
Insulin Resistance Is Cheerfully Hitched with Hypertension


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Objective of This Study
1.2. Materials and Methods
1.3. Insulin and Its Effects
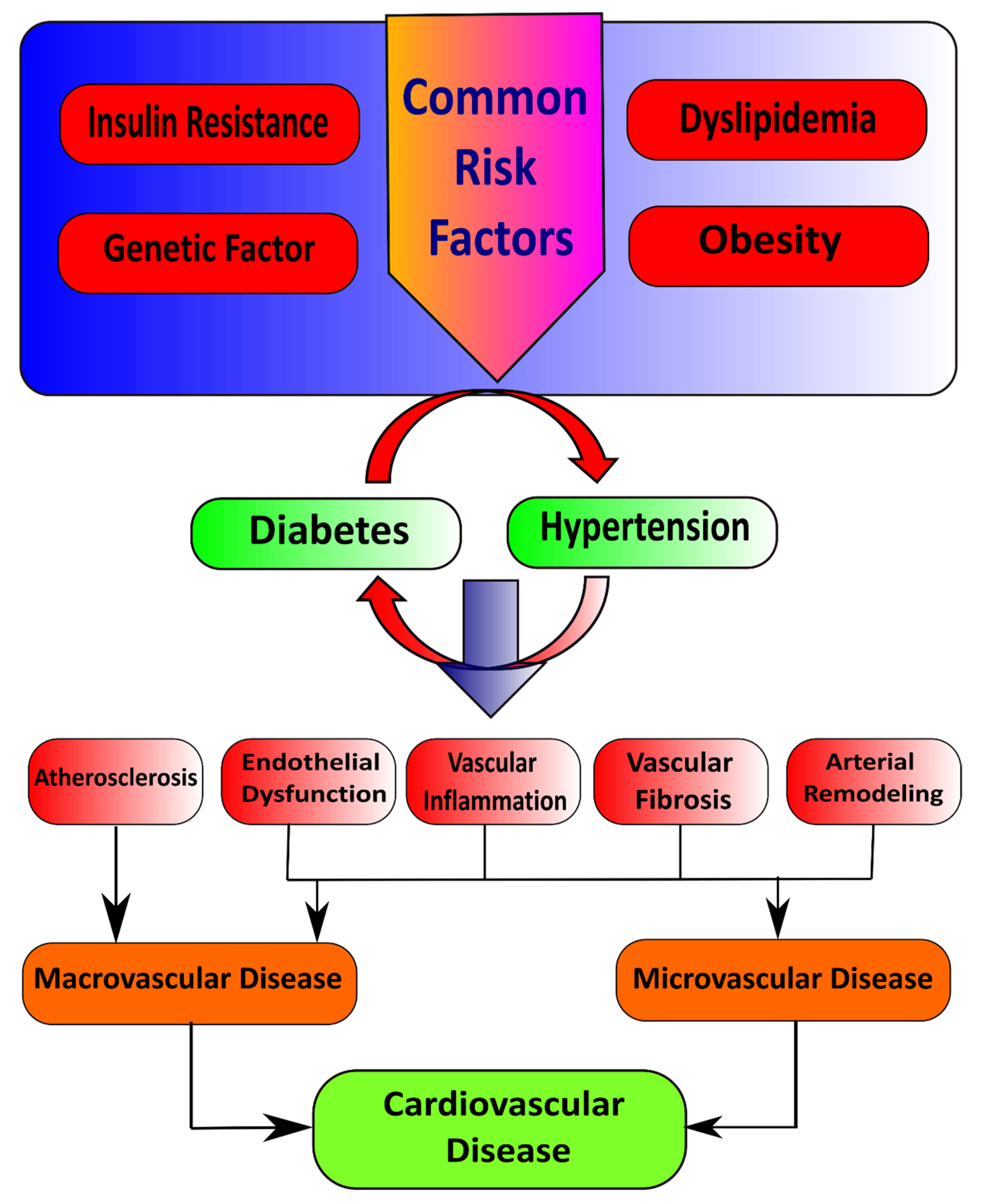
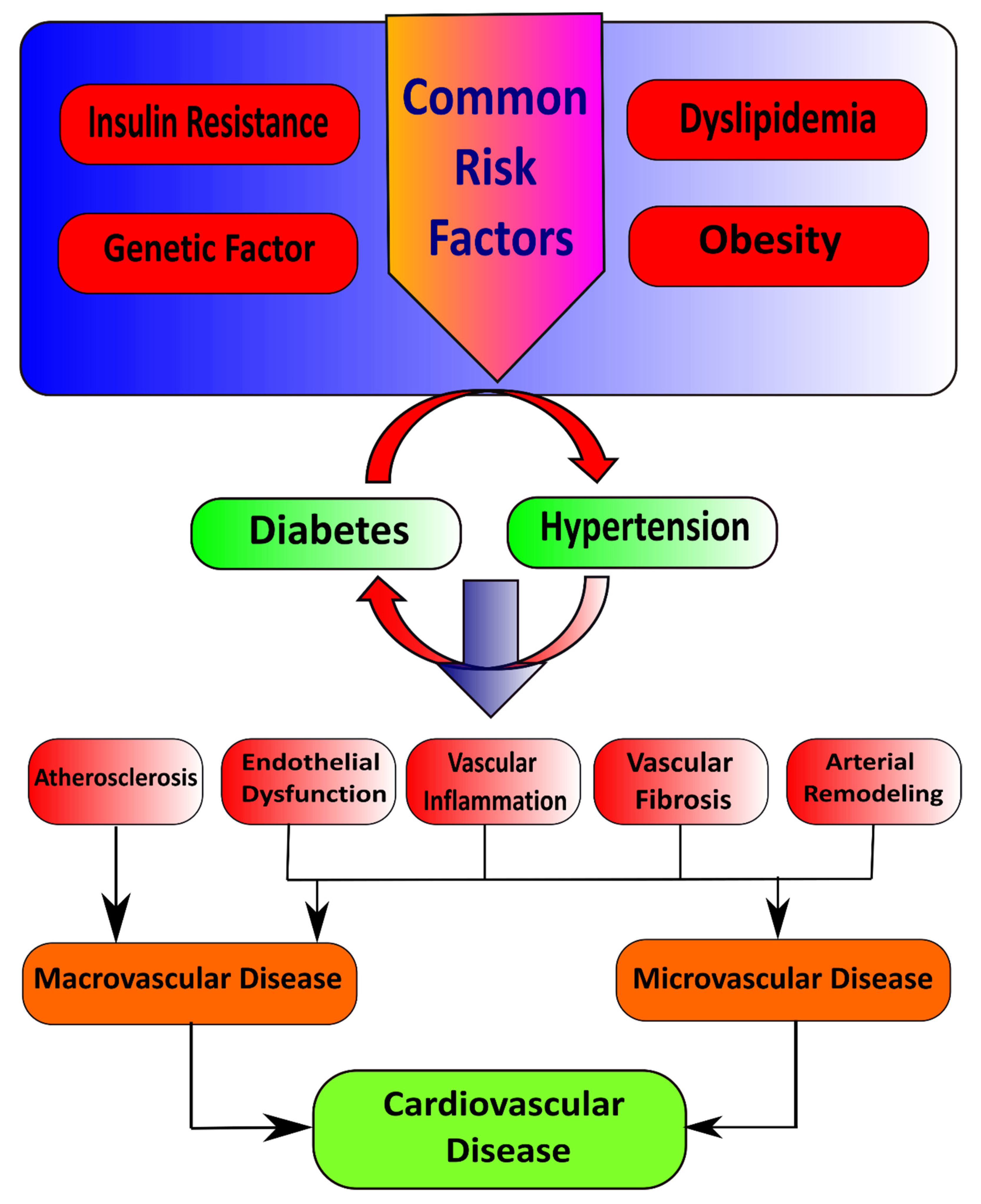
2. Type 2 Diabetes Mellitus and Hypertension
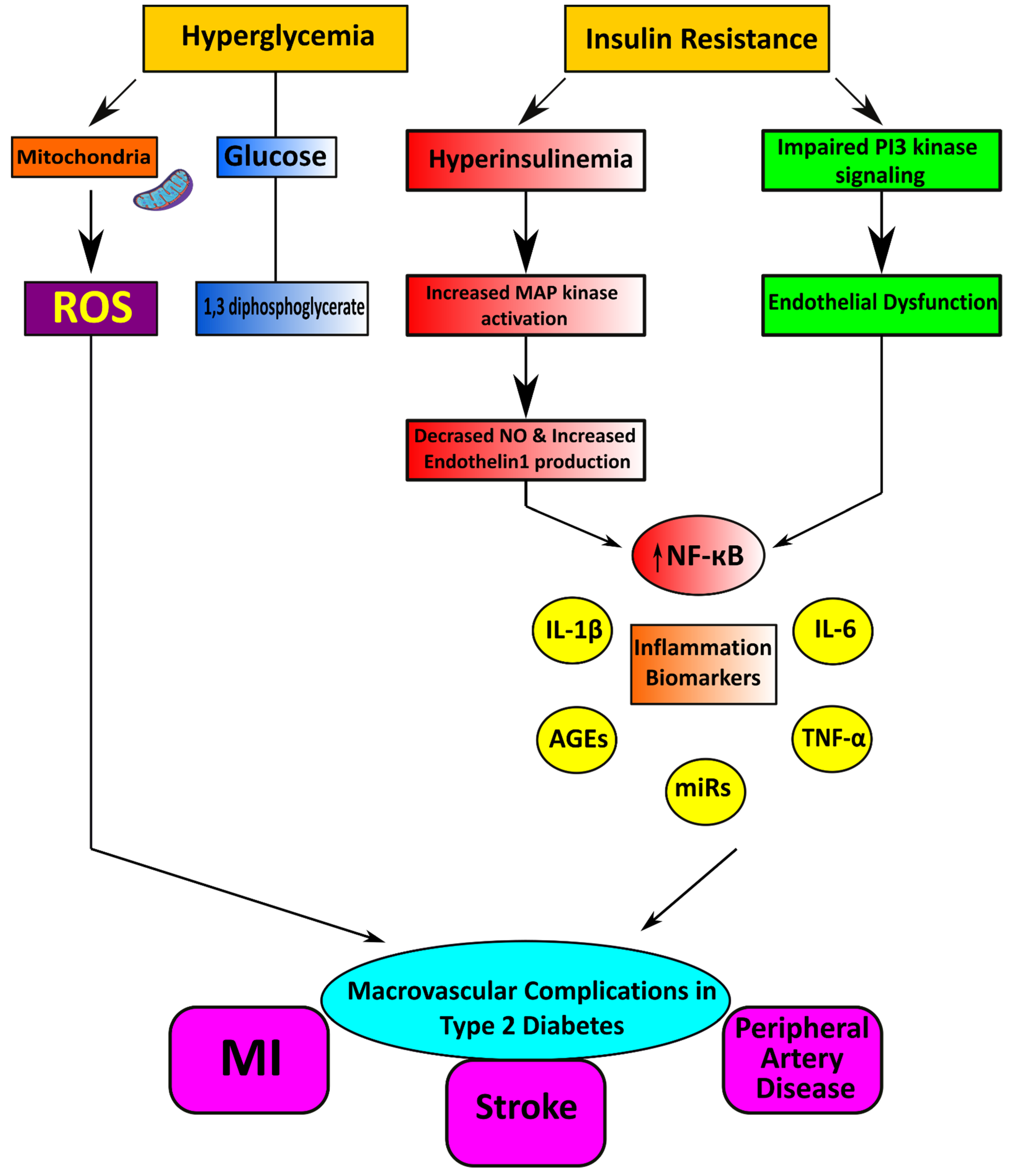
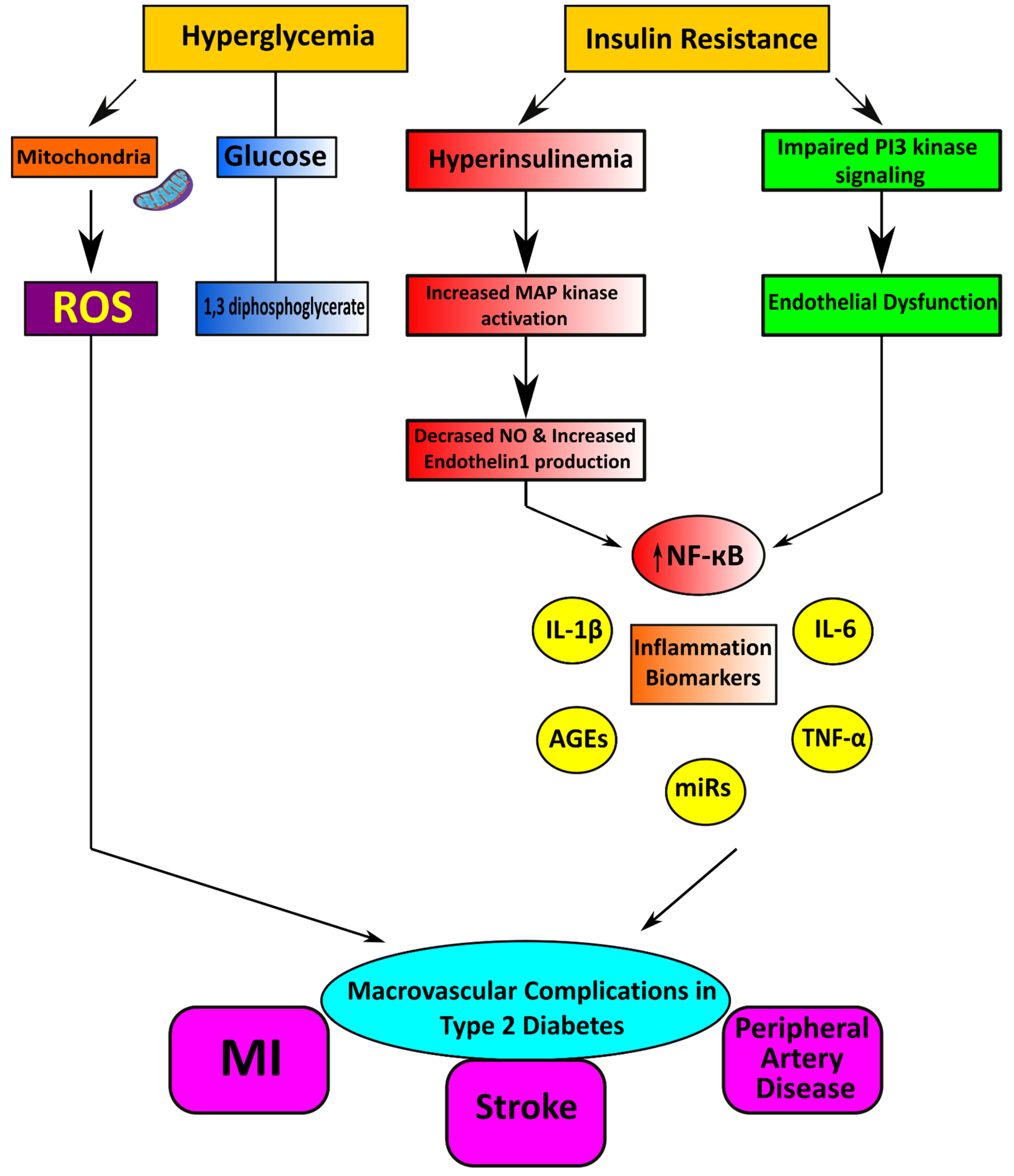
2.1. Macrovascular Diseases in Diabetes
Pathophysiological Features of Macrovascular Disease
2.2. Microvascular Diseases in Diabetes
Pathophysiological Features of Microvascular Disease
3. Interacting Mechanisms in T2DM and Hypertension
3.1. AGE–RAGE Axis
3.2. Oxidative Stress and Nox
3.3. Role of Inflammation and the Immune System
4. Insulin Resistance
4.1. Insulin Resistance and Hypertension
4.2. Blood Pressure and Insulin Resistance
4.3. Molecular Mechanism of Insulin Resistance
4.4. Neuro-Hormonal Activities and Insulin Resistance
4.5. Role of Prorenin
4.6. Sympathetic Nervous System
5. Target Organ Damage in Insulin Resistance and Hypertension
5.1. Left Ventricular Hypertrophy
5.2. Carotid Atherosclerosis
5.3. Renal Dysfunction
6. Conclusions and Recommendation
Author Contributions
Funding
Conflicts of Interest
References
- Petrie, J.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.; Ali, W.; Parati, G. ACC/AHA Versus ESC/ESH on Hypertension Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Li, A.-L.; Peng, Q.; Shao, Y.-Q.; Fang, X.; Zhang, Y.-Y. The interaction on hypertension between family history and diabetes and other risk factors. Sci. Rep. 2021, 11, 4716. [Google Scholar] [CrossRef]
- American Diabetes Association Addendum. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43, 1979. [Google Scholar] [CrossRef]
- Wu, W.-C.; Wei, J.-N.; Chen, S.-C.; Fan, K.-C.; Lin, C.-H.; Yang, C.-Y.; Lin, M.-S.; Shih, S.-R.; Hua, C.-H.; Hsein, Y.-C.; et al. Progression of insulin resistance: A link between risk factors and the incidence of diabetes. Diabetes Res. Clin. Pract. 2020, 161, 108050. [Google Scholar] [CrossRef]
- Lin, C.-H.; Wei, J.-N.; Fan, K.-C.; Fang, C.-T.; Wu, W.-C.; Yang, C.-Y.; Lin, M.-S.; Shih, S.-R.; Hua, C.-H.; Hsein, Y.-C.; et al. Different cutoffs of hypertension, risk of incident diabetes and progression of insulin resistance: A prospective cohort study. J. Formos. Med Assoc. 2022, 121, 193–201. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J. Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef]
- Posner, B.I. Insulin Signalling: The Inside Story. Can. J. Diabetes 2017, 41, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Dong, H.H. FoxO integration of insulin signaling with glucose and lipid metabolism. J. Endocrinol. 2017, 233, R67–R79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Santoro, A.; Peroni, O.D.; Nelson, A.T.; Saghatelian, A.; Siegel, D.; Kahn, B.B. PAHSAs enhance hepatic and systemic insulin sensitivity through direct and indirect mechanisms. J. Clin. Investig. 2019, 129, 4138–4150. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2017, 1411, 21–35. [Google Scholar] [CrossRef]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias, D.D.S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Oktay, A.A.; Akturk, H.K.; Jahangir, E. Diabetes mellitus and hypertension. Curr. Opin. Cardiol. 2016, 31, 402–409. [Google Scholar] [CrossRef]
- Ogurtsova, K.; Da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Oguoma, V.M.; Nwose, E.U.; Ulasi, I.I.; Akintunde, A.A.; Chukwukelu, E.E.; Bwititi, P.T.; Richards, R.S.; Skinner, T.C. Cardiovascular disease risk factors in a Nigerian population with impaired fasting blood glucose level and diabetes mellitus. BMC Public Health 2017, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, G.; Wang, W.; Best, L.G.; Yeh, F.; Izzo, R.; Mancusi, C.; Roman, M.J.; Lee, E.T.; Howard, B.V.; Devereux, R.B. Target organ damage and incident type 2 diabetes mellitus: The Strong Heart Study. Cardiovasc. Diabetol. 2017, 16, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Akram, R.; Sheikh, N.; Sarker, A.R.; Sultana, M. Sex-specific prevalence, inequality and associated predictors of hypertension, diabetes, and comorbidity among Bangladeshi adults: Results from a nationwide cross-sectional demographic and health survey. BMJ Open 2019, 9, e029364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visca, D.; Pignatti, P.; Spanevello, A.; Lucini, E.; La Rocca, E. Relationship between diabetes and respiratory diseases—Clinical and therapeutic aspects. Pharmacol. Res. 2018, 137, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Climie, R.E.; van Sloten, T.T.; Bruno, R.-M.; Taddei, S.; Empana, J.-P.; Stehouwer, C.D.; Sharman, J.E.; Boutouyrie, P.; Laurent, S. Macrovasculature and Microvasculature at the Crossroads Between Type 2 Diabetes Mellitus and Hypertension. Hypertension 2019, 73, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.-I.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, X.; Icli, B.; Feinberg, M.W. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr. Rev. 2017, 38, 145–168. [Google Scholar] [CrossRef] [Green Version]
- Strain, W.D.; Paldanius, P.M. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc. Diabetol. 2018, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Refaat, M.; Mohammedi, K.; Jayyousi, A.; Al Suwaidi, J.; Khalil, C.A. Macrovascular Complications in Patients with Diabetes and Prediabetes. BioMed Res. Int. 2017, 2017, 7839101. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Hamburg, N.M.; Creager, M.A. Contemporary Medical Management of Peripheral Artery Disease. Circ. Res. 2021, 128, 1868–1884. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-W.; Chang, H.-H.; Yang, K.-C.; Kuo, C.-S.; Lee, L.-T.; Huang, K.-C. High serum selenium levels are associated with increased risk for diabetes mellitus independent of central obesity and insulin resistance. BMJ Open Diabetes Res. Care 2016, 4, e000253. [Google Scholar] [CrossRef] [Green Version]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Antoniades, C. ‘Dysfunctional’ adipose tissue in cardiovascular disease: A reprogrammable target or an innocent bystander? Cardiovasc. Res. 2017, 113, 997–998. [Google Scholar] [CrossRef]
- Camastra, S.; Vitali, A.; Anselmino, M.; Gastaldelli, A.; Bellini, R.; Berta, R.D.; Severi, I.; Baldi, S.; Astiarraga, B.; Barbatelli, G.; et al. Muscle and adipose tissue morphology, insulin sensitivity and beta-cell function in diabetic and nondiabetic obese patients: Effects of bariatric surgery. Sci. Rep. 2017, 7, 9007. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Husmann, M.; Meyer, M.R. Accelerated Vascular Aging as a Paradigm for Hypertensive Vascular Disease: Prevention and Therapy. Can. J. Cardiol. 2016, 32, 680–686.e4. [Google Scholar] [CrossRef]
- Qiu, C.; Fratiglioni, L. A major role for cardiovascular burden in age-related cognitive decline. Nat. Rev. Cardiol. 2015, 12, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. Aging, arterial stiffness, and hypertension. Hypertension 2015, 65, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, M.; Zhang, Z.; Mima, A.; King, G.L. Molecular mechanisms of diabetic vascular complications. J. Diabetes Investig. 2010, 1, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammedi, K.; Woodward, M.; Marre, M.; Colagiuri, S.; Cooper, M.; Harrap, S.; Mancia, G.; Poulter, N.; Williams, B.; Zoungas, S.; et al. Comparative effects of microvascular and macrovascular disease on the risk of major outcomes in patients with type 2 diabetes. Cardiovasc. Diabetol. 2017, 16, 95. [Google Scholar] [CrossRef]
- Kibel, A.; Selthofer-Relatic, K.; Drenjancevic, I.; Bacun, T.; Bosnjak, I.; Kibel, D.; Gros, M. Coronary microvascular dysfunction in diabetes mellitus. J. Int. Med Res. 2017, 45, 1901–1929. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Madonna, R.; Balistreri, C.R.; Geng, Y.-J.; De Caterina, R. Diabetic microangiopathy: Pathogenetic insights and novel therapeutic approaches. Vasc. Pharmacol. 2017, 90, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sheetz, M.J. Molecular Understanding of Hyperglycemia’s Adverse Effects for Diabetic Complications. JAMA 2002, 288, 2579–2588. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van Den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic Pseudohypoxia and Diabetic Complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. Intracellular Signaling by Hydrolysis of Phospholipids and Activation of Protein Kinase C. Science 1992, 258, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Das Evcimen, N.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 2007, 55, 498–510. [Google Scholar] [CrossRef]
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, J.; Yang, Y.; Zhang, X. An update on the potential role of advanced glycation end products in glycolipid metabolism. Life Sci. 2020, 245, 117344. [Google Scholar] [CrossRef]
- Haque, E.; Kamil, M.; Hasan, A.; Irfan, S.; Sheikh, S.; Khatoon, A.; Nazir, A.; Mir, S.S. Advanced glycation end products (AGEs), protein aggregation and their cross talk: New insight in tumorigenesis. Glycobiology 2019, 30, 49–57. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Uribarri, J. Advanced Glycation End Products (AGE) and Diabetes: Cause, Effect, or Both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Wake, H.; Nishinaka, T.; Hatipoglu, O.F.; Liu, K.; Watanabe, M.; Toyomura, T.; Mori, S.; Yoshino, T.; Nishibori, M.; et al. Involvement of multiple scavenger receptors in advanced glycation end product-induced vessel tube formation in endothelial cells. Exp. Cell Res. 2021, 408, 112857. [Google Scholar] [CrossRef] [PubMed]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of RAGE in diabetic microvascular complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular Calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Koulis, C.; Watson, A.; Gray, S.; Jandeleit-Dahm, K. Linking RAGE and Nox in diabetic micro- and macrovascular complications. Diabetes Metab. 2015, 41, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Frimat, M.; Daroux, M.; Litke, R.; Nevière, R.; Tessier, F.J.; Boulanger, E. Kidney, heart and brain: Three organs targeted by ageing and glycation. Clin. Sci. 2017, 131, 1069–1092. [Google Scholar] [CrossRef]
- Schmidt, A.M. 2016 ATVB Plenary Lecture: Receptor for Advanced Glycation Endproducts and Implications for the Pathogenesis and Treatment of Cardiometabolic Disorders: Spotlight on the Macrophage. Arter. Thromb. Vasc. Biol. 2017, 37, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Spadaccio, C.; Nenna, A.; Nappi, F.; Singh, S.S.A.; Sutherland, F.W.; Di Domenico, F.; Chello, M. Pharmacologic approaches against Advanced Glycation End Products (AGEs) in diabetic cardiovascular disease. Res. Cardiovasc. Med. 2015, 4, e26949. [Google Scholar] [CrossRef]
- Mallat, R.K.; John, C.M.; Kendrick, D.J.; Braun, A. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar] [PubMed]
- Darenskaya, M.A.; Kolesnikova, L.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, T.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced Oxidative Stress and its Role in Diabetes Mellitus Related Cardiovascular Diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Sada, L.; Zezza, L.; Pucci, L.; Lauri, F.M.; Befani, A.; Alonzo, A.; Volpe, M. Vascular Inflammation and Endothelial Dysfunction in Experimental Hypertension. Int. J. Hypertens. 2011, 2011, 1–8. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.-H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Ortega-Lozano, A.J.; Pedraza-Chaverri, J. Redox signaling pathways in unilateral ureteral obstruction (UUO)-induced renal fibrosis. Free Radic. Biol. Med. 2021, 172, 65–81. [Google Scholar] [CrossRef]
- Sedeek, M.; Montezano, A.C.; Hebert, R.L.; Gray, S.P.; Di Marco, E.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.; Schiffrin, E.L.; Wilkinson-Berka, J.L.; et al. Oxidative Stress, Nox Isoforms and Complications of Diabetes—Potential Targets for Novel Therapies. J. Cardiovasc. Transl. Res. 2012, 5, 509–518. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Guzik, T.J.; Cosentino, F. Epigenetics and Immunometabolism in Diabetes and Aging. Antioxid. Redox Signal. 2018, 29, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.J.; Sie, C.; Schumann, E.; Witte, A.-K.; Dressel, R.; Brandt, J.V.D.; Reichardt, H.M. The Insulin Receptor Plays a Critical Role in T Cell Function and Adaptive Immunity. J. Immunol. 2017, 198, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Pérez-García, A.; Torrecilla-Parra, M.; Frutos, M.F.-D.; Martín-Martín, Y.; Pardo-Marqués, V.; Ramírez, C.M. Posttranscriptional Regulation of Insulin Resistance: Implications for Metabolic Diseases. Biomolecules 2022, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.M.; Pennings, N. Insulin Resistance; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507839/ (accessed on 6 March 2022).
- Emoto, M.; Nishizawa, Y.; Maekawa, K.; Hiura, Y.; Kanda, H.; Kawagishi, T.; Shoji, T.; Okuno, Y.; Morii, H. Homeostasis model assessment as a clinical index of insulin resistance in type 2 diabetic patients treated with sulfonylureas. Diabetes Care 1999, 22, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Mancusi, C.; Izzo, R.; Di Gioia, G.; Losi, M.A.; Barbato, E.; Morisco, C. Insulin Resistance the Hinge Between Hypertension and Type 2 Diabetes. High Blood Press. Cardiovasc. Prev. 2020, 27, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Randin, D.; Vollenweider, P.; Vollenweider, L.; Nicod, P. Nitric oxide release accounts for insulin’s vascular effects in humans. J. Clin. Investig. 1994, 94, 2511–2515. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Maratou, E.; Kountouri, A.; Board, M.; Lambadiari, V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients 2021, 13, 159. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Hill, M.A.; Jaisser, F.; Sowers, J.R. Role of the vascular endothelial sodium channel activation in the genesis of pathologically increased cardiovascular stiffness. Cardiovasc. Res. 2022, 118, 130–140. [Google Scholar] [CrossRef]
- Mansley, M.K.; Watt, G.B.; Francis, S.L.; Walker, D.J.; Land, S.C.; Bailey, M.A.; Wilson, S.M. Dexamethasone and insulin activate serum and glucocorticoid-inducible kinase 1 (SGK1) via different molecular mechanisms in cortical collecting duct cells. Physiol. Rep. 2016, 4, e12792. [Google Scholar] [CrossRef]
- Sierra-Ramos, C.; Velazquez-Garcia, S.; Vastola-Mascolo, A.; Hernández, G.; Faresse, N.; de la Rosa, D.A. SGK1 activation exacerbates diet-induced obesity, metabolic syndrome and hypertension. J. Endocrinol. 2020, 244, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Kunes, J.; Zicha, J. The interaction of genetic and environmental factors in the etiology of hypertension. Physiol. Res. 2009, 58, S33–S42. [Google Scholar] [CrossRef] [PubMed]
- Benite-Ribeiro, S.A.; Rodrigues, V.A.d.L.; Machado, M.R.F. Food intake in early life and epigenetic modifications of pro-opiomelanocortin expression in arcuate nucleus. Mol. Biol. Rep. 2021, 48, 3773–3784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pradhan, S. Mammalian epigenetic mechanisms. IUBMB Life 2014, 66, 240–256. [Google Scholar] [CrossRef]
- Sylow, L.; Nielsen, I.L.; Kleinert, M.; Møller, L.L.V.; Ploug, T.; Schjerling, P.; Bilan, P.J.; Klip, A.; Jensen, T.; Richter, E.A. Rac1 governs exercise-stimulated glucose uptake in skeletal muscle through regulation of GLUT4 translocation in mice. J. Physiol. 2016, 594, 4997–5008. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; DeMarco, V.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Ji, Y.; Wu, Z.; Dai, Z.; Sun, K.; Wang, J.; Wu, G. Nutritional epigenetics with a focus on amino acids: Implications for the development and treatment of metabolic syndrome. J. Nutr. Biochem. 2016, 27, 1–8. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Yang, M.; Shi, L.; Tao, W.; Shen, K.; Li, X.; Wang, X.; Yang, Y.; Yao, Y. Association of single nucleotide polymorphisms of miRNAs involved in the GLUT4 pathway in T2DM in a Chinese population. Mol. Genet. Genom. Med. 2019, 7, e907. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, R.; Bahiraee, A.; Niazpour, F.; Emamgholipour, S.; Meshkani, R. The role of microRNAs in the regulation of insulin signaling pathway with respect to metabolic and mitogenic cascades: A review. J. Cell. Biochem. 2019, 120, 19290–19309. [Google Scholar] [CrossRef]
- Stols-Gonçalves, D.; Tristão, L.S.; Henneman, P.; Nieuwdorp, M. Epigenetic Markers and Microbiota/Metabolite-Induced Epigenetic Modifications in the Pathogenesis of Obesity, Metabolic Syndrome, Type 2 Diabetes, and Non-alcoholic Fatty Liver Disease. Curr. Diabetes Rep. 2019, 19, 31. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, J.M.; Moreli, M.; Tewari, S.; Benite-Ribeiro, S.A. The effect of exercise on skeletal muscle glucose uptake in type 2 diabetes: An epigenetic perspective. Metabolism 2015, 64, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Pravenec, M.; Křen, V.; Landa, V.; Mlejnek, P.; Musilová, A.; Šilhavý, J.; Šimáková, M.; Zídek, V. Recent Progress in the Genetics of Spontaneously Hypertensive Rats. Physiol. Res. 2014, 63, S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Masodsai, K.; Lin, Y.-Y.; Chaunchaiyakul, R.; Su, C.-T.; Lee, S.-D.; Yang, A.-L. Twelve-Week Protocatechuic Acid Administration Improves Insulin-Induced and Insulin-Like Growth Factor-1-Induced Vasorelaxation and Antioxidant Activities in Aging Spontaneously Hypertensive Rats. Nutrients 2019, 11, 699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, G.; Morisco, C.; Lembo, V.; D’Argenio, G.; D’Armiento, M.; Rossi, A.; Del Giudice, C.; Trimarco, B.; Caporaso, N.; Morisco, F. Dietary supplementation of vitamin D prevents the development of western diet-induced metabolic, hepatic and cardiovascular abnormalities in rats. United Eur. Gastroenterol. J. 2018, 6, 1056–1064. [Google Scholar] [CrossRef]
- Haeusler, R.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Hubbard, S.R. The Insulin Receptor: Both a Prototypical and Atypical Receptor Tyrosine Kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- De Meyts, P.; Feingold, K.R.; Anawalt, B.; Boyce, A.; Chrousos, G.; de Herder, W.W.; Dhatariya, K.; Dungan, K.; Hershman, J.M.; Hofland, J.; et al. (Eds.) The Insulin Receptor and Its Signal Transduction Network. Endotext. Available online: https://www.ncbi.nlm.nih.gov/books/NBK378978/ (accessed on 6 March 2022).
- Klip, A.; Sun, Y.; Chiu, T.T.; Foley, K.P. Signal transduction meets vesicle traffic: The software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 2014, 306, C879–C886. [Google Scholar] [CrossRef] [Green Version]
- Escribano, O.; Beneit, N.; Rubio-Longás, C.; López-Pastor, A.R.; Gómez-Hernández, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2018, 234, 8152–8161. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Han, L.; Hu, D. Fasting insulin, insulin resistance and risk of hypertension in the general population: A meta-analysis. Clin. Chim. Acta 2017, 464, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.F.; Cunha, A.R.; Cunha, M.R.; Gismondi, R.A.; Oigman, W. The Role of Renin–Angiotensin–Aldosterone System and Its New Components in Arterial Stiffness and Vascular Aging. High Blood Press. Cardiovasc. Prev. 2018, 25, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.J.; Toma, I.; Sipos, A.; Meer, E.J.; Vargas, S.L.; Peti-Peterdi, J. The Collecting Duct Is the Major Source of Prorenin in Diabetes. Hypertension 2008, 51, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, N.; Kohan, D.E. The (pro)renin receptor: An emerging player in hypertension and metabolic syndrome. Kidney Int. 2019, 95, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Ramkumar, N.; Kohan, D.E. Role of the Collecting Duct Renin Angiotensin System in Regulation of Blood Pressure and Renal Function. Curr. Hypertens. Rep. 2016, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ledford, K.J.; Pitkin, W.B.; Russo, L.; Najjar, S.M.; Siragy, H.M. Targeted deletion of murine CEACAM 1 activates PI3K-Akt signaling and contributes to the expression of (Pro)renin receptor via CREB family and NF-κB transcription factors. Hypertension 2013, 62, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, A.A.; Carmo, J.M.D.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Komici, K.; Femminella, G.D.; de Lucia, C.; Cannavo, A.; Bencivenga, L.; Corbi, G.; Leosco, D.; Ferrara, N.; Rengo, G. Predisposing factors to heart failure in diabetic nephropathy: A look at the sympathetic nervous system hyperactivity. Aging Clin. Exp. Res. 2019, 31, 321–330. [Google Scholar] [CrossRef]
- Mangmool, S.; Denkaew, T.; Parichatikanond, W.; Kurose, H. β-Adrenergic Receptor and Insulin Resistance in the Heart. Biomol. Ther. 2017, 25, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Moody, W.E.; Edwards, N.C.; Madhani, M.; Chue, C.D.; Steeds, R.P.; Ferro, C.J.; Townend, J.N. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: Cause or association? Atherosclerosis 2012, 223, 86–94. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buono, F.; Crispo, S.; Pagano, G.; Rengo, G.; Petitto, M.; Grieco, F.; Trimarco, B.; Morisco, C. Determinants of left ventricular hypertrophy in patients with recent diagnosis of essential hypertension. J. Hypertens. 2014, 32, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Izzo, R.; Losi, M.-A.; Stabile, E.; Lönnebakken, M.T.; Canciello, G.; Esposito, G.; Barbato, E.; De Luca, N.; Trimarco, B.; de Simone, G. Development of Left Ventricular Hypertrophy in Treated Hypertensive Outpatients: The Campania Salute Network. Hypertension 2017, 69, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G. Sympathetic Overdrive and Cardiovascular Risk in the Metabolic Syndrome. Hypertens. Res. 2006, 29, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yuan, M.; Bradley, K.M.; Dong, F.; Anversa, P.; Ren, J. Insulin-Like Growth Factor 1 Alleviates High-Fat Diet–Induced Myocardial Contractile Dysfunction: Role of insulin signaling and mitochondrial function. Hypertension 2012, 59, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiee, A.; Krüger, M.; Ardenkjær-Larsen, J.; Kahn, C.R.; Emanuelli, B. Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell. Signal. 2018, 47, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.E.; Crook, E.D.; Jones, D.W.; Wofford, M.R.; Dubbert, P.M. Mechanisms of Obesity-Associated Cardiovascular and Renal Disease. Am. J. Med Sci. 2002, 324, 127–137. [Google Scholar] [CrossRef]
- El-Atat, F.A.; Stas, S.N.; McFarlane, S.I.; Sowers, J.R. The Relationship between Hyperinsulinemia, Hypertension and Progressive Renal Disease. J. Am. Soc. Nephrol. 2004, 15, 2816–2827. [Google Scholar] [CrossRef] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G. Insulin Resistance and Coronary Heart Disease in Nondiabetic Individuals. Arter. Thromb. Vasc. Biol. 2012, 32, 1754–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, M.M.; Cortese, F.; Gesualdo, M.; Donvito, I.; Carbonara, S.; De Pergola, G. A Glycemic Threshold of 90 mg/dl Promotes Early Signs of Atherosclerosis in Apparetly Healthy Overweight/Obese Subjects. Endocrine Metab. Immune Disord. Drug Targets 2016, 16, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Okura, T.; Kitami, Y.; Hiwada, K. Carotid hemodynamic alterations in hypertensive patients with insulin resistance. Am. J. Hypertens. 2002, 15, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Eddy, A.A. Progression in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2005, 12, 353–365. [Google Scholar] [CrossRef]
- E Kosmas, C.; Silverio, D.; Tsomidou, C.; Salcedo, M.D.; Montan, P.D.; Guzman, E. The Impact of Insulin Resistance and Chronic Kidney Disease on Inflammation and Cardiovascular Disease. Clin. Med. Insights: Endocrinol. Diabetes 2018, 11, 1179551418792257. [Google Scholar] [CrossRef]
- Satoh, M. Endothelial dysfunction as an underlying pathophysiological condition of chronic kidney disease. Clin. Exp. Nephrol. 2012, 16, 518–521. [Google Scholar] [CrossRef]
- Yatabe, M.S.; Yatabe, J.; Yoneda, M.; Watanabe, T.; Otsuki, M.; A Felder, R.; A Jose, P.; Sanada, H. Salt sensitivity is associated with insulin resistance, sympathetic overactivity, and decreased suppression of circulating renin activity in lean patients with essential hypertension. Am. J. Clin. Nutr. 2010, 92, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Komnenov, D.; Levanovich, P.E.; Rossi, N.F. Hypertension Associated with Fructose and High Salt: Renal and Sympathetic Mechanisms. Nutrients 2019, 11, 569. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.; Haque, M. Insulin Resistance Is Cheerfully Hitched with Hypertension. Life 2022, 12, 564. https://doi.org/10.3390/life12040564
Sinha S, Haque M. Insulin Resistance Is Cheerfully Hitched with Hypertension. Life. 2022; 12(4):564. https://doi.org/10.3390/life12040564
Chicago/Turabian StyleSinha, Susmita, and Mainul Haque. 2022. "Insulin Resistance Is Cheerfully Hitched with Hypertension" Life 12, no. 4: 564. https://doi.org/10.3390/life12040564
APA StyleSinha, S., & Haque, M. (2022). Insulin Resistance Is Cheerfully Hitched with Hypertension. Life, 12(4), 564. https://doi.org/10.3390/life12040564