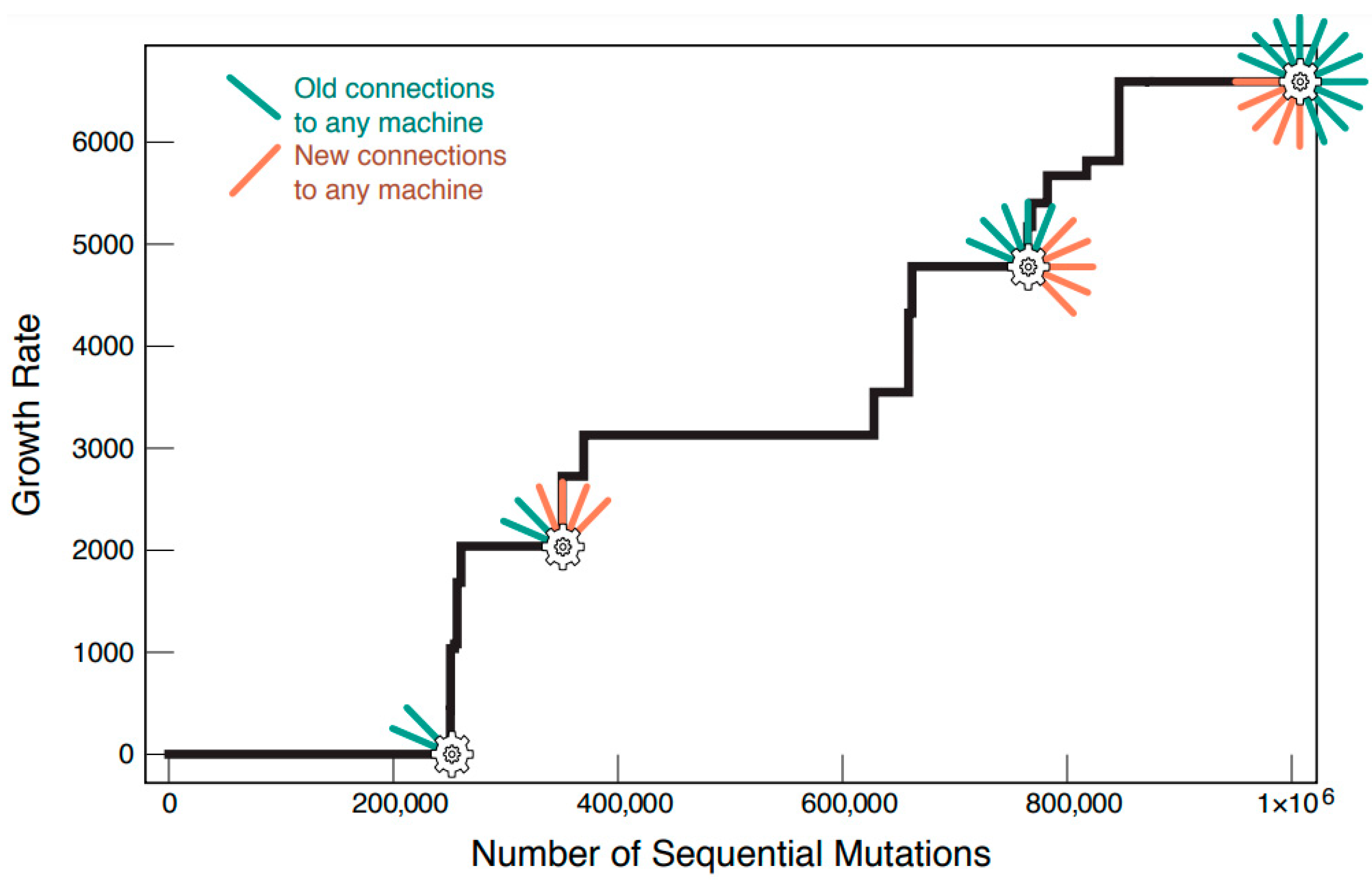
3.2. Networks Evolve to Become Bigger and More Complex
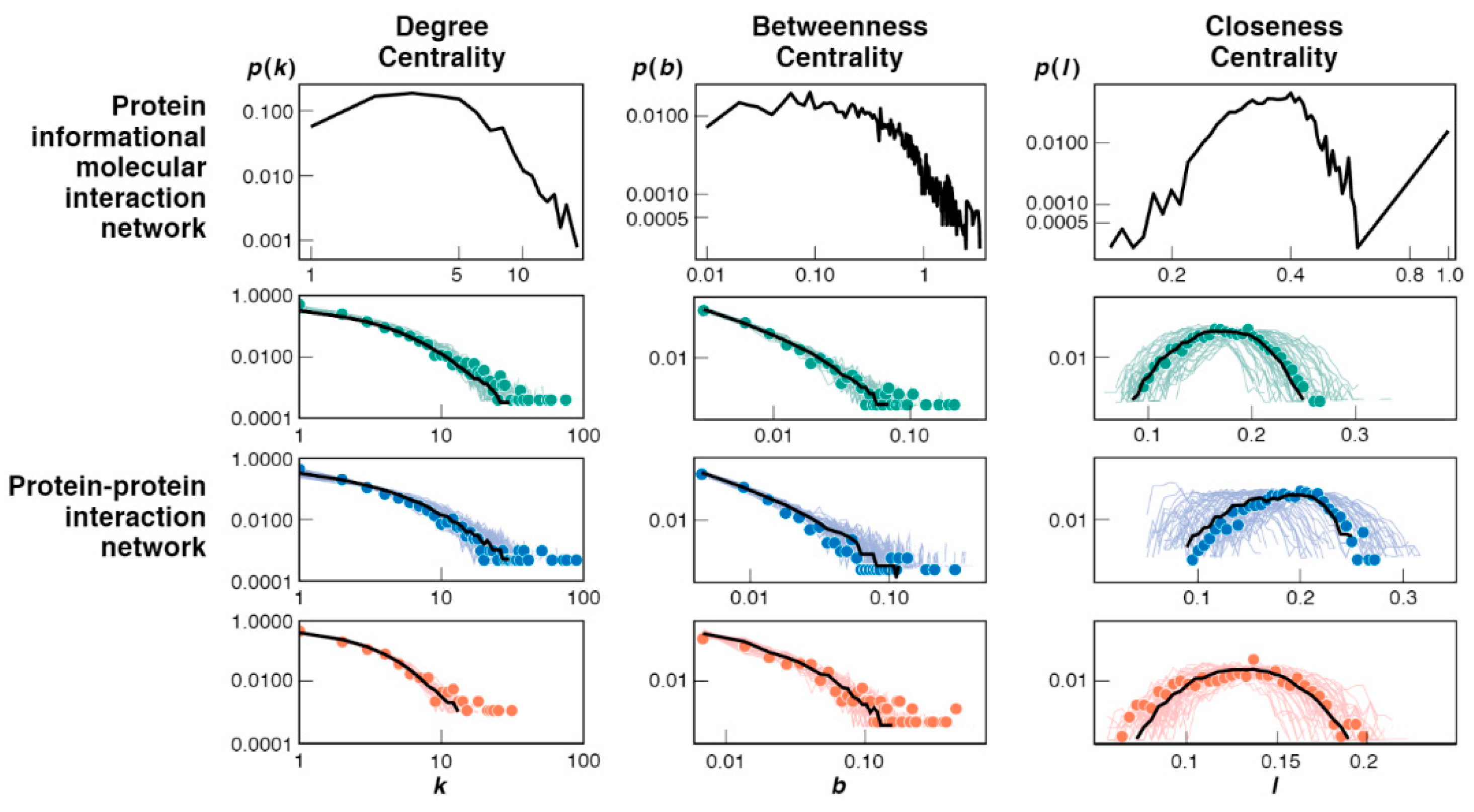
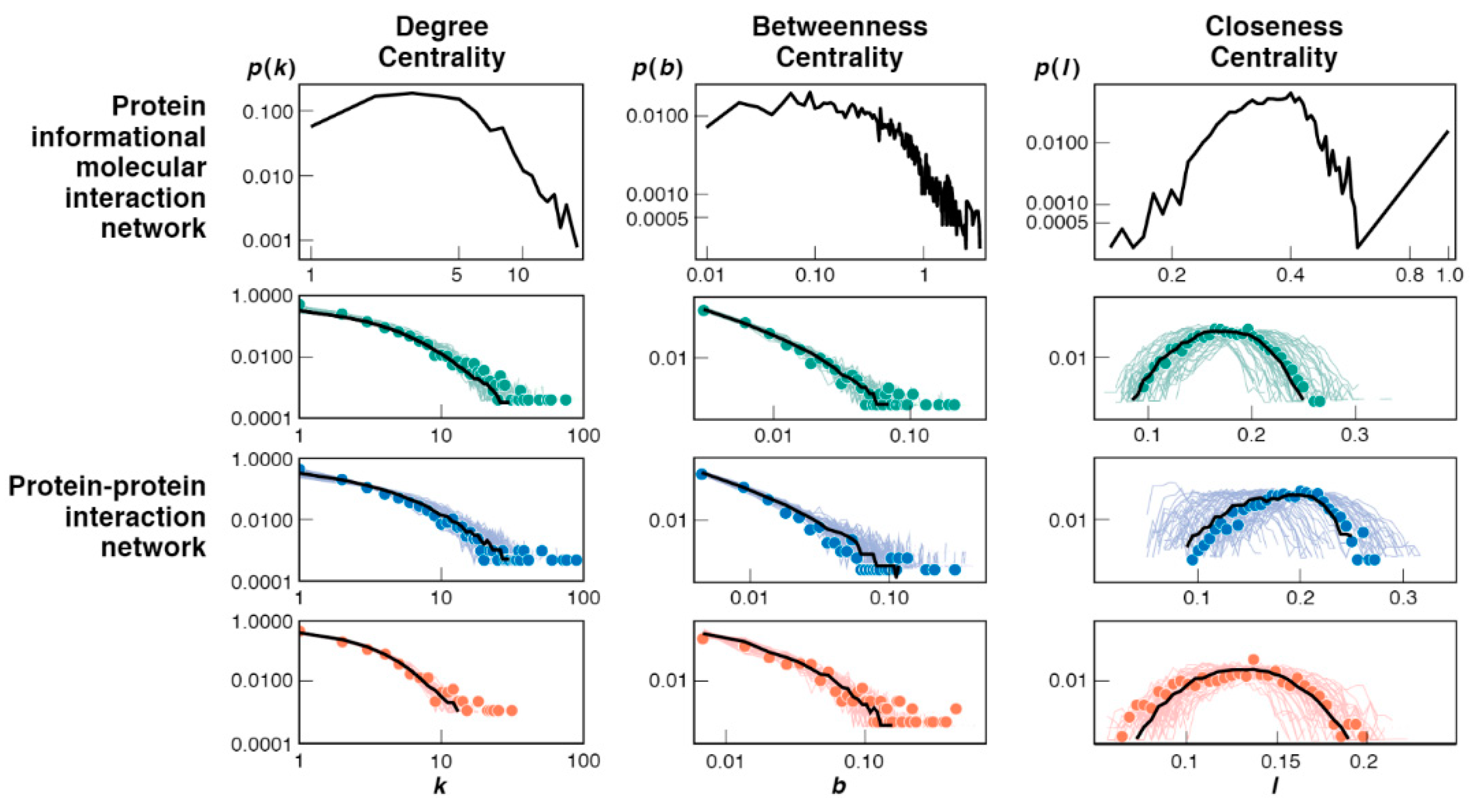
3.3. Bootstrap Model Network Topologies Resemble Today’s PPI Networks
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | M | |||||
|---|---|---|---|---|---|---|
| Static Presets | 25 | 25 | 0.143 | 0.303 | 10 | 10 |
References
- Gilbert, W. Origin of life: The RNA world. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [CrossRef]
- Dyson, F.J. Origins of Life, 2nd ed.; Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Segré, D.; Ben-Eli, D.; Deamer, D.W.; Lancet, D. The lipid world. Orig. Life Evol. Biosph. 2001, 31, 119–145. [Google Scholar] [CrossRef]
- Carter, C.W. What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life 2015, 5, 294–320. [Google Scholar] [CrossRef]
- Bowman, J.C.; Hud, N.V.; Williams, L.D. The ribosome challenge to the RNA world. J. Mol. Evol. 2015, 80, 143–161. [Google Scholar] [CrossRef]
- Carter, C.W., Jr. An Alternative to the RNA World. Nat. Hist. 2016, 125, 28–33. [Google Scholar]
- Cech, T.R. Crawling out of the RNA world. Cell 2009, 136, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Frenkel-Pinter, M.; Haynes, J.W.; Mohyeldin, A.M.; Martin, C.; Sargon, A.B.; Petrov, A.S.; Krishnamurthy, R.; Hud, N.V.; Williams, L.D.; Leman, L.J. Mutually Stabilizing Interactions Between Proto-Peptides and RNA. Nat. Commun. 2020, 11, 3137. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Haynes, J.W.; Martin, C.; Petrov, A.S.; Burcar, B.T.; Krishnamurthy, R.; Hud, N.V.; Leman, L.J.; Williams, L.D. Selective Incorporation of Proteinaceous over Nonproteinaceous Cationic Amino Acids in Model Prebiotic Oligomerization Reactions. Proc. Natl. Acad. Sci. USA 2019, 116, 16338–16346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guseva, E.; Zuckermann, R.N.; Dill, K.A. Foldamer Hypothesis for the Growth and Sequence Differentiation of Prebiotic Polymers. Proc. Natl. Acad. Sci. USA 2017, 114, 7460–7468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordijk, W. A History of Autocatalytic Sets. Biol. Theory 2019, 14, 224–246. [Google Scholar] [CrossRef]
- Smith, J.I.; Steel, M.; Hordijk, W. Autocatalytic Sets in a Partitioned Biochemical Network. J. Syst. Chem. 2014, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Higgs, P.G. Origin of Self-Replicating Biopolymers: Autocatalytic Feedback Can Jump-Start the RNA World. J. Mol. Evol. 2009, 69, 541–554. [Google Scholar] [CrossRef]
- Tkachenko, A.V.; Maslov, S. Onset of autocatalysis of information-coding polymers. arXiv 2014, arXiv:1405.2888. [Google Scholar] [CrossRef]
- Lee, D.; Granja, J.; Martinez, J.; Severin, K.; Ghadiri, M.R. A Self-Replicating Peptide. Nature 1996, 382, 525–528. [Google Scholar] [CrossRef]
- Rubinov, B.; Wagner, N.; Rapaport, H.; Ashkenasy, G. Self-Replicating Amphiphilic β-Sheet Peptides. Angew. Chem. Int. Ed. 2009, 48, 6683–6686. [Google Scholar] [CrossRef]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing Life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef]
- Monnard, P.A.; Deamer, D.W. Membrane Self-Assembly Processes: Steps towards the First Cellular Life. Anat. Rec. 2002, 268, 196–207. [Google Scholar] [CrossRef]
- Hanczyc, M.M.; Fujikawa, S.M.; Szostak, J.W. Experimental Models of Primitive Cellular Compartments: Encapsulation, Growth, and Division. Science 2003, 302, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.A.; Roberts, R.W.; Szostak, J.W. The Emergence of Competition Between Model Protocells. Science 2004, 305, 1474–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, G.F.; Szostak, J.W. Protocells and RNA Self-Replication. Cold Spring Harb. Perspect. Biol. 2018, 10, a034801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walde, P.; Goto, A.; Monnard, P.A.; Wessicken, M.; Luisi, P.L. Oparins Reactions Revisted-Enzymatic-Synthesis of Poly (adenylic acid) in MICELLES and Self-Reproducing Vesicles. J. Am. Chem. Soc. 1994, 116, 7541–7547. [Google Scholar] [CrossRef]
- Chakrabarti, A.C.; Deamer, D.W. Permeation of Membranes by the Neutral Form of Amino-acids and Peptides—Relevance to the Origin of Peptide Translocation. J. Mol. Evol. 1994, 39, 1–5. [Google Scholar] [CrossRef]
- Carter, C.W., Jr.; Wills, P.R. Reciprocally-Coupled Gating: Strange Loops in Bioenergetics, Genetics, and Catalysis. Biomolecules 2021, 11, 265. [Google Scholar] [CrossRef]
- Horowitz, E.D.; Engelhart, A.E.; Chen, M.C.; Quarles, K.A.; Smith, M.W.; Lynn, D.G.; Hud, N.V. Intercalation as a Means to Suppress Cyclization and Promote Polymerization of Base-Pairing Oligonucleotides in a Prebiotic World. Proc. Natl. Acad. Sci. USA 2010, 107, 5288–5293. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.S.; Anet, F.A.L.; Stahle, C.J.; Hud, N.V. Enzymatic Behavior by Intercalating Molecules in a Template-Directed Ligation Reaction. Angew. Chem. Int. Ed. 2004, 43, 2004–2008. [Google Scholar] [CrossRef]
- Hud, N.V.; Cafferty, B.J.; Krishnamurthy, R.; Williams, L.D. The Origin of RNA and “My Grandfather’s Axe”. Biol. Chem. 2013, 20, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Dill, K.A.; Agozzino, L. Driving Forces in the Origins of Life. Open Biol. 2021, 11, 200324. [Google Scholar] [CrossRef]
- Maibaum, L.; Dinner, A.R.; Chandler, D. Micelle Formation and the Hydrophobic Effect. J. Phys. Chem. 2004, 108, 6778–6781. [Google Scholar] [CrossRef] [Green Version]
- Tanford, C. Theory of Micelle Formation in Aqueous Solutions. J. Phys. Chem. 1974, 78, 2469–2479. [Google Scholar] [CrossRef]
- Feng, B.; Sosa, R.P.; Martensson, A.K.F.; Jiang, K.; Tong, A.; Dorfman, K.D.; Takahashi, M.; Lincoln, P.; Bustamante, C.J.; Westerlund, F.; et al. Hydrophobic Catalysis and a Potential Biological Role of DNA Unstacking Induced by Environment Effects. Proc. Natl. Acad. Sci. USA 2019, 116, 17169–17174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marat, Y.M.; Gulnara, Y.Z.; Baucom, A.; Lieberman, K.; Earnest, T.N.; Cate, J.H.D.; Noller, H.F. Crystal Structure of the Ribosome at 5.5 Å Resolution. Science 2001, 292, 883–896. [Google Scholar] [CrossRef]
- Moore, P.; Steitz, T. The Involvement of RNA in Ribosome Function. Nature 2002, 418, 229–235. [Google Scholar] [CrossRef]
- Campbell, J.H. An RNA Replisome as the Ancestor of the Ribosome. J. Mol. Evol. 1991, 32, 3–5. [Google Scholar] [CrossRef]
- Ferreira-Cerca, S.; Pöll, G.; Gleizes, P.E.; Tschochner, H.; Milkereit, P. Roles of Eukaryotic Ribosomal Proteins in Maturation and Transport of Pre-18S rRNA and Ribosome Function. Mol. Cell 2005, 20, 263–275. [Google Scholar] [CrossRef]
- Venema, J.; Tollervey, D. Ribosome Synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 1999, 33, 261–311. [Google Scholar] [CrossRef]
- Reimer, J.M.; Haque, A.S.; Tarry, M.J.; Schmeing, M.T. Piecing Together Nonribosomal Peptide Synthesis. Curr. Opin. Struct. Biol. 2018, 49, 104–113. [Google Scholar] [CrossRef]
- Kimura, M. On the Probability of Fixation of Mutant Genes in a Population. Genetics 1962, 47, 713–719. [Google Scholar] [CrossRef]
- Peterson, G.J.; Pressé, S.; Peterson, K.S.; Dill, K.A. Simulated Evolution of Protein-Protein Interaction Networks with Realistic Topology. PLoS ONE 2012, 7, e39052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdős, P.; Rényi, A. On the Evolution of Random Graphs. Publ. Math. Inst. Hung. Acad. Sci. 1960, 7, 17–61. [Google Scholar]
- Fujimori, S.; Hino, K.; Saito, A.; Miyano, S.; Miyamoto-Sato, E. PRD: A Protein-RNA Interaction Database. J. Bioinform. 2012, 8, 729–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.; Zhao, Y.; Li, C.; Zhang, L.; Huang, H.; Li, Y.; Liu, L.; Hou, P.; Cui, T.; Tan, P.; et al. RAID v2.0: An Updated Resource of RNA-Associated Interactions Across Organisms. Nucleic Acids Res. 2016, 45, D115–D118. [Google Scholar] [CrossRef]
- Yi, Y.; Zhao, Y.; Huang, Y.; Wang, D. A Brief Review of RNA-Protein Interaction Database Resources. Noncoding RNA 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Kirsanov, D.D.; Zanegina, O.N.; Aksianov, E.A.; Spirin, S.A.; Karyagina, A.S.; Alexeevski, A.V. NPIDB: Nucleic Acid-Protein Interaction Database. Nucleic Acids Res. 2013, 41, D517–D523. [Google Scholar] [CrossRef] [Green Version]
- Panni, S.; Prakash, A.; Bateman, A.; Orchard, S. The Yeast Noncoding RNA Interaction Network. RNA 2017, 23, 1479–1492. [Google Scholar] [CrossRef] [Green Version]
- Perkins, S.J. The Calculations of Partial Specific Volumes, Neutron Scattering Matchpoints and 280-nm Absorption Coefficients for Proteins and Glycoproteins from Amino Acid Sequences. Eur. J. Chem. 1986, 157, 169–180. [Google Scholar] [CrossRef]
- Voss, N.R.; Gerstein, M. Calculation of Standard Atomic Volumes for RNA and Comparison with Proteins: RNA Is Packed More Tightly. J. Mol. Biol. 2005, 346, 477–492. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farquharson, T.; Agozzino, L.; Dill, K. The Bootstrap Model of Prebiotic Networks of Proteins and Nucleic Acids. Life 2022, 12, 724. https://doi.org/10.3390/life12050724
Farquharson T, Agozzino L, Dill K. The Bootstrap Model of Prebiotic Networks of Proteins and Nucleic Acids. Life. 2022; 12(5):724. https://doi.org/10.3390/life12050724
Chicago/Turabian StyleFarquharson, Thomas, Luca Agozzino, and Ken Dill. 2022. "The Bootstrap Model of Prebiotic Networks of Proteins and Nucleic Acids" Life 12, no. 5: 724. https://doi.org/10.3390/life12050724
APA StyleFarquharson, T., Agozzino, L., & Dill, K. (2022). The Bootstrap Model of Prebiotic Networks of Proteins and Nucleic Acids. Life, 12(5), 724. https://doi.org/10.3390/life12050724