
Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ligand Generation
2.2. Drug Likeliness and ADME and Toxicity Calculations
2.3. Preparation of Receptors and Their Binding Site
2.4. Molecular Docking
2.4.1. Protein Preparation
2.4.2. Active Site and Grid Generation
2.4.3. Genetic Algorithm
2.4.4. Lamarckian Genetic Algorithm
2.4.5. Ligand–Receptor Interactions
2.4.6. Molecular Dynamics Simulations
3. Results and Discussion
3.1. Drug Likeliness Prediction [ADME] Calculations
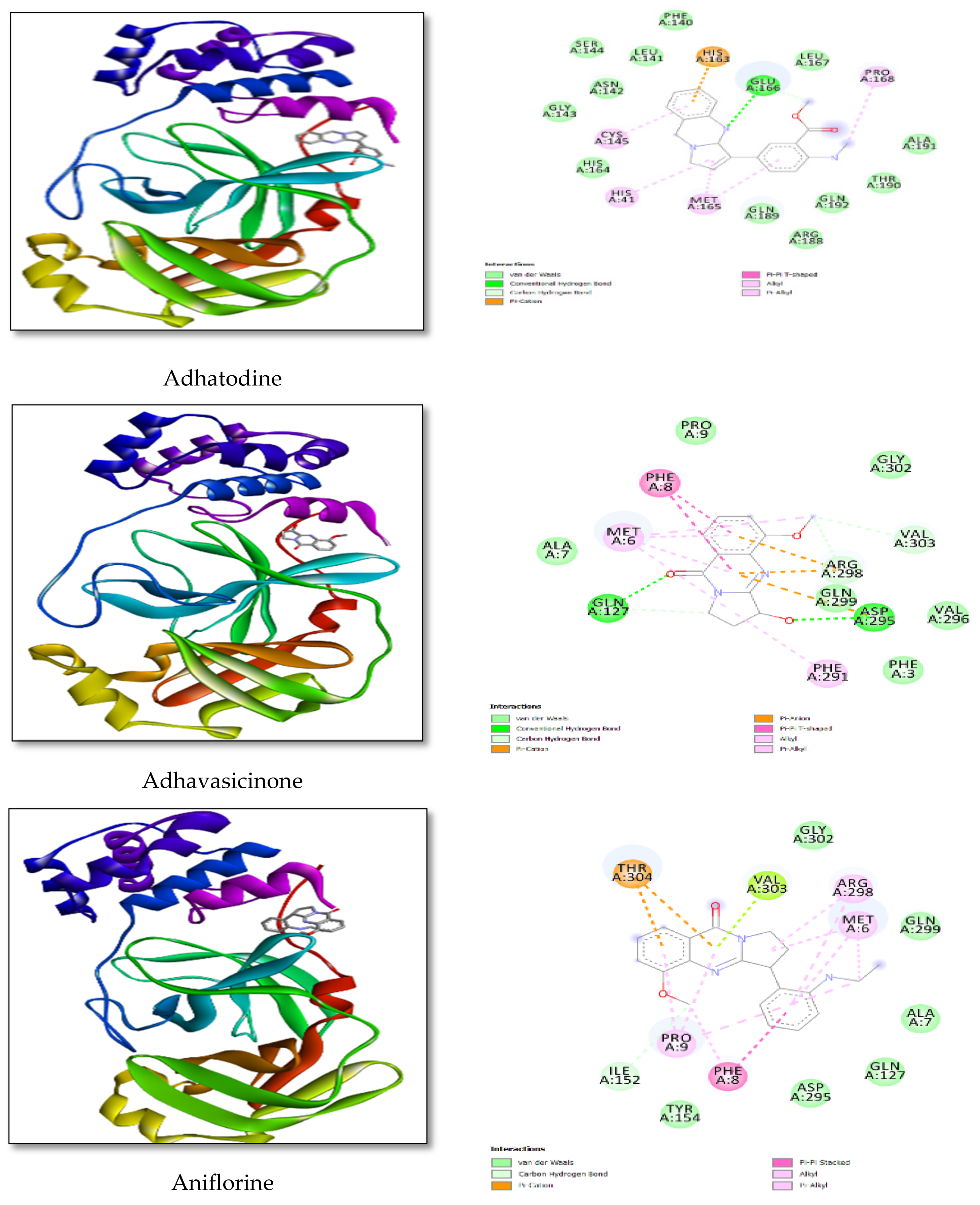
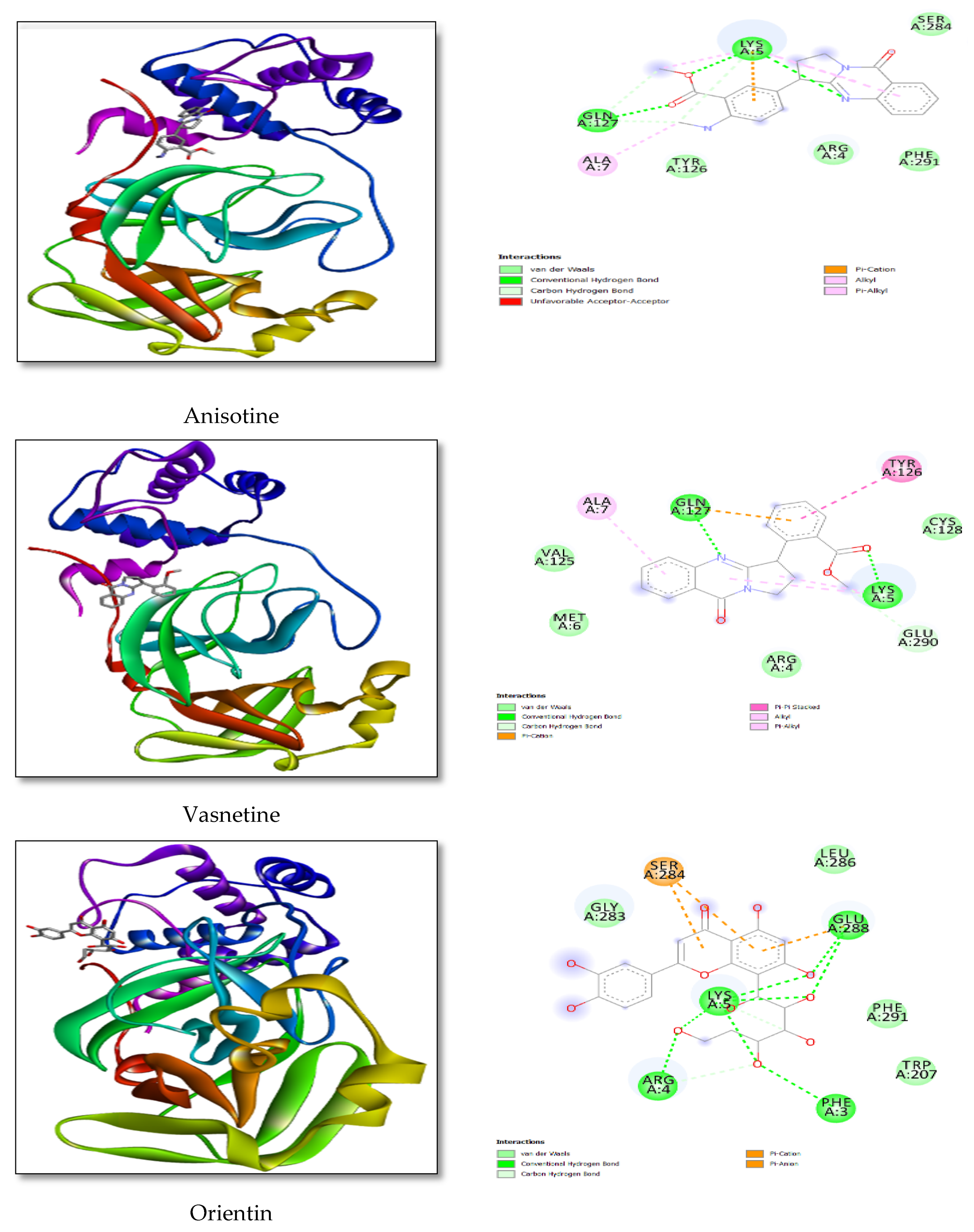
3.2. Docking Study
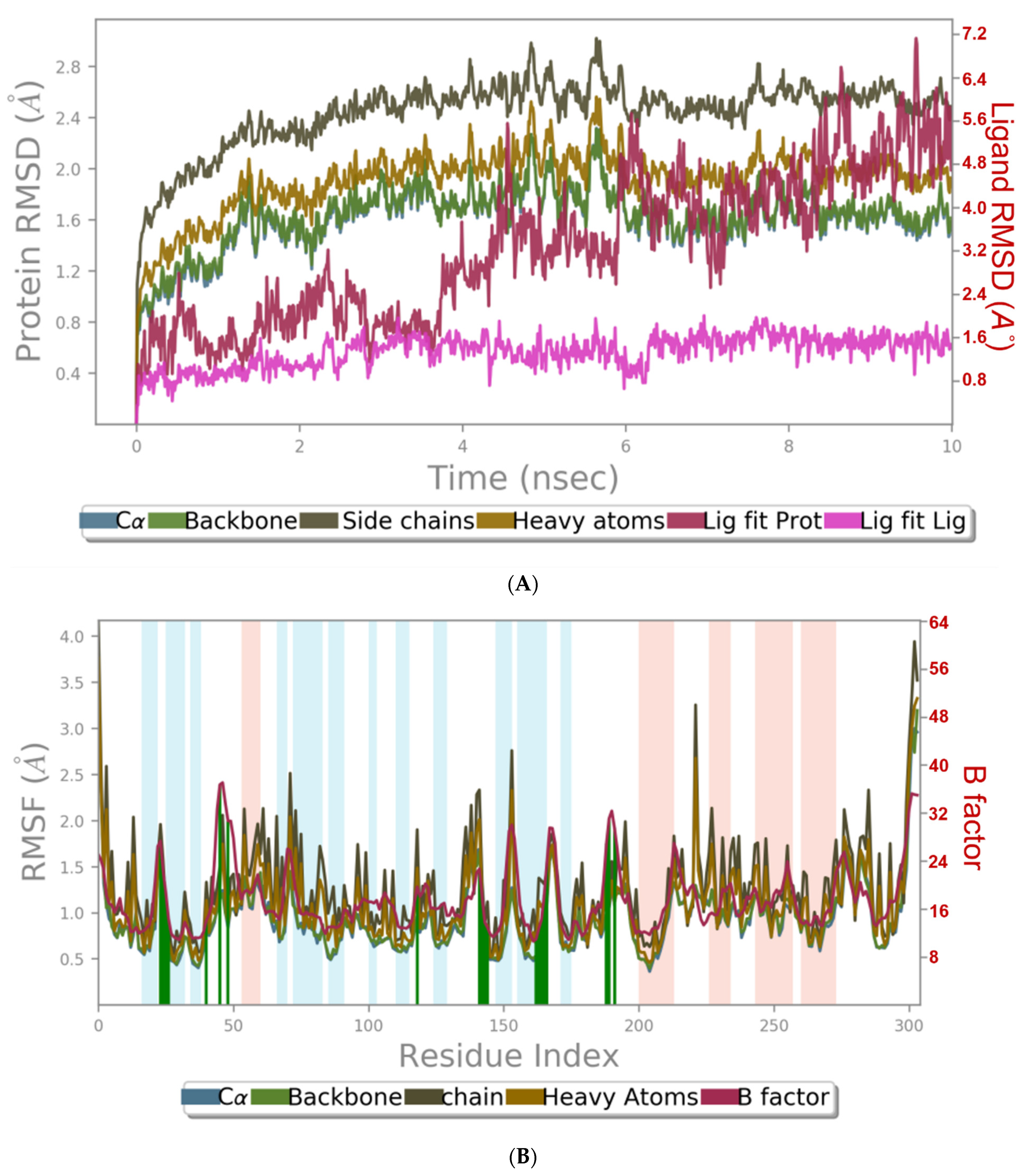
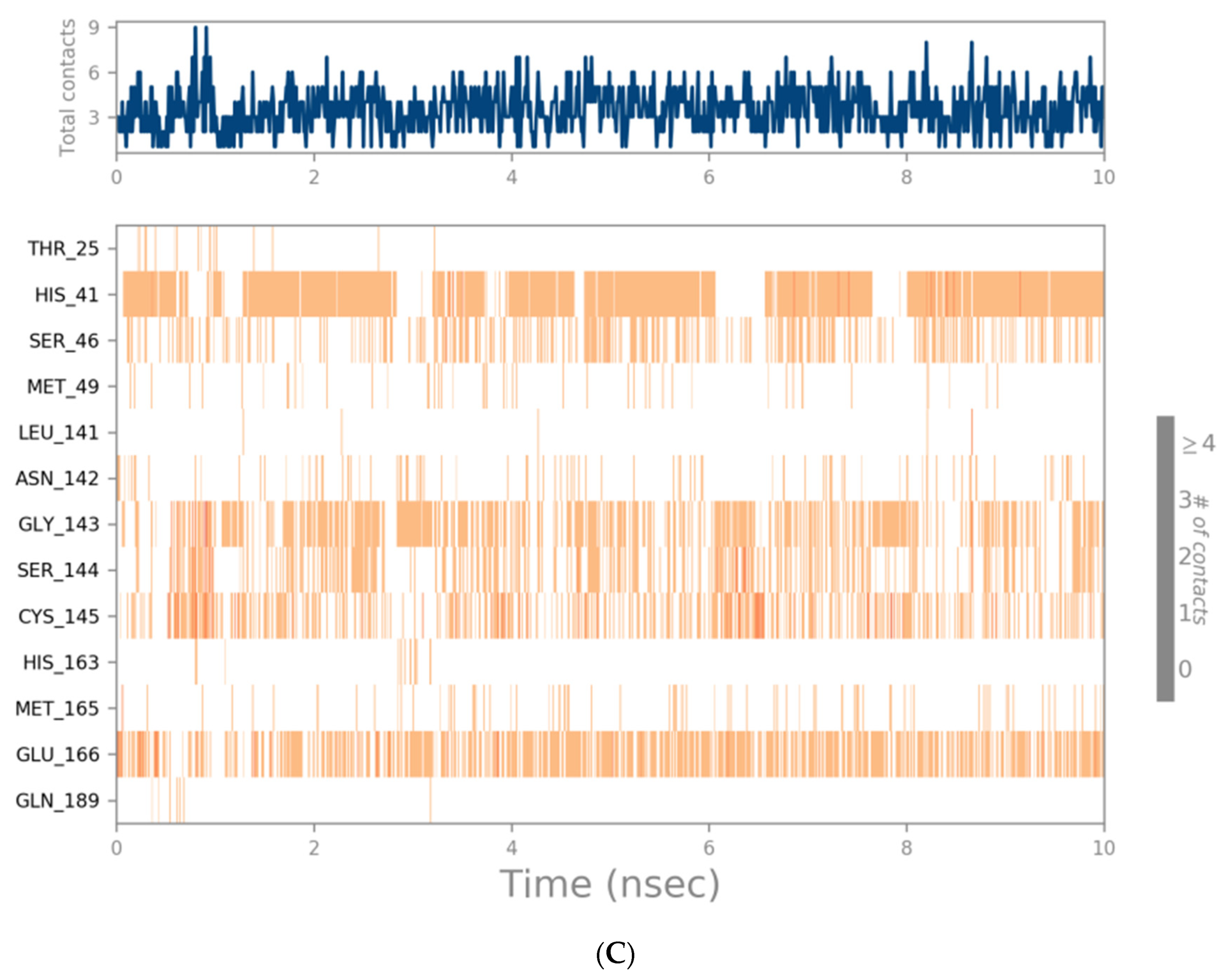
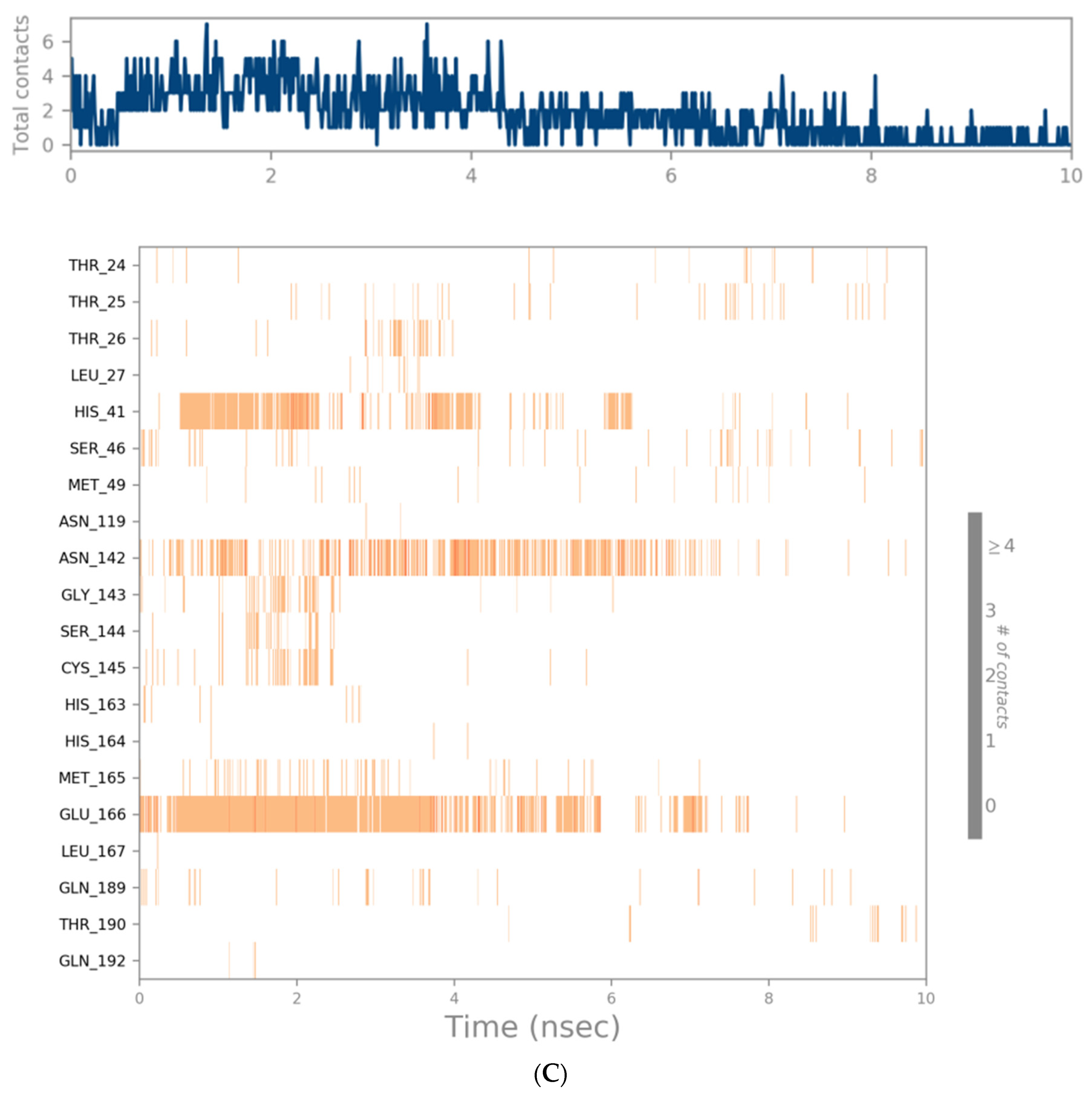
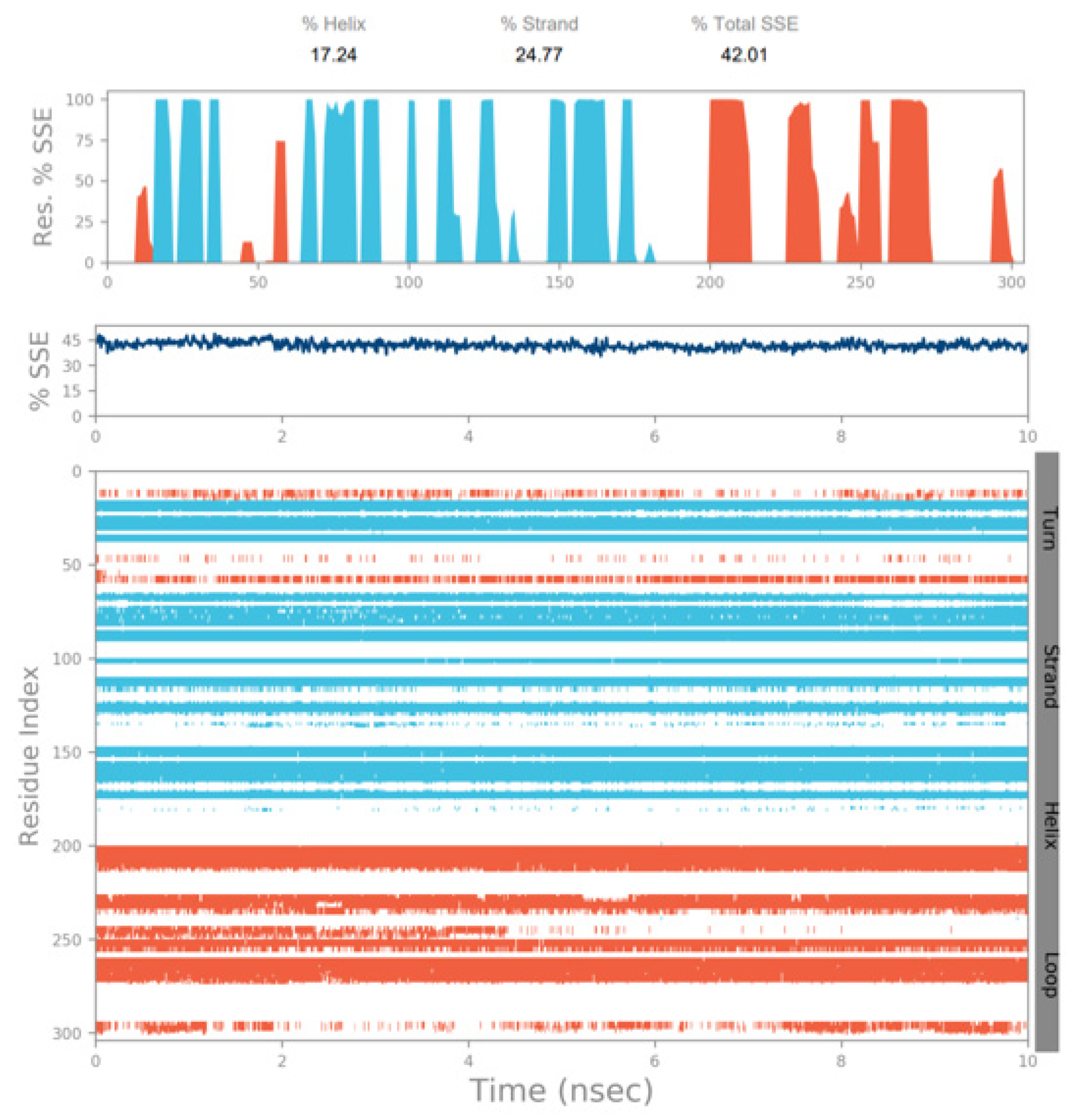
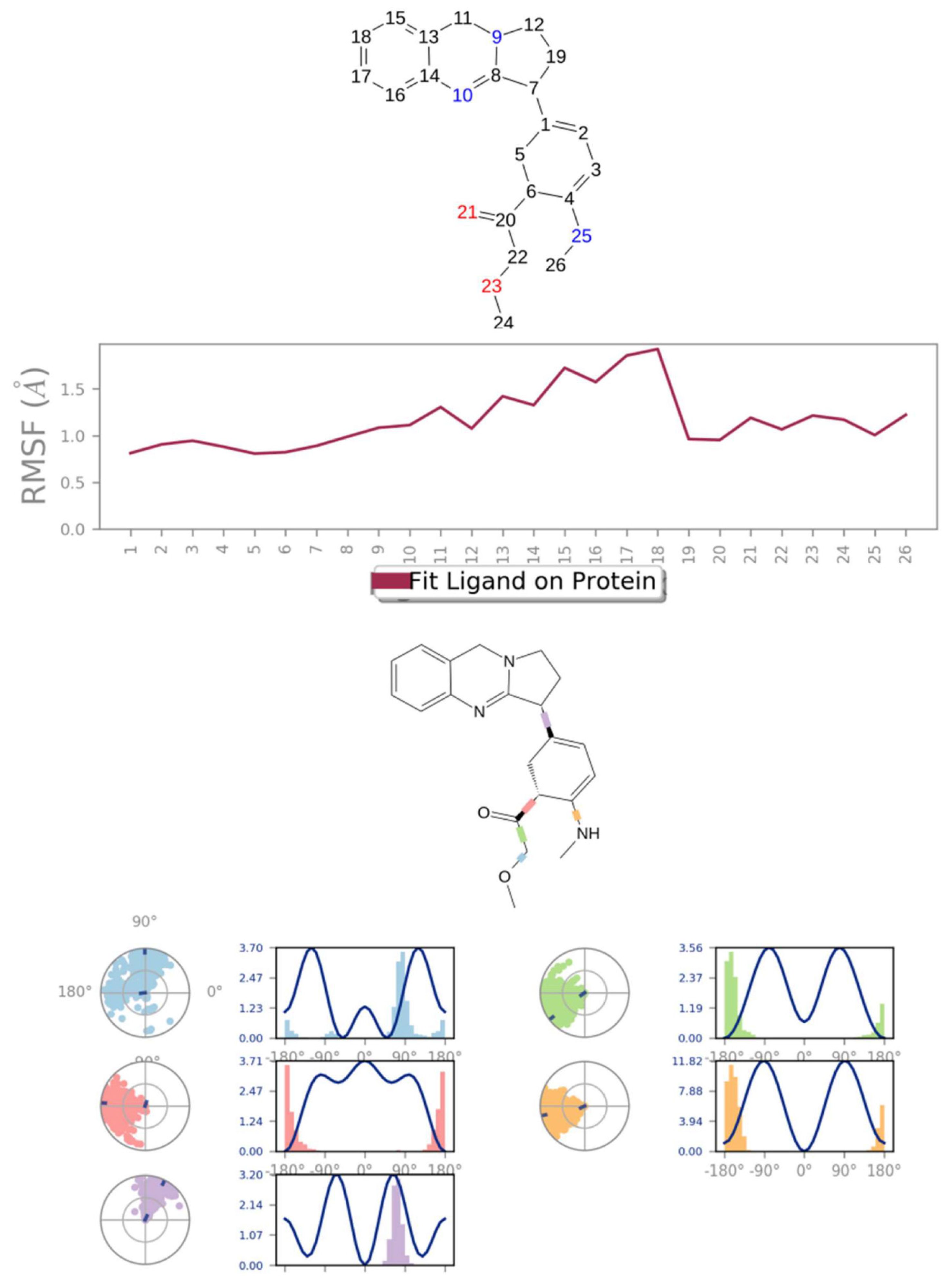
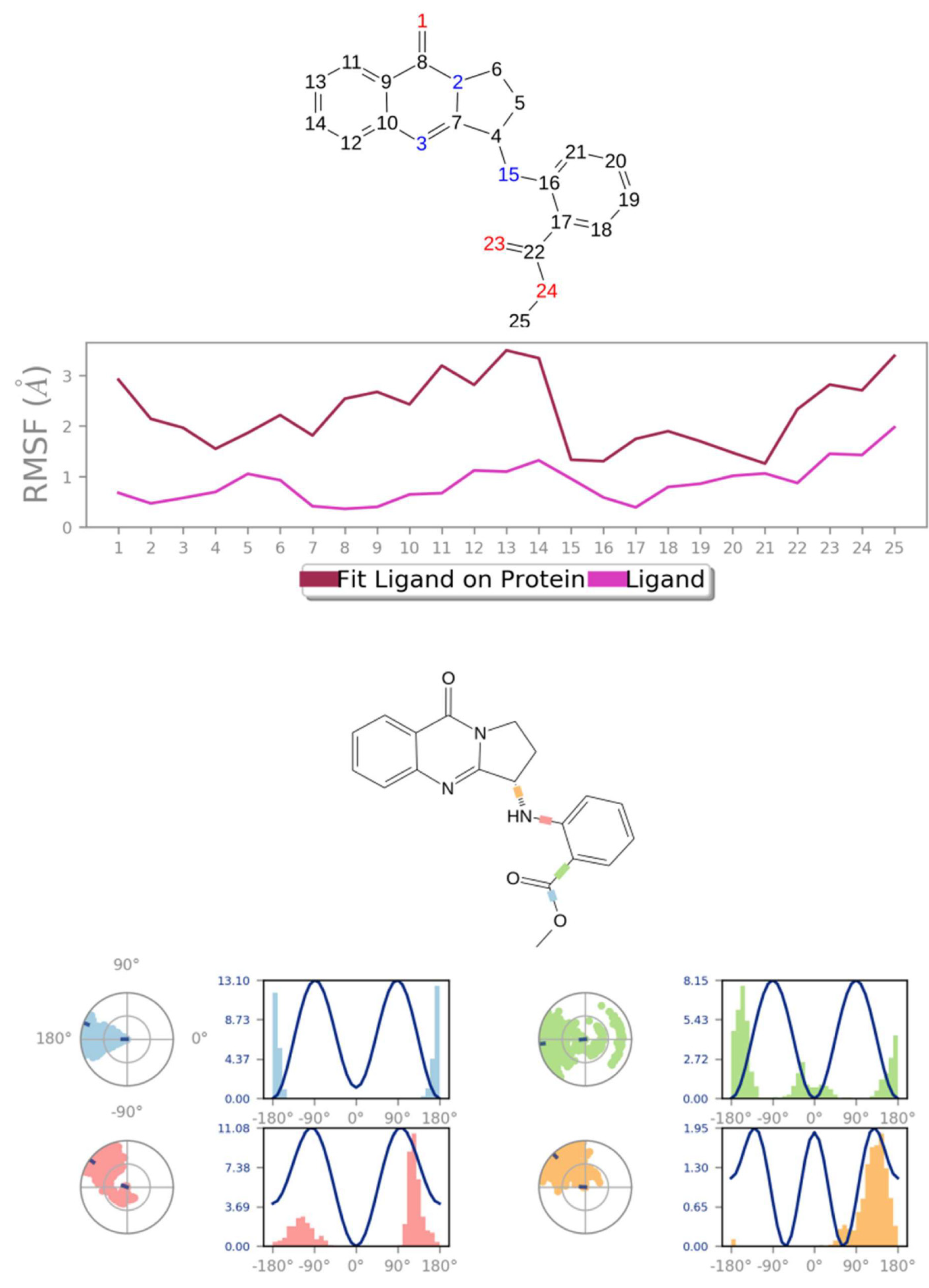
3.3. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Contini, C.; Di Nuzzo, M.; Barp, N.; Bonazza, A.; De Giorgio, R.; Tognon, M.; Rubino, S. The novel zoonotic COVID-19 pandemic: An expected global health concern. J. Infect. Dev. Ctries 2020, 14, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taefehshokr, N.; Taefehshokr, S.; Heit, B. Mechanisms of Dysregulated Humoral and Cellular Immunity by SARS-CoV-2. Pathogens 2020, 9, 1027. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 28 January 2022).
- Pallivalappil, B.; Manappallil, R.G.; Pallivalappil, S.; Josephine, B. Corona Virus disease-2019 (Covid-19) an insight into the pandemic. BMH. Medical. J. 2020, 8, 8–23. [Google Scholar]
- Bchetnia, M.; Girard, C.; Duchaine, C.; Laprise, C. The outbreak of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): A review of the current global status. J. Infect. Public Health 2020, 13, 1601–1610. [Google Scholar] [CrossRef]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and Immunotherapeutic. Sig. Transduct. Target Ther. 2020, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.; Wymant, C.; Kendall, M.; Zhao, L.; Nurtay, A.; Abeler-Dörner, L.; Parker, M.; Bonsall, D.; Fraser, C. Quantifying SARS-CoV-2 transmission suggests epidemic control with digital contact tracing. Science 2020, 368, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.T.; Hoq, M.O. Therapeutic use of Adhatoda vasica. Asian J. Med. Biol. Res. 2016, 2, 156–163. [Google Scholar] [CrossRef]
- Muthu, C.; Ayyanar, M.; Raja, N.; Ignacimuthu, S. Medicinal plants used by traditional healers in Kancheepuram district of Tamil Nadu India. J. Ethnobiol. Bio. Med. 2006, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Sivasubramanian, S. Influence of drying temperature on quality parameters of Adhatoda vasica leaves. Int. J. Life Sci. Biotechnol. Pharma Res. 2014, 2, 280–285. [Google Scholar]
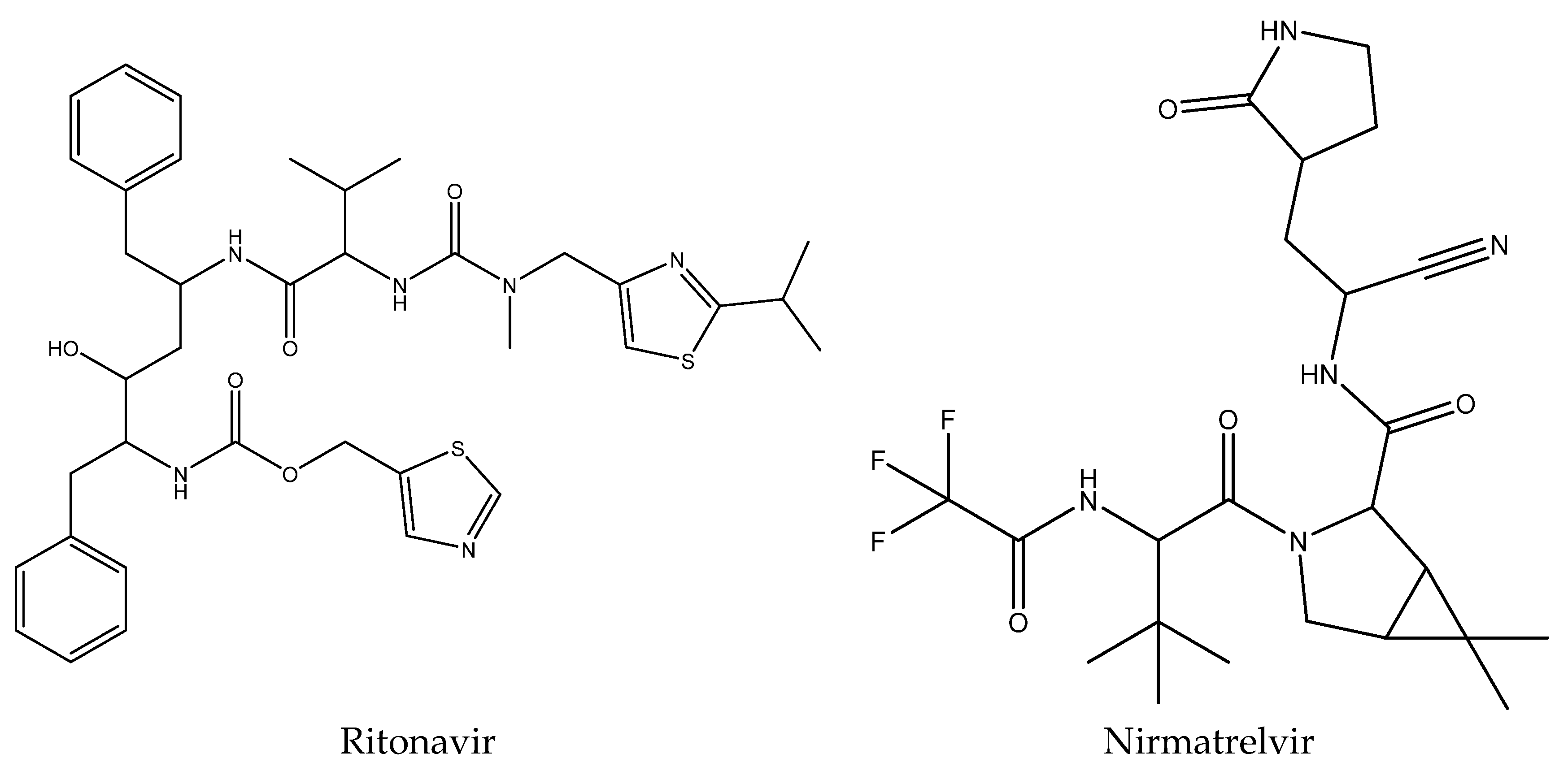
- Owen, D.R.; Allerton, C.M.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Pfizer’s Nirmatrelvir is 89% effective in patients at risk of serious illness company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef]
- Weininger, D.; Smiles, A. Chemical language and information system introduction to the methodology and encoding rules. J. Chem. Inf. Comp. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.N.; Shindyalov, I.N.; Bourne, P.E. Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurie, A.T.; Jackson, R.M. Q-Site Finder an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands applications of Autodock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Stierand, K.; Maab, P.C.; Rarey, M. Molecular complexes at a glance Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef]
- Shivanika, C.; Kumar, D.; Ragunathan, V.; Tiwari, P.; Sumitha, A. Molecular docking, validation dynamics simulations and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2020, 40, 585–611. [Google Scholar]
- Biovia, D.S. Discovery Studio Modeling Environment. 2017-Release. Available online: https://www.3ds.com/products-services/biovia (accessed on 25 January 2022).
- Sharma, A.; Vora, J.; Patel, D.; Sinha, S.; Jha, P.C.; Shrivastava, N. Identification of natural inhibitors against prime targets of SARS-CoV-2 using molecular docking molecular dynamics simulation and MM-PBSA approaches. J. Biomol. Struct. Dyn. 2020, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 2904–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umesh; Kundu, D.; Selvaraj, C.; Singh, S.K.; Dubey, V.K. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J. Biomol. Struct. Dyn. 2021, 39, 3428–3434. [Google Scholar]
- Tripathi, P.; Ghosh, S.; Talapatra, S.N. Bioavailability prediction of phytochemicals present in Calotropis procera (Aiton) R Br by using Swiss-ADME tool. World Sci. News 2019, 131, 147–163. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. Swiss ADME a free web tool to evaluate pharmacokinetics drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717–42727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, A.; Mohite, S. ADME analysis of phytochemical constituents of Psidium guajava. Asian. J. Release Chem. 2020, 13, 373–375. [Google Scholar] [CrossRef]
- Rathinavel, T.; Palanisamy, M.; Palanisamy, S.; Subramanian, A.; Thangaswamy, S. Phytochemical 6-Gingerol–A promising Drug of choice for COVID-19. Int. J. Adv. Sci. Eng. 2020, 6, 1482–1489. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | MW | iLOGP | HBD | HBA | TPSA | RB | nAH | MR |
|---|---|---|---|---|---|---|---|---|
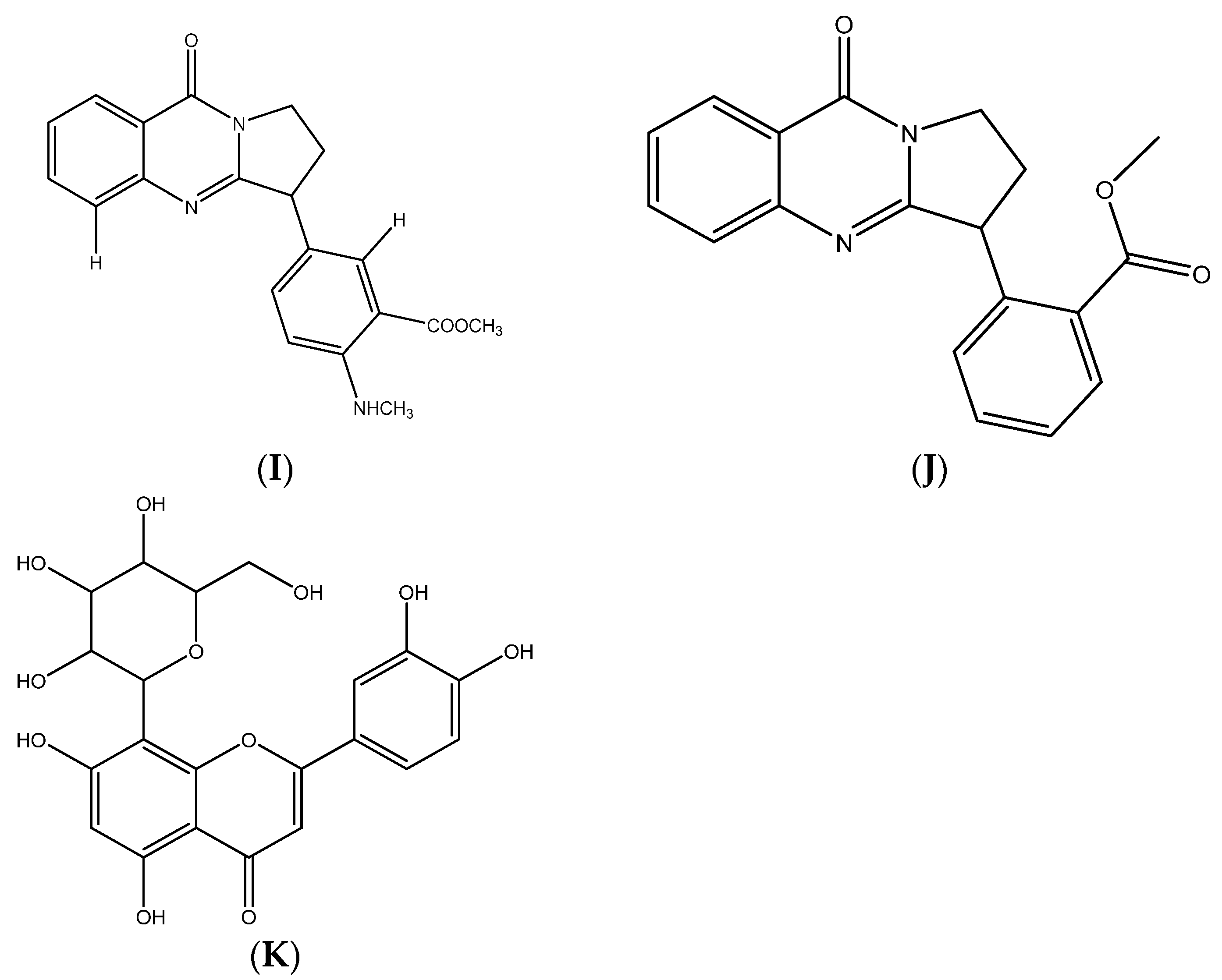
| Vasicine | 188.23 | 1.64 | 1 | 2 | 35.83 | - | 14 | 62.11 |
| Vasicinone | 202.21 | 1.61 | 1 | 3 | 55.12 | - | 15 | 56.09 |
| Vasicinolone | 218.21 | 1.52 | 2 | 4 | 75.35 | - | 16 | 58.11 |
| Vasicol | 206.24 | 1.64 | 2 | 2 | 66.56 | 2 | 15 | 61.10 |
| Vasicolinone | 305.37 | 2.86 | - | 2 | 38.13 | 2 | 23 | 93.62 |
| Adhatodine | 335.40 | 3.20 | 1 | 3 | 53.93 | 4 | 25 | 106.2 |
| Adhavasicinone | 232.24 | 1.78 | 1 | 4 | 64.35 | 1 | 7 | 62.58 |
| Aniflorine | 335.40 | 3.05 | 1 | 3 | 56.15 | 4 | 25 | 100.02 |
| Anisotine | 349.38 | 3.21 | 1 | 4 | 73.22 | 4 | 26 | 100.00 |
| Vasnetine | 320.34 | 2.91 | - | 3 | 61.19 | 3 | 24 | 90.69 |
| Orientin | 448.38 | 1.27 | 8 | 11 | 201.28 | 3 | 32 | 108.63 |
| Compounds Name | Binding Affinity (KJ/mol) | Amino Acid-Distance |
|---|---|---|
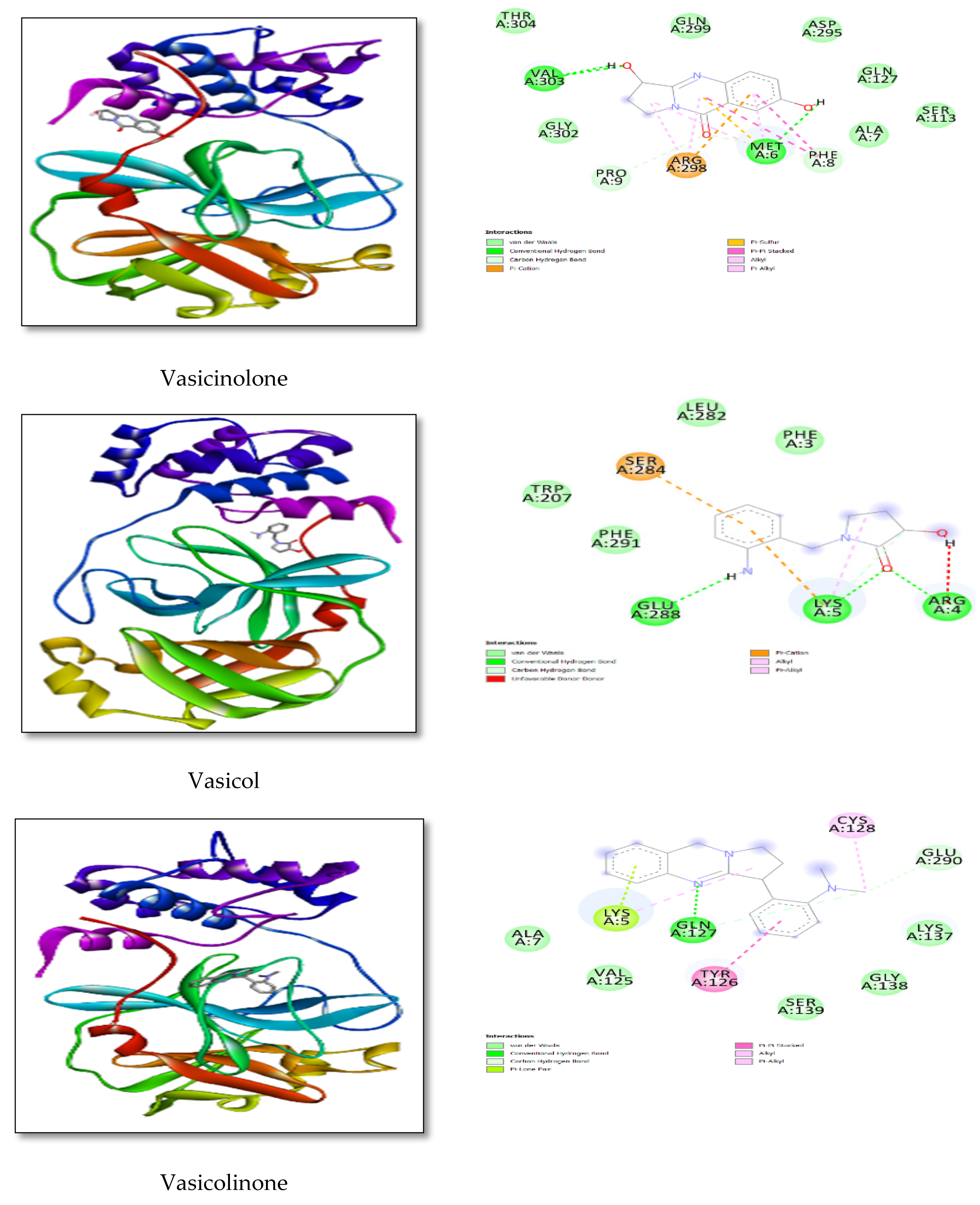
| Vasicine | −6.90 | 06-MET-3.29 |
| 07-ALA-3.20 | ||
| 295-ASP-3.59 | ||
| Vasicinone | −7.19 | 8-PHE-3.99 |
| 9-PRO-3.30 | ||
| 127-GLN-3.83 | ||
| 295-ASP-3.43 | ||
| Vasicinolone | −6.81 | 05-LYS-3.68 |
| 207-TRP-3.89 | ||
| 282-LEU-3.96 | ||
| Vasicol | −7.19 | 8-PHE-3.13 |
| 152-ILE-3.72 | ||
| 298-ARG-3.64 | ||
| Vasicolinone | −9.06 | 06-MET-3.65 |
| 08-PHE-3.68 | ||
| 09-PRO-3.84 | ||
| 152-ILE-3.65 | ||
| 298-ARG-3.32 | ||
| 303-VAL-3.97 | ||
| Adhatodine | −9.60 | 8-PHE-3.99 |
| 9-PRO-3.30 | ||
| 127-GLN-3.83 | ||
| 295-ASP-3.43 | ||
| Adhavasicinone | −6.83 | 8-PHE-3.11 |
| 291-PHE-3.11 | ||
| 295-ASP-3.02 | ||
| Aniflorine | −7.78 | 09-PRO-3.51 |
| 304-THR-3.39 | ||
| Anisotine | −8.23 | 5-LYS-3.31 |
| 288-GLU-3.88 | ||
| 291-PHE-3.15 | ||
| Vasnetine | −8.78 | 05-LYS-3.4 |
| 07-ALA-3.52 | ||
| 126-TYR-3.37 | ||
| Orientin | −6.06 | 03-PHE-2.88 |
| 04-ARG-2.93 | ||
| 05-LYS-2.78 | ||
| 288-GLU-3.75 | ||
| Ritonavir | −7.25 | 5-LYS-3.85 |
| 126-TYR-3.28 | ||
| 137-LYS-2.29 | ||
| 284-SER-3.57 | ||
| Nirmatrelvir | −8.10 | 5-LYS-3.40 |
| 128-CYS-3.65 | ||
| 291-PHE-3.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siva Kumar, B.; Anuragh, S.; Kammala, A.K.; Ilango, K. Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Life 2022, 12, 315. https://doi.org/10.3390/life12020315
Siva Kumar B, Anuragh S, Kammala AK, Ilango K. Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Life. 2022; 12(2):315. https://doi.org/10.3390/life12020315
Chicago/Turabian StyleSiva Kumar, Bathula, Singh Anuragh, Ananth Kumar Kammala, and Kaliappan Ilango. 2022. "Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme" Life 12, no. 2: 315. https://doi.org/10.3390/life12020315
APA StyleSiva Kumar, B., Anuragh, S., Kammala, A. K., & Ilango, K. (2022). Computer Aided Drug Design Approach to Screen Phytoconstituents of Adhatoda vasica as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Life, 12(2), 315. https://doi.org/10.3390/life12020315