
Regulation of Oxidative Stress by Long Non-Coding RNAs in Vascular Complications of Diabetes




Abstract
:1. Diabetes Mellitus and Oxidative Stress
2. Long Non-Coding RNA
3. Diabetic Nephropathy
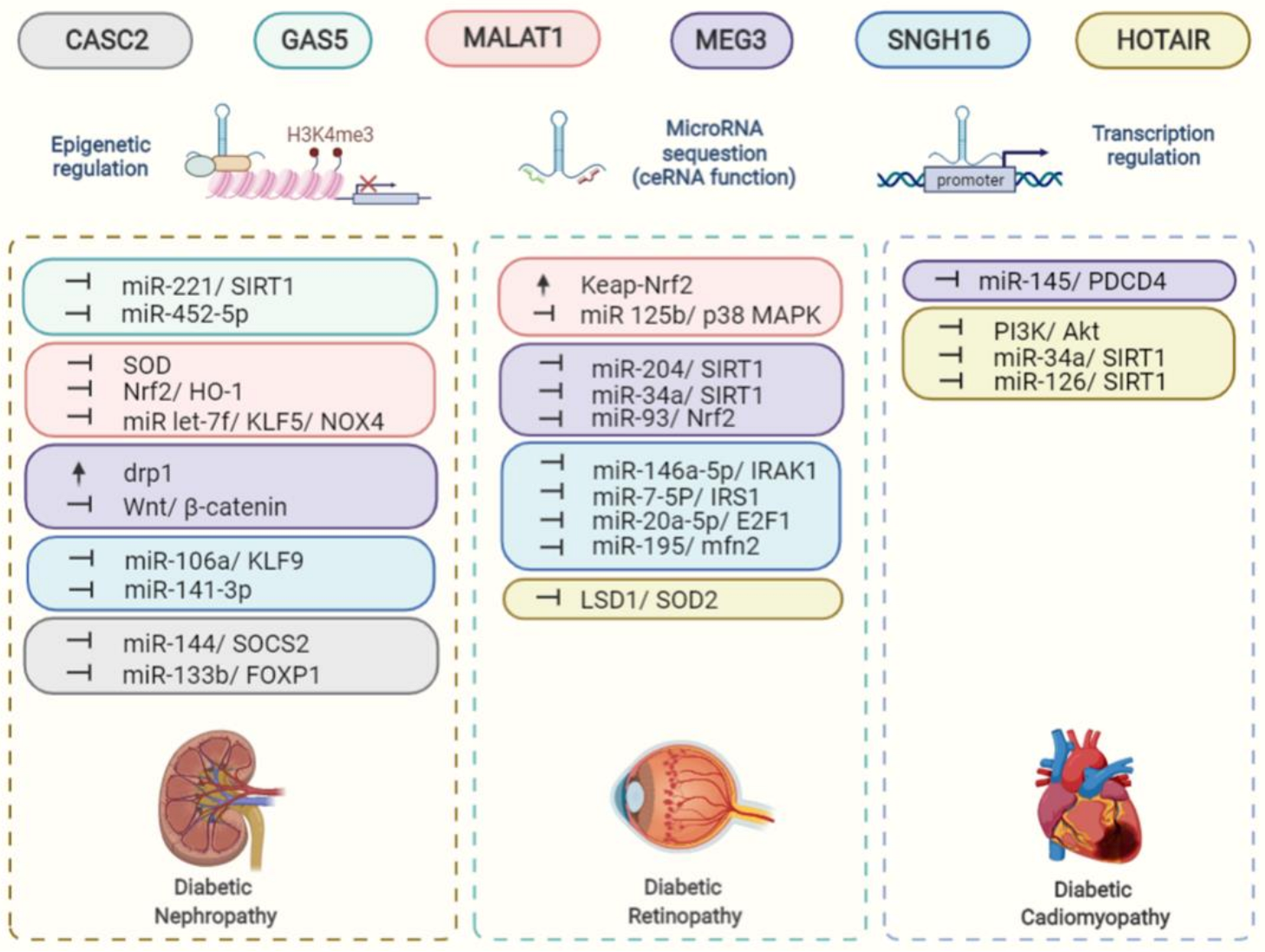
3.1. HOTAIR
3.2. MALAT1
3.3. MEG3
3.4. GAS5
3.5. SNHG16
3.6. CASC2
4. Diabetic Cardiomyopathy and Vascular Complications
4.1. HOTAIR
4.2. MEG3
5. Diabetic Retinopathy and Diabetic Cataract
5.1. HOTAIR
5.2. MALAT1
5.3. MEG3
5.4. SNHG16
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar]
- Yang, J.S.; Lu, C.C.; Kuo, S.C.; Hsu, Y.M.; Tsai, S.C.; Chen, S.Y.; Chen, Y.T.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; et al. Autophagy and its link to type II diabetes mellitus. Biomedicine 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson-Stuttard, J.; Bennett, J.; Cheng, Y.J.; Vamos, E.P.; Cross, A.J.; Ezzati, M.; Gregg, E.W. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: An epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol. 2021, 9, 165–173. [Google Scholar] [CrossRef]
- Gregg, E.W.; Cheng, Y.J.; Srinivasan, M.; Lin, J.; Geiss, L.S.; Albright, A.L.; Imperatore, G. Trends in cause-specific mortality among adults with and without diagnosed diabetes in the USA: An epidemiological analysis of linked national survey and vital statistics data. Lancet 2018, 391, 2430–2440. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. Non-coding RNAs are involved in the response to oxidative stress. Biomed. Pharmacother. 2020, 127, 110228. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Dieter, C.; Lemos, N.E.; Corrêa, N.R.F.; Assmann, T.S.; Crispim, D. The Impact of lncRNAs in Diabetes Mellitus: A Systematic Review and In Silico Analyses. Front. Endocrinol. 2021, 12, 602597. [Google Scholar] [CrossRef]
- Tello-Flores, V.A.; Beltrán-Anaya, F.O.; Ramírez-Vargas, M.A.; Esteban-Casales, B.E.; Navarro-Tito, N.; Alarcón-Romero, L.D.C.; Luciano-Villa, C.A.; Ramírez, M.; Del Moral-Hernández, Ó.; Flores-Alfaro, E. Role of Long Non-Coding RNAs and the Molecular Mechanisms Involved in Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 7256. [Google Scholar] [CrossRef]
- Molitch, M.E.; DeFronzo, R.A.; Franz, M.J.; Keane, W.F.; Mogensen, C.E.; Parving, H.H.; Steffes, M.W. Nephropathy in diabetes. Diabetes Care 2004, 27 (Suppl. 1), S79–S83. [Google Scholar]
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vasc. Pharmacol. 2013, 58, 259–271. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Majumder, S.; Hadden, M.J.; Thieme, K.; Batchu, S.N.; Niveditha, D.; Chowdhury, S.; Yerra, V.G.; Advani, S.L.; Bowskill, B.B.; Liu, Y.; et al. Dysregulated expression but redundant function of the long non-coding RNA HOTAIR in diabetic kidney disease. Diabetologia 2019, 62, 2129–2142. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Wang, R.; Li, X.; Fan, M.; Lin, J.; Zhen, J.; Chen, L.; Lv, Z. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with β-catenin. J. Cell. Mol. Med. 2017, 21, 2732–2747. [Google Scholar] [CrossRef]
- Zhou, L.J.; Yang, D.W.; Ou, L.N.; Guo, X.R.; Wu, B.L. Circulating Expression Level of LncRNA Malat1 in Diabetic Kidney Disease Patients and Its Clinical Significance. J. Diabetes Res. 2020, 2020, 4729019. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, Y.; Hu, Q.; Zhang, X. LncRNA MALAT1/microRNA let-7f/KLF5 axis regulates podocyte injury in diabetic nephropathy. Life Sci. 2021, 266, 118794. [Google Scholar] [CrossRef]
- Kyriazis, I.D.; Hoffman, M.; Gaignebet, L.; Lucchese, A.M.; Markopoulou, E.; Palioura, D.; Wang, C.; Bannister, T.D.; Christofidou-Solomidou, M.; Oka, S.I.; et al. KLF5 Is Induced by FOXO1 and Causes Oxidative Stress and Diabetic Cardiomyopathy. Circ. Res. 2021, 128, 335–357. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, L.; He, X.; Ye, Z.; Li, L.; Liu, Z.; Zhou, S. Atorvastatin Regulates MALAT1/miR-200c/NRF2 Activity to Protect Against Podocyte Pyroptosis Induced by High Glucose. Diabetes Metab. Syndr. Obes. 2021, 14, 1631–1645. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Schmidt, J.V.; Matteson, P.G.; Jones, B.K.; Guan, X.J.; Tilghman, S.M. The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 2000, 14, 1997–2002. [Google Scholar] [CrossRef]
- Kagami, M.; Sekita, Y.; Nishimura, G.; Irie, M.; Kato, F.; Okada, M.; Yamamori, S.; Kishimoto, H.; Nakayama, M.; Tanaka, Y.; et al. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat. Genet. 2008, 40, 237–242. [Google Scholar] [CrossRef]
- Cheng, X.; Ali, M.S.S.H.; Moran, M.; Viana, M.P.; Schlichte, S.L.; Zimmerman, M.C.; Khalimonchuk, O.; Feinberg, M.W.; Sun, X. Long non-coding RNA Meg3 deficiency impairs glucose homeostasis and insulin signaling by inducing cellular senescence of hepatic endothelium in obesity. Redox Biol. 2021, 40, 101863. [Google Scholar] [CrossRef]
- Deng, Q.; Wen, R.; Liu, S.; Chen, X.; Song, S.; Li, X.; Su, Z.; Wang, C. Increased long noncoding RNA maternally expressed gene 3 contributes to podocyte injury induced by high glucose through regulation of mitochondrial fission. Cell Death Dis. 2020, 11, 814. [Google Scholar] [CrossRef]
- Zha, F.; Qu, X.; Tang, B.; Li, J.; Wang, Y.; Zheng, P.; Ji, T.; Zhu, C.; Bai, S. Long non-coding RNA MEG3 promotes fibrosis and inflammatory response in diabetic nephropathy via miR-181a/Egr-1/TLR4 axis. Aging 2019, 11, 3716–3730. [Google Scholar] [CrossRef]
- Li, J.; Jiang, X.; Duan, L.; Wang, W. Long non-coding RNA MEG3 impacts diabetic nephropathy progression through sponging miR-145. Am. J. Transl. Res. 2019, 11, 6691–6698. [Google Scholar]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Li, T.; Wei, J.; Zhang, Y.; Zuo, Q.; Lin, Y. Identification of miR-145 as a regulator of the cardiomyocyte inflammatory response and oxidative stress under hyperglycemia. Exp. Ther. Med. 2021, 21, 467. [Google Scholar] [CrossRef]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef] [Green Version]
- Che, X.; Deng, X.; Xie, K.; Wang, Q.; Yan, J.; Shao, X.; Ni, Z.; Ying, L. Long noncoding RNA MEG3 suppresses podocyte injury in diabetic nephropathy by inactivating Wnt/β-catenin signaling. PeerJ 2019, 7, e8016. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; King, R.M.; Philipson, L. Genes specifically expressed at growth arrest of mammalian cells. Cell 1988, 54, 787–793. [Google Scholar] [CrossRef]
- Xie, C.; Wu, W.; Tang, A.; Luo, N.; Tan, Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes Metab. Syndr. Obes. 2019, 12, 2609–2617. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Xu, B.; Xu, W.; Xia, L.; Xu, Z.; Shen, L.; Peng, W.; Huang, S. Long noncoding RNA GAS5 inhibits cell proliferation and fibrosis in diabetic nephropathy by sponging miR-221 and modulating SIRT1 expression. Aging 2019, 11, 8745–8759. [Google Scholar] [CrossRef]
- Wang, W.; Jia, Y.-J.; Yang, Y.-L.; Xue, M.; Zheng, Z.-J.; Wang, L.; Xue, Y.-M. LncRNA GAS5 exacerbates renal tubular epithelial fibrosis by acting as a competing endogenous RNA of miR-96-5p. Biomed. Pharmacother. 2020, 121, 109411. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, S.; Zhu, Y. Long noncoding RNA growth arrest-specific transcript 5 alleviates renal fibrosis in diabetic nephropathy by downregulating matrix metalloproteinase 9 through recruitment of enhancer of zeste homolog 2. FASEB J. 2020, 34, 2703–2714. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Meng, T.; Qin, W.; Liu, B. SIRT1 Antagonizes Oxidative Stress in Diabetic Vascular Complication. Front. Endocrinol. 2020, 11, 568861. [Google Scholar] [CrossRef]
- Zimta, A.-A.; Tigu, A.B.; Braicu, C.; Stefan, C.; Ionescu, C.; Berindan-Neagoe, I. An Emerging Class of Long Non-coding RNA With Oncogenic Role Arises From the snoRNA Host Genes. Front. Oncol. 2020, 10, 389. [Google Scholar] [CrossRef]
- Yu, M.; Ohira, M.; Li, Y.; Niizuma, H.; Oo, M.L.; Zhu, Y.; Ozaki, T.; Isogai, E.; Nakamura, Y.; Koda, T.; et al. High expression of ncRAN, a novel non-coding RNA mapped to chromosome 17q25.1, is associated with poor prognosis in neuroblastoma. Int. J. Oncol. 2009, 34, 931–938. [Google Scholar]
- He, X.; Zeng, X. LncRNA SNHG16 Aggravates High Glucose-Induced Podocytes Injury in Diabetic Nephropathy Through Targeting miR-106a and Thereby Up-Regulating KLF9. Diabetes Metab. Syndr. Obes. 2020, 13, 3551–3560. [Google Scholar] [CrossRef]
- Jiang, X.; Ru, Q.; Li, P.; Ge, X.; Shao, K.; Xi, L.; Xu, B.; Wang, Q.; Huang, S. LncRNA SNHG16 induces proliferation and fibrogenesis via modulating miR-141-3p and CCND1 in diabetic nephropathy. Gene Ther. 2020, 27, 557–566. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Baldinu, P.; Cossu, A.; Manca, A.; Satta, M.P.; Sini, M.C.; Rozzo, C.; Dessole, S.; Cherchi, P.; Gianfrancesco, F.; Pintus, A.; et al. Identification of a novel candidate gene, CASC2, in a region of common allelic loss at chromosome 10q26 in human endometrial cancer. Hum. Mutat. 2004, 23, 318–326. [Google Scholar] [CrossRef]
- Palmieri, G.; Paliogiannis, P.; Sini, M.C.; Manca, A.; Palomba, G.; Doneddu, V.; Tanda, F.; Pascale, M.R.; Cossu, A. Long non-coding RNA CASC2 in human cancer. Crit. Rev. Oncol. Hematol. 2017, 111, 31–38. [Google Scholar] [CrossRef]
- Yang, H.; Kan, Q.E.; Su, Y.; Man, H. Long Non-Coding RNA CASC2 Improves Diabetic Nephropathy by Inhibiting JNK Pathway. Exp. Clin. Endocrinol. Diabetes 2019, 127, 533–537. [Google Scholar] [CrossRef]
- Min, X.Q.; Xie, Y. LncRNA CASC2 Alleviates the Progression of Diabetic Nephropathy by Regulating the miR-144/SOCS2 Signalling Axis. Kidney Blood Press. Res. 2020, 45, 837–849. [Google Scholar] [CrossRef]
- Li, F.; Dai, B.; Ni, X. Long non-coding RNA cancer susceptibility candidate 2 (CASC2) alleviates the high glucose-induced injury of CIHP-1 cells via regulating miR-9-5p/PPARγ axis in diabetes nephropathy. Diabetol. Metab. Syndr. 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-L.; Zhu, H.-Q.; Zhang, Y.; Zhang, C.-Y.; Jiao, J.-S.; Xing, X.-Y. LncRNA CASC2 regulates high glucose-induced proliferation, extracellular matrix accumulation and oxidative stress of human mesangial cells via miR-133b/FOXP1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 802–812. [Google Scholar] [PubMed]
- Monti-Rocha, R.; Cramer, A.; Leite, P.G.; Antunes, M.M.; Pereira, R.V.S.; Barroso, A.; Queiroz-Junior, C.M.; David, B.A.; Teixeira, M.M.; Menezes, G.B.; et al. SOCS2 Is Critical for the Balancing of Immune Response and Oxidate Stress Protecting Against Acetaminophen-Induced Acute Liver Injury. Front. Immunol. 2018, 9, 3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.; Xue, W.; Wu, X.; Zheng, J.; Ding, C.; Li, Y.; Dou, M. FOXP1 inhibits high glucose-induced ECM accumulation and oxidative stress in mesangial cells. Chem. Biol. Interact. 2019, 313, 108818. [Google Scholar] [CrossRef]
- Tarquini, R.; Lazzeri, C.; Pala, L.; Rotella, C.M.; Gensini, G.F. The diabetic cardiomyopathy. Acta Diabetol. 2011, 48, 173–181. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Yao, B.; Xu, W.; Chen, J.; Zhou, X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci. Rep. 2016, 6, 36340. [Google Scholar] [CrossRef] [Green Version]
- Kuster, G.M.; Lancel, S.; Zhang, J.; Communal, C.; Trucillo, M.P.; Lim, C.C.; Pfister, O.; Weinberg, E.O.; Cohen, R.A.; Liao, R.; et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 2010, 48, 1182–1187. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; DeMarco, V.G.; Lee, L.E.; Ma, L.; Barron, B.J.; Whaley-Connell, A.; Sowers, J.R. Enhanced endothelium epithelial sodium channel signaling prompts left ventricular diastolic dysfunction in obese female mice. Metabolism 2018, 78, 69–79. [Google Scholar] [CrossRef]
- Qu, X.; Alsager, S.; Zhuo, Y.; Shan, B. HOX transcript antisense RNA (HOTAIR) in cancer. Cancer Lett. 2019, 454, 90–97. [Google Scholar] [CrossRef]
- Qi, K.; Zhong, J. LncRNA HOTAIR improves diabetic cardiomyopathy by increasing viability of cardiomyocytes through activation of the PI3K/Akt pathway. Exp. Ther. Med. 2018, 16, 4817–4823. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.C.; Zhang, Y.F.; Liu, S.S.; Cheng, X.J.; Yang, X.; Cui, X.G.; Zhao, X.R.; Zhao, H.; Hao, M.F.; Li, M.D.; et al. Curcumin alleviates oxidative stress and inhibits apoptosis in diabetic cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt signalling pathways. J. Cell. Mol. Med. 2020, 24, 12355–12367. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L. Calycosin ameliorates diabetes-induced cognitive impairments in rats by reducing oxidative stress via the PI3K/Akt/GSK-3β signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 428–434. [Google Scholar] [CrossRef]
- Gao, L.; Wang, X.; Guo, S.; Xiao, L.; Liang, C.; Wang, Z.; Li, Y.; Liu, Y.; Yao, R.; Liu, Y.; et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J. Cell. Physiol. 2019, 234, 4944–4958. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, C.; Zhu, S.Y.; Zou, H.C.; Xu, C.Y.; Chen, Y.X. Overexpression of HOTAIR attenuates Pi-induced vascular calcification by inhibiting Wnt/β-catenin through regulating miR-126/Klotho/SIRT1 axis. Mol. Cell. Biochem. 2021, 476, 3551–3561. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Z.; Zhu, D.; Zhao, W.; Li, F. Long non-coding RNA MEG3 serves as a ceRNA for microRNA-145 to induce apoptosis of AC16 cardiomyocytes under high glucose condition. Biosci. Rep. 2019, 39, BSR20190444. [Google Scholar] [CrossRef] [Green Version]
- Hui, Y.; Yin, Y. MicroRNA-145 attenuates high glucose-induced oxidative stress and inflammation in retinal endothelial cells through regulating TLR4/NF-κB signaling. Life Sci. 2018, 207, 212–218. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R. Retinopathy in diabetes. Diabetes Care 2004, 27 (Suppl. 1), S84–S87. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.A.; Biswas, S.; Feng, B.; Chen, S.; Gonder, J.; Chakrabarti, S. lncRNA H19 prevents endothelial-mesenchymal transition in diabetic retinopathy. Diabetologia 2019, 62, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Xiao, F.; Wang, P.; Hu, Y.X. lncRNA H19 sponging miR-93 to regulate inflammation in retinal epithelial cells under hyperglycemia via XBP1s. Inflamm. Res. 2020, 69, 255–265. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Tang, J.; Kern, T.S. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes 2001, 50, 1938–1942. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Koppolu, P.; Chakrabarti, S.; Chen, S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic. Res. 2003, 37, 1169–1180. [Google Scholar] [CrossRef]
- Shaker, O.G.; Abdelaleem, O.O.; Mahmoud, R.H.; Abdelghaffar, N.K.; Ahmed, T.I.; Said, O.M.; Zaki, O.M. Diagnostic and prognostic role of serum miR-20b, miR-17-3p, HOTAIR, and MALAT1 in diabetic retinopathy. IUBMB Life 2019, 71, 310–320. [Google Scholar] [CrossRef]
- Biswas, S.; Feng, B.; Chen, S.; Liu, J.; Aref-Eshghi, E.; Gonder, J.; Ngo, V.; Sadikovic, B.; Chakrabarti, S. The Long Non-Coding RNA HOTAIR Is a Critical Epigenetic Mediator of Angiogenesis in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 20. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, Y.; Wang, J.; Wu, L.; Liu, Y.; Zhao, S.; Guo, F.; Ma, X.; Zhang, H.; Li, Z.; et al. Long noncoding RNA Hotair facilitates retinal endothelial cell dysfunction in diabetic retinopathy. Clin. Sci. 2020, 134, 2419–2434. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: Role of histone methylation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Tao, Z.F.; Li, X.M.; Zhang, H.; Yao, J.; Jiang, Q. Aberrant expression of long noncoding RNAs in early diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ke, S.; Zhong, L.; Wu, J.; Tseng, A.; Morpurgo, B.; Golovko, A.; Wang, G.; Cai, J.J.; Ma, X.; et al. Long noncoding RNA MALAT1 regulates generation of reactive oxygen species and the insulin responses in male mice. Biochem. Pharmacol. 2018, 152, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, R.; Kowluru, R.A. Long Noncoding RNA MALAT1 and Regulation of the Antioxidant Defense System in Diabetic Retinopathy. Diabetes 2021, 70, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jia, S.B.; Shi, J.M.; Li, W.J.; Tang, L.S.; Zhu, X.H.; Tong, P. LncRNA-MALAT1 promotes neovascularization in diabetic retinopathy through regulating miR-125b/VE-cadherin axis. Biosci. Rep. 2019, 39, BSR20181469. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Xu, F.; Zhang, C.; Shuai, J.; Huang, Z.; Liang, Y.; Xu, X. Dexmedetomidine exerts neuroprotective effects during high glucose-induced neural injury by inhibiting miR-125b. Biosci. Rep. 2020, 40, BSR20200394. [Google Scholar] [CrossRef]
- Gong, W.; Zhu, G.; Li, J.; Yang, X. LncRNA MALAT1 promotes the apoptosis and oxidative stress of human lens epithelial cells via p38MAPK pathway in diabetic cataract. Diabetes Res. Clin. Pract. 2018, 144, 314–321. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Tian, W.; Fu, H.T.; Li, C.P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Tu, Y.; Zhu, M.; Wang, Z.; Wang, K.; Chen, L.; Liu, W.; Shi, Q.; Zhao, Q.; Sun, Y.; Wang, X.; et al. Melatonin inhibits Müller cell activation and pro-inflammatory cytokine production via upregulating the MEG3/miR-204/Sirt1 axis in experimental diabetic retinopathy. J. Cell. Physiol. 2020, 235, 8724–8735. [Google Scholar] [CrossRef]
- Tu, Y.; Song, E.; Wang, Z.; Ji, N.; Zhu, L.; Wang, K.; Sun, H.; Zhang, Y.; Zhu, Q.; Liu, X.; et al. Melatonin attenuates oxidative stress and inflammation of Müller cells in diabetic retinopathy via activating the Sirt1 pathway. Biomed. Pharmacother. 2021, 137, 111274. [Google Scholar] [CrossRef]
- Luo, R.; Jin, H.; Li, L.; Hu, Y.X.; Xiao, F. Long Noncoding RNA MEG3 Inhibits Apoptosis of Retinal Pigment Epithelium Cells Induced by High Glucose via the miR-93/Nrf2 Axis. Am. J. Pathol. 2020, 190, 1813–1822. [Google Scholar] [CrossRef]
- Tong, P.; Peng, Q.H.; Gu, L.M.; Xie, W.W.; Li, W.J. LncRNA-MEG3 alleviates high glucose induced inflammation and apoptosis of retina epithelial cells via regulating miR-34a/SIRT1 axis. Exp. Mol. Pathol. 2019, 107, 102–109. [Google Scholar] [CrossRef]
- Cai, F.; Jiang, H.; Li, Y.; Li, Q.; Yang, C. Upregulation of long non-coding RNA SNHG16 promotes diabetes-related RMEC dysfunction via activating NF-κB and PI3K/AKT pathways. Mol. Ther. Nucleic Acids 2021, 24, 512–527. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, X.; Jiang, L.; Xia, W.; Li, H.; Zhao, N.; Cui, X.; Zhang, N.; Zhou, H.; Xu, S. Decreased lncRNA SNHG16 Accelerates Oxidative Stress Induced Pathological Angiogenesis in Human Retinal Microvascular Endothelial Cells by Regulating miR-195/mfn2 Axis. Curr. Pharm. Des. 2021, 27, 3047–3060. [Google Scholar] [CrossRef]
- Li, X.; Guo, C.; Chen, Y.; Yu, F. Long non-coding RNA SNHG16 regulates E2F1 expression by sponging miR-20a-5p and aggravating proliferative diabetic retinopathy. Can. J. Physiol. Pharmacol. 2021, 99, 1207–1216. [Google Scholar] [CrossRef]
- Blanchet, E.; Annicotte, J.S.; Lagarrigue, S.; Aguilar, V.; Clapé, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, Y.; Xiao, L.; Zheng, S.; Yan, N.; Chen, D. E2f1 mediates high glucose-induced neuronal death in cultured mouse retinal explants. Cell Cycle 2017, 16, 1824–1834. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef]

{kind=link}
| Diabetic Complications | LncRNA | Expression Level | Cells or Tissues | Interacting Factors/Pathways | Ref. |
|---|---|---|---|---|---|
| Diabetic Nephropathy | MALAT1 | Upregulated | Kidney cortices in STZ-injected C57BL/6 mice | [18] | |
| Diabetic Nephropathy | MALAT1 | Patients with DN | SOD | [19] | |
| Diabetic Nephropathy | MALAT1 | Mouse podocyte MPC-5 cells | KLF5 | [20] | |
| Diabetic Nephropathy | MALAT1 | Mouse podocyte MPC-5 cells | Nrf2/HO-1 signaling | [22] | |
| Diabetic Nephropathy | MEG3 | Upregulated | Podocytes of STZ-induced mice | drp1 | [27] |
| Diabetic Nephropathy | MEG3 | Upregulated | HG-treated mesangial cells | miR-181a | [28] |
| Diabetic Nephropathy | MEG3 | Upregulated | Serum of patients with DN | miR-145 | [29] |
| Diabetic Nephropathy | MEG3 | Downregulated | Renal tissues of patients with DN and HG-treated podocytes | Wnt/β-catenin signaling | [34] |
| Diabetic Nephropathy | GAS5 | Downregulated | HG-induced proximal tubular cells | miR-452-5p/SOD axis | [36] |
| Diabetic Nephropathy | GAS5 | Downregulated | HG-treated mesangial cells | miR-221/SIRT1 axis | [37] |
| Diabetic Nephropathy | GAS5 | Upregulated | Kidneys of the HFD/ STZ-induced diabetic mice | [38] | |
| Diabetic Nephropathy | SNHG16 | Upregulated | Serum of DN patients and HG-treated podocytes | miR-106a/KLF9 axis | [44] |
| Diabetic Nephropathy | SNHG16 | Upregulated | HG-treated mesangial cells | [45] | |
| Diabetic Nephropathy | CASC2 | Downregulated | Serum of DN patients and podocyte cells | [49] | |
| Diabetic Nephropathy | CASC2 | Downregulated | Tissues in db/db diabetic mouse and HG-treated mesangial cells and podocytes | [50] | |
| Diabetic Nephropathy | CASC2 | Downregulated | HG-treated podocyte cells | miR-133b/FOXP1 axis | [52] |
| Diabetic Cardiomyopathy | HOTAIR | Downregulated | Myocardial tissues and serum of diabetic cardiomyopathy patients | [61] | |
| Diabetic Cardiomyopathy | HOTAIR | Downregulated | Diabetic hearts in STZ-injected C57/B6 mice andHG-stimulated H9c2 cells | miR-34a/SIRT1 axis | [64] |
| Diabetic Cardiomyopathy | MEG3 | Upregulated | HG-treated cardiomyocytes | miR-145 | [66] |
| Diabetic Retinopathy | HOTAIR | Upregulated | HG-treated human retinal endothelial cells | [74] | |
| Diabetic Retinopathy | HOTAIR | HG-treated human retinal endothelial cells | LSD1/MnSOD axis | [75,76] | |
| Diabetic Retinopathy | MALAT1 | Upregulated | Diabetic retinas using clinical samples, STZ-induced type I DM mice | [77] | |
| Diabetic Retinopathy | MALAT1 | db/db type 2 DM mice | [78] | ||
| Diabetic Retinopathy | MALAT1 | Keap1-Nrf2 axis | [79,80] | ||
| Diabetic Retinopathy | MALAT1 | HG-treated human lens epithelial cells | SP1 and p38MAPK pathway | [83] | |
| Diabetic Retinopathy | MEG3 | Downregulated | HG-treated Müller cells | miR-204/SIRT1 axis | [85] |
| Diabetic Retinopathy | MEG3 | HG-treated retinal pigment epithelium cells | miR-93/Nrf2 axis | [87] | |
| Diabetic Retinopathy | MEG3 | HG-treated retina epithelial cells | miR-34a/SIRT1 axis | [88] | |
| Diabetic Retinopathy | SNHG16 | Upregulated | HG-treated retinal microvascular endothelial cells | [89] | |
| Diabetic Retinopathy | SNHG16 | HG-treated retinal microvascular endothelial cells | miR-195/mfn2 axis | [90] | |
| Diabetic Retinopathy | SNHG16 | HG-treated retinal microvascular endothelial cells | miR-20a-5p/E2F1 axis | [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, P.-M.; Yu, C.-C.; Tsai, K.-L.; Hsieh, P.-L. Regulation of Oxidative Stress by Long Non-Coding RNAs in Vascular Complications of Diabetes. Life 2022, 12, 274. https://doi.org/10.3390/life12020274
Chu P-M, Yu C-C, Tsai K-L, Hsieh P-L. Regulation of Oxidative Stress by Long Non-Coding RNAs in Vascular Complications of Diabetes. Life. 2022; 12(2):274. https://doi.org/10.3390/life12020274
Chicago/Turabian StyleChu, Pei-Ming, Cheng-Chia Yu, Kun-Ling Tsai, and Pei-Ling Hsieh. 2022. "Regulation of Oxidative Stress by Long Non-Coding RNAs in Vascular Complications of Diabetes" Life 12, no. 2: 274. https://doi.org/10.3390/life12020274
APA StyleChu, P.-M., Yu, C.-C., Tsai, K.-L., & Hsieh, P.-L. (2022). Regulation of Oxidative Stress by Long Non-Coding RNAs in Vascular Complications of Diabetes. Life, 12(2), 274. https://doi.org/10.3390/life12020274