
Searching for Low Probability Opening Events in a DNA Sliding Clamp



Abstract
:
1. Introduction
2. Materials and Methods
2.1. All Atom Simulations
2.2. Coarse-Grained Simulations
2.3. Basics of Replica Exchange Molecular Dynamics and MELD
2.4. MELD Setup
2.5. Indicators for Conformational Fluctuations
3. Results
3.1. Validation of the Methods
3.1.1. Coarse-Grained Simulations of Three Transcription Factors Bound to DNA Remain Bound throughout the Simulation Timescale
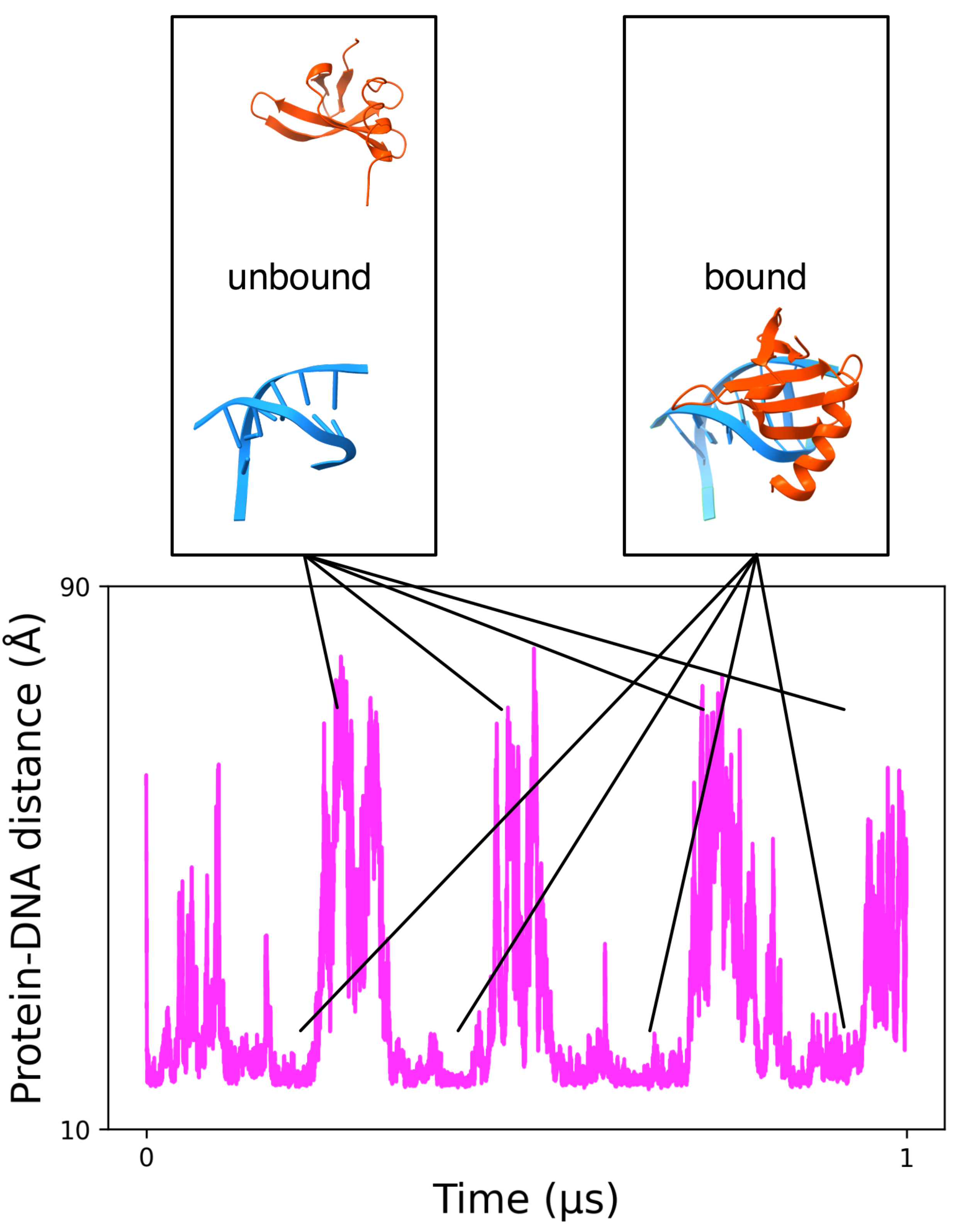
3.1.2. MELD Simulations Capture Multiple Binding/Unbinding Events of Proteins to DNA
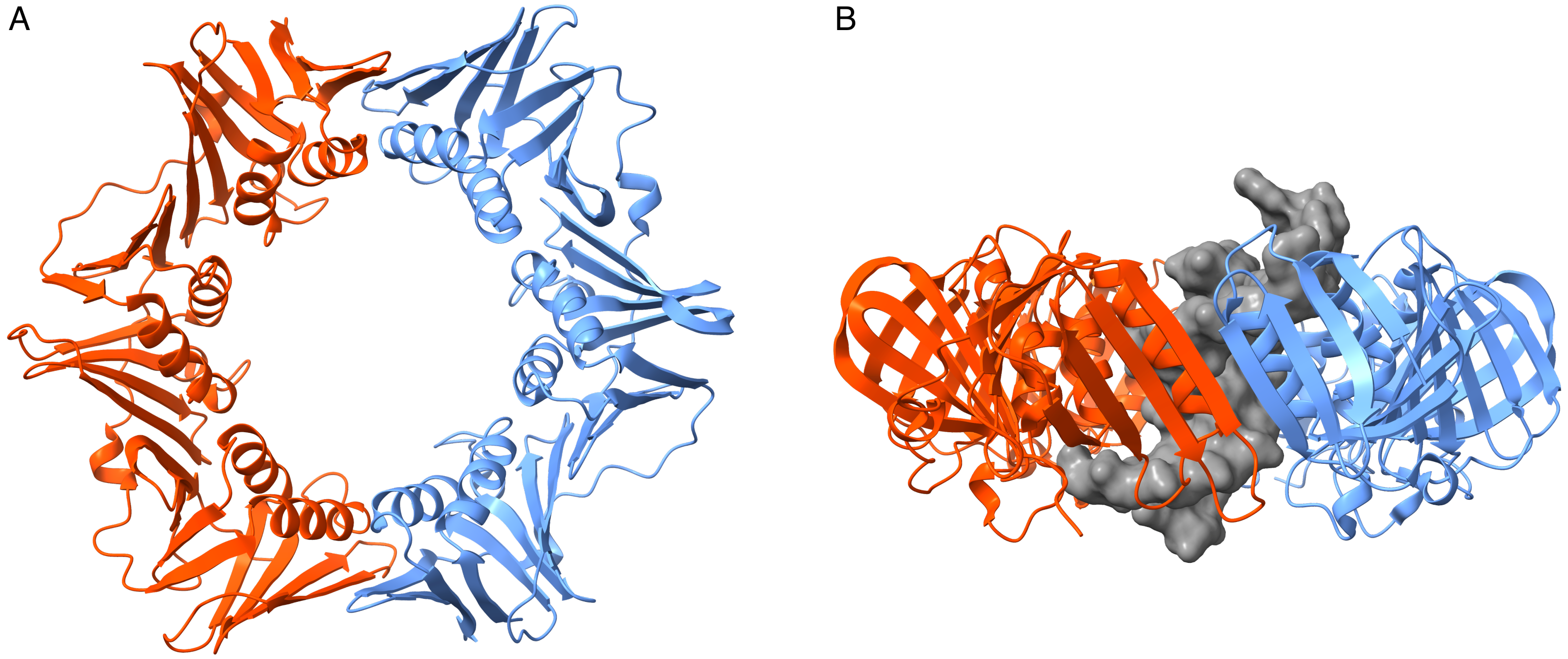
3.2. Simulations of the Homodimer DNA Clamp
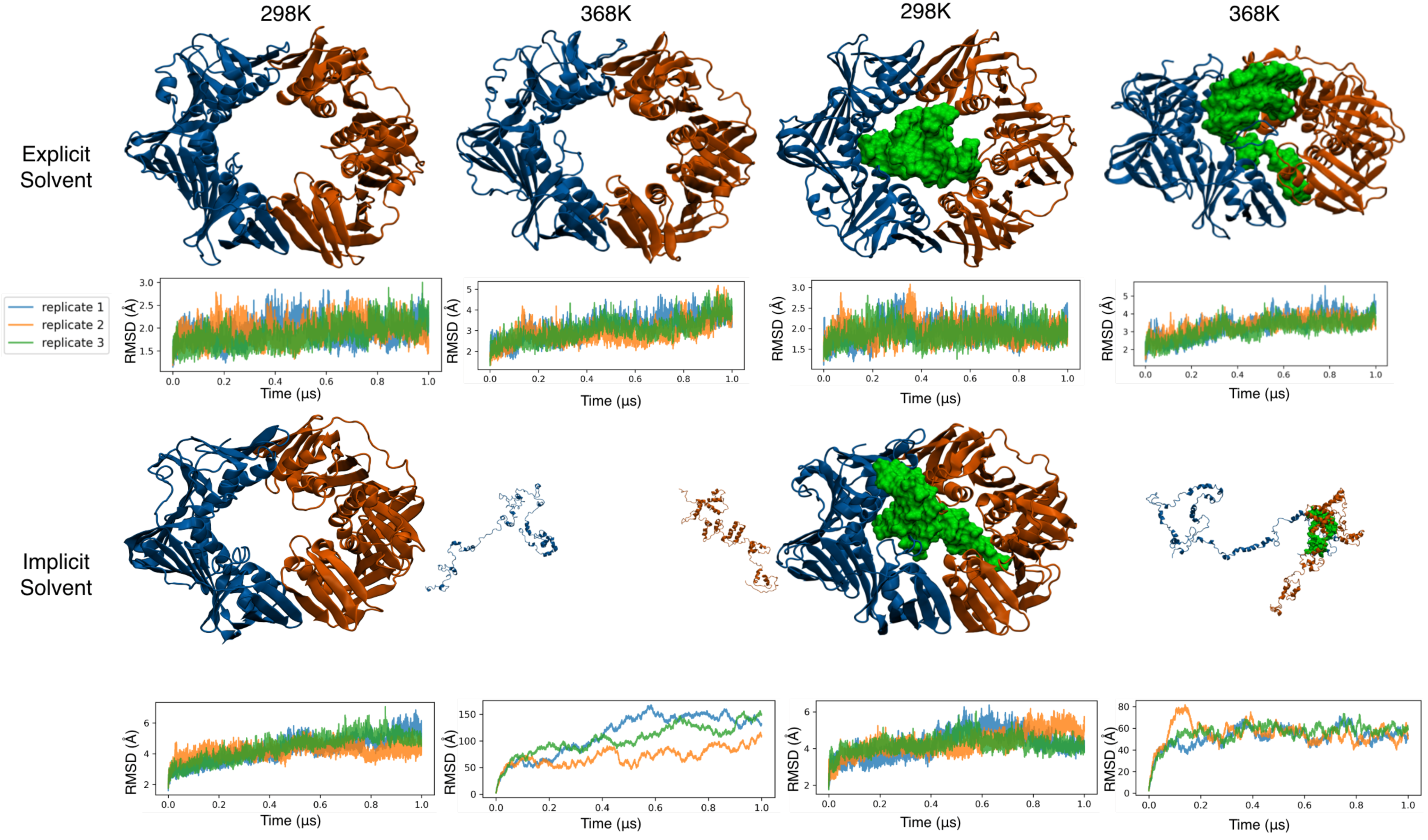
3.3. All-Atom Simulations of the Clamp in Explicit Solvent Showed a Stable Clamp Structure
3.4. All-Atom Simulations of the Clamp in Implicit Solvent Accelerate Heat Denaturation
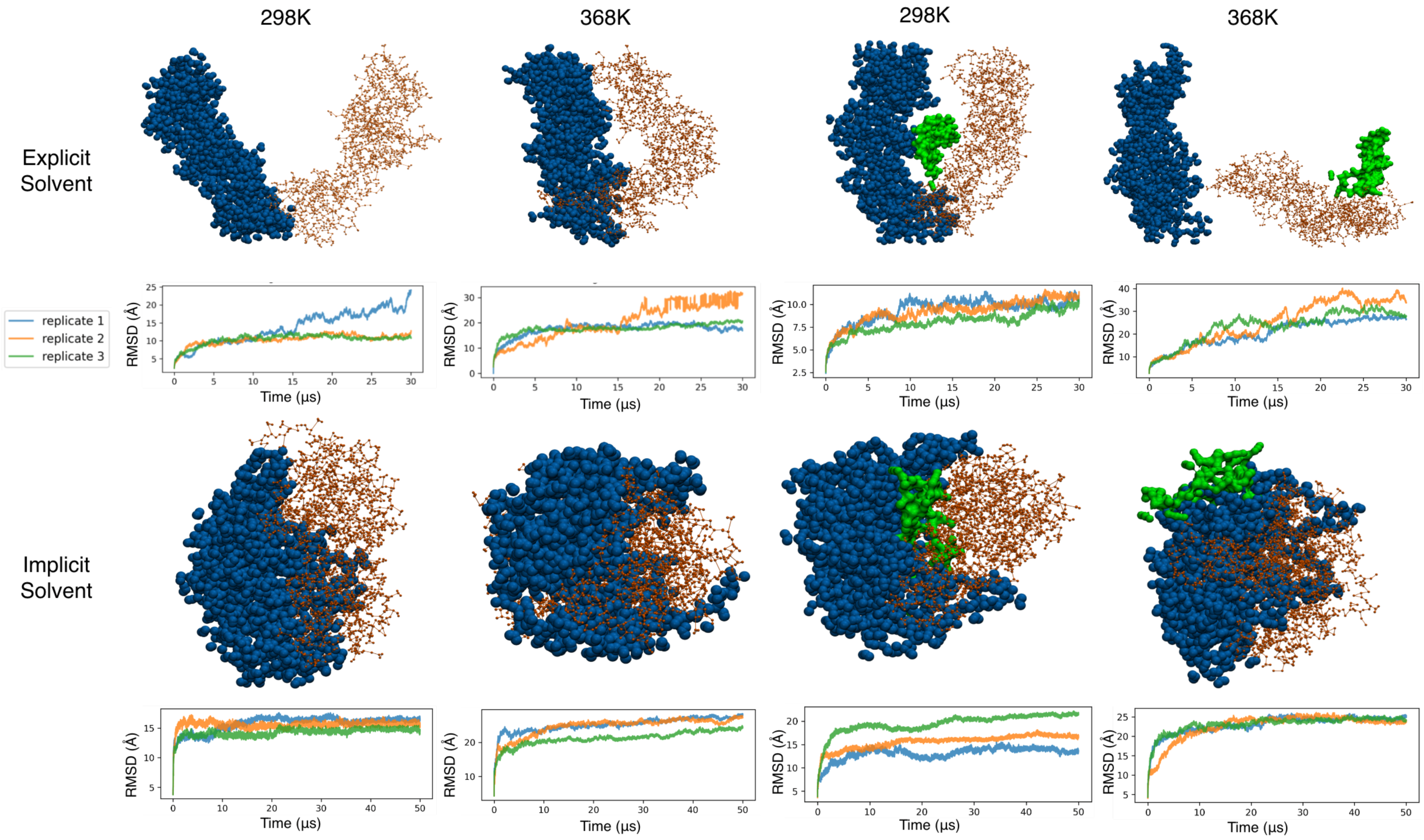
3.5. Coarse-Grained Simulations of the Clamp Spontaneously Sample Open States
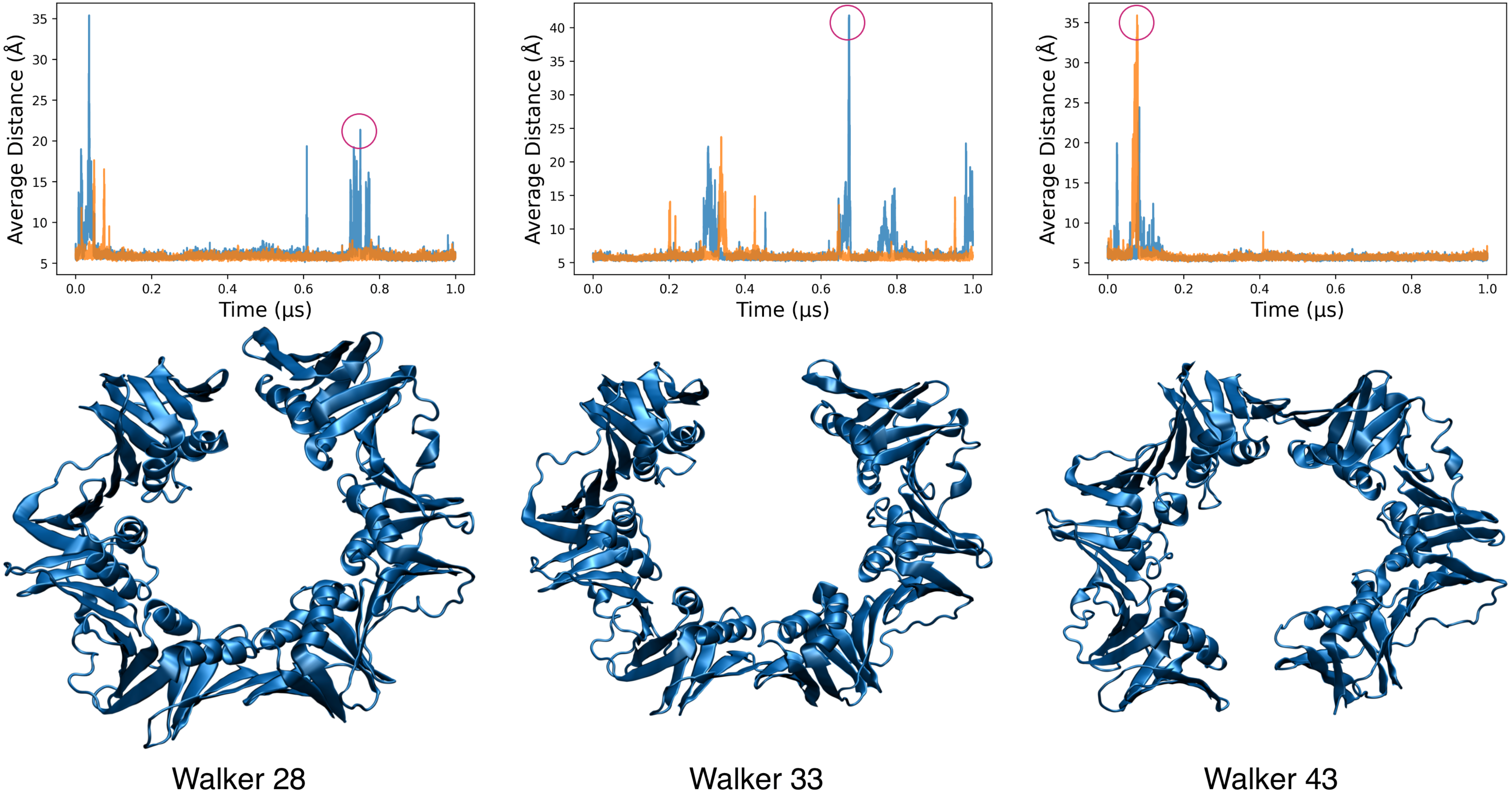
3.6. MELD Samples Show Frequent Spontaneous Opening and Closing of the Clamp
3.7. Comparison of Sampling Efficiency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MELD | Modeling Employing Limited Data |
| RMSD | Root mean square deviation |
| AA | All-Atom |
| CG | Coarse Grain |
References
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC’14: International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014; pp. 41–53. [Google Scholar]
- Perez, A.; Marchan, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E. On the absence of intrahelical DNA dynamics on the μs to ms timescale. Nat. Commun. 2014, 5, 5152. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Murillo, R.; Cheatham, T.E., III. Lessons learned in atomistic simulation of double-stranded DNA: Solvation and salt concerns [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1. [Google Scholar] [CrossRef] [Green Version]
- Pasi, M.; Maddocks, J.H.; Beveridge, D.; Bishop, T.C.; Case, D.A.; Cheatham, T.; Dans, P.D.; Jayaram, B.; Lankas, F.; Laughton, C.; et al. μABC: A systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Res. 2014, 42, 12272–12283. [Google Scholar] [CrossRef]
- Rosa, G.d.; Grille, L.; Calzada, V.; Ahmad, K.; Arcon, J.P.; Battistini, F.; Bayarri, G.; Bishop, T.; Carloni, P.; Cheatham, T.C., III; et al. Sequence-dependent structural properties of B-DNA: What have we learned in 40 years? Biophys. Rev. 2021, 13, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Dans, P.D.; Balaceanu, A.; Pasi, M.; Patelli, A.S.; Petkevičiūtė, D.; Walther, J.; Hospital, A.; Bayarri, G.; Lavery, R.; Maddocks, J.H.; et al. The static and dynamic structural heterogeneities of B-DNA: Extending Calladine–Dickerson rules. Nucleic Acids Res. 2019, 47, 11090–11102. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Nishima, W.; Daniels, M.; Bascom, G.; Kobayashi, C.; Adedoyin, A.; Wall, M.; Lappala, A.; Phillips, D.; Fischer, W.; et al. Scaling molecular dynamics beyond 100,000 processor cores for large-scale biophysical simulations. J. Comput. Chem. 2019, 40, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Pyne, A.L.B.; Noy, A.; Main, K.H.S.; Velasco-Berrelleza, V.; Piperakis, M.M.; Mitchenall, L.A.; Cugliandolo, F.M.; Beton, J.G.; Stevenson, C.E.M.; Hoogenboom, B.W.; et al. Base-pair resolution analysis of the effect of supercoiling on DNA flexibility and major groove recognition by triplex-forming oligonucleotides. Nat. Commun. 2021, 12, 1053. [Google Scholar] [CrossRef]
- Mitchell, B.P.; Hsu, R.V.; Medrano, M.A.; Zewde, N.T.; Narkhede, Y.B.; Palermo, G. Spontaneous Embedding of DNA Mismatches Within the RNA:DNA Hybrid of CRISPR-Cas9. Front. Mol. Biosci. 2020, 7, 39. [Google Scholar] [CrossRef]
- Poppleton, E.; Bohlin, J.; Matthies, M.; Sharma, S.; Zhang, F.; Šulc, P. Design, optimization and analysis of large DNA and RNA nanostructures through interactive visualization, editing and molecular simulation. Nucleic Acids Res. 2020, 48, e72. [Google Scholar] [CrossRef]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Bock, L.V.; Kolář, M.H.; Grubmüller, H. Molecular simulations of the ribosome and associated translation factors. Curr. Opin. Struct. Biol. 2018, 49, 27–35. [Google Scholar] [CrossRef]
- Ray, A.; Felice, R.D. Protein-Mutation-Induced Conformational Changes of the DNA and Nuclease Domain in CRISPR/Cas9 Systems by Molecular Dynamics Simulations. J. Phys. Chem. B 2020, 124, 2168–2179. [Google Scholar] [CrossRef]
- East, K.W.; Newton, J.C.; Morzan, U.N.; Narkhede, Y.B.; Acharya, A.; Skeens, E.; Jogl, G.; Batista, V.S.; Palermo, G.; Lisi, G.P. Allosteric Motions of the CRISPR–Cas9 HNH Nuclease Probed by NMR and Molecular Dynamics. J. Am. Chem. Soc. 2020, 142, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Banáš, P.; Jurečka, P.; Zgarbová, M.; Kührová, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular Dynamics Simulations of Nucleic Acids. From Tetranucleotides to the Ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, K.; Essuman, B.; He, Y.; Coutsias, E.; Garcia-Diaz, M.; Simmerling, C. A human transcription factor in search mode. Nucleic Acids Res. 2015, 44, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Tuszynska, I.; Magnus, M.; Jonak, K.; Dawson, W.; Bujnicki, J.M. NPDock: A web server for protein-nucleic acid docking. Nucleic Acids Res. 2015, 43, W425–W430. [Google Scholar] [CrossRef] [Green Version]
- Honorato, R.V.; Roel-Touris, J.; Bonvin, A.M.J.J. MARTINI-Based Protein-DNA Coarse-Grained HADDOCKing. Front. Mol. Biosci. 2019, 6, 102. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Bonvin, A.M.J.J. Pre- and post-docking sampling of conformational changes using ClustENM and HADDOCK for protein-protein and protein-DNA systems. Proteins Struct. Funct. Bioinform. 2019, 88, 292–306. [Google Scholar] [CrossRef]
- Dijk, M.v.; Dijk, A.D.J.v.; Hsu, V.; Boelens, R.; Bonvin, A.M.J.J. Information-driven protein-DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef]
- Dijk, M.v.; Bonvin, A.M.J.J. A protein-DNA docking benchmark. Nucleic Acids Res. 2008, 36, e88. [Google Scholar] [CrossRef] [Green Version]
- Dijk, M.v.; Bonvin, A.M.J.J. Pushing the limits of what is achievable in protein-DNA docking: Benchmarking HADDOCK’s performance. Nucleic Acids Res. 2010, 38, 5634–5647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijk, M.v.; Visscher, K.M.; Kastritis, P.L.; Bonvin, A.M.J.J. Solvated protein–DNA docking using HADDOCK. J. Biomol. NMR 2013, 56, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dans, P.D.; Walther, J.; Gómez, H.; Orozco, M. Multiscale simulation of DNA. Curr. Opin. Struct. Biol. 2016, 37, 29–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanbonmatsu, K.Y. Large-scale simulations of nucleoprotein complexes: Ribosomes, nucleosomes, chromatin, chromosomes and CRISPR. Curr. Opin. Struct. Biol. 2019, 55, 104–113. [Google Scholar] [CrossRef]
- Ozturk, M.A.; De, M.; Cojocaru, V.; Wade, R.C. Chromatosome Structure and Dynamics from Molecular Simulations. Annu. Rev. Phys. Chem. 2020, 71, 101–119. [Google Scholar] [CrossRef] [Green Version]
- Schlick, T.; Portillo-Ledesma, S.; Myers, C.G.; Beljak, L.; Chen, J.; Dakhel, S.; Darling, D.; Ghosh, S.; Hall, J.; Jan, M.; et al. Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field. Annu. Rev. Biophys. 2021, 50, 267–301. [Google Scholar] [CrossRef]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef]
- Dans, P.D.; Zeida, A.; Machado, M.R.; Pantano, S. A Coarse Grained Model for Atomic-Detailed DNA Simulations with Explicit Electrostatics. J. Chem. Theory Comput. 2010, 6, 1711–1725. [Google Scholar] [CrossRef]
- Machado, M.R.; Barrera, E.E.; Klein, F.; Sáñora, M.; Silva, S.; Pantano, S. The SIRAH 2.0 Force Field: Altius, Fortius, Citius. J. Chem. Theory Comput. 2019, 15, 2719–2733. [Google Scholar] [CrossRef]
- Gopal, S.M.; Mukherjee, S.; Cheng, Y.; Feig, M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins Struct. Funct. Bioinform. 2010, 78, 1266–1281. [Google Scholar] [CrossRef]
- Nguyen, H.; Perez, A.; Bermeo, S.; Simmerling, C. Refinement of Generalized Born Implicit Solvation Parameters for Nucleic Acids and Their Complexes with Proteins. J. Chem. Theory Comput. 2015, 11, 3714–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCallum, J.L.; Perez, A.; Dill, K. Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc. Natl. Acad. Sci. USA 2015, 112, 6985–6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, A.; MacCallum, J.L.; Dill, K.A. Accelerating molecular simulations of proteins using Bayesian inference on weak information. Proc. Natl. Acad. Sci. USA 2015, 112, 11846–11851. [Google Scholar] [CrossRef] [Green Version]
- Bauzá, A.; Pérez, A. MELD-DNA: A new tool for capturing protein-DNA binding. bioRxiv 2021. [Google Scholar] [CrossRef]
- O’Donnell, M.; Kuriyan, J.; Kong, X.P.; Stukenberg, P.T.; Onrust, R. The sliding clamp of DNA polymerase III holoenzyme encircles DNA. Mol. Biol. Cell 1992, 3, 953–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyan, J.; O’Donnell, M. Sliding Clamps of DNA Polymerases. J. Mol. Biol. 1993, 234, 915–925. [Google Scholar] [CrossRef]
- Hingorani, M.M.; O’Donnell, M. Sliding clamps: A (tail)ored fit. Curr. Biol. 2000, 10, R25–R29. [Google Scholar] [CrossRef] [Green Version]
- Alley, S.C.; Abel-Santos, E.; Benkovic, S.J. Tracking Sliding Clamp Opening and Closing during Bacteriophage T4 DNA Polymerase Holoenzyme Assembly. Biochemistry 2000, 39, 3076–3090. [Google Scholar] [CrossRef]
- Douma, L.G.; Yu, K.K.; England, J.K.; Levitus, M.; Bloom, L.B. Mechanism of opening a sliding clamp. Nucleic Acids Res. 2017, 45, 10178–10189. [Google Scholar] [CrossRef] [Green Version]
- Hedglin, M.; Kumar, R.; Benkovic, S.J. Replication Clamps and Clamp Loaders. Cold Spring Harb. Perspect. Biol. 2013, 5, a010165. [Google Scholar] [CrossRef] [Green Version]
- Tainer, J.A.; McCammon, J.A.; Ivanov, I. Recognition of the Ring-Opened State of Proliferating Cell Nuclear Antigen by Replication Factor C Promotes Eukaryotic Clamp-Loading. J. Am. Chem. Soc. 2010, 132, 7372–7378. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-acid specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengfei, L.; Song, L.F.; Merz, K.M. Parameterization of Highly Charged Metal Ions Using the 12-6-4 LJ-Type Nonbonded Model in Explicit Water. J. Phys. Chem. B 2015, 119, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Roe, D.R.; Simmerling, C. Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput. 2013, 9, 2020–2034. [Google Scholar] [CrossRef] [Green Version]
- Curry, H.B. The method of steepest descent for non-linear minimization problems. Q. Appl. Math. 1944, 2, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Hestenes, M.R.; Stiefel, E. Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bur. Stand. 1952, 49, 409. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Darré, L.; Machado, M.R.; Dans, P.D.; Herrera, F.E.; Pantano, S. Another Coarse Grain Model for Aqueous Solvation: WAT FOUR? J. Chem. Theory Comput. 2010, 6, 3793–3807. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar] [CrossRef]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Parametrized Models of Aqueous Free Energies of Solvation Based on Pairwise Descreening of Solute Atomic Charges from a Dielectric Medium. J. Phys. Chem. 1996, 100, 19824–19839. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Morrone, J.A.; Perez, A.; MacCallum, J.; Dill, K. Computed Binding of Peptides to Proteins with MELD-Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2017, 13, 870–876. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Maier, J.; Huang, H.; Perrone, V.; Simmerling, C. Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent. J. Am. Chem. Soc. 2014, 136, 13959–13962. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.W.; Greenall, R.J.; Probert, M.I.J.; Noy, A.; Leake, M.C. The emergence of sequence-dependent structural motifs in stretched, torsionally constrained DNA. Nucleic Acids Res. 2020, 48, 1748–1763. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Zakrevsky, P.; Kasprzak, W.K.; Heinz, W.F.; Wu, W.; Khant, H.; Bindewald, E.; Dorjsuren, N.; Fields, E.A.; Val, N.d.; Jaeger, L.; et al. Truncated tetrahedral RNA nanostructures exhibit enhanced features for delivery of RNAi substrates. Nanoscale 2020, 12, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Brini, E.; Perez, A.; Dill, K.A. Computing Ligands Bound to Proteins Using MELD-Accelerated MD. J. Chem. Theory Comput. 2020, 16, 6377–6382. [Google Scholar] [CrossRef] [PubMed]
- Morrone, J.A.; Perez, A.; Deng, Q.; Ha, S.N.; Holloway, M.K.; Sawyer, T.K.; Sherborne, B.S.; Brown, F.K.; Dill, K. Molecular Simulations Identify Binding Poses and Approximate Affinities of Stapled α-Helical Peptides to MDM2 and MDMX. J. Chem. Theory Comput. 2017, 13, 863–869. [Google Scholar] [CrossRef]
- Lang, L.; Perez, A. Binding Ensembles of p53-MDM2 Peptide Inhibitors by Combining Bayesian Inference and Atomistic Simulations. Molecules 2021, 26, 198. [Google Scholar] [CrossRef]
- Brini, E.; Kozakov, D.; Dill, K. Predicting Protein Dimer Structures Using MELD × MD. J. Chem. Theory Comput. 2019, 15, 3381–3389. [Google Scholar] [CrossRef]
- Zagrovic, B.; Pande, V. Solvent viscosity dependence of the folding rate of a small protein: Distributed computing study. J. Comput. Chem. 2003, 24, 1432–1436. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Pande, V.S.; Beauchamp, K.; Bowman, G.R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, N.; Turner, J.; Kelman, Z.; Stukenberg, P.T.; Dean, F.; Shechter, D.; Pan, Z.Q.; Hurwitz, J.; O’Donnell, M. Clamp loading, unloading and intrinsic stability of the PCNA, β and gp45 sliding clamps of human, E. coli and T4 replicases. Genes Cells 1996, 1, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, A.G.; Baldi, P.; Chauvin, Y.; Brunak, S. DNA structure in human RNA polymerase II promoters 1 1Edited by J. Karn. J. Mol. Biol. 1998, 281, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Várnai, P.; Zakrzewska, K. DNA and its counterions: A molecular dynamics study. Nucleic Acids Res. 2004, 32, 4269–4280. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.; Latzer, J.; Case, D.A. Revised AMBER Parameters for Bioorganic Phosphates. J. Chem. Theory Comput. 2012, 8, 4405–4412. [Google Scholar] [CrossRef] [Green Version]
- Bergonzo, C.; Cheatham, T.E. Improved Force Field Parameters Lead to a Better Description of RNA Structure. J. Chem. Theory Comput. 2015, 11, 3969–3972. [Google Scholar] [CrossRef]
- Esadze, A.; Chen, C.; Zandarashvili, L.; Roy, S.; Pettitt, B.M.; Iwahara, J. Changes in conformational dynamics of basic side chains upon protein–DNA association. Nucleic Acids Res. 2016, 44, 6961–6970. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Lee, H.G.; Kim, K.; Yoo, J. Improved Parameterization of Protein–DNA Interactions for Molecular Dynamics Simulations of PCNA Diffusion on DNA. J. Chem. Theory Comput. 2020, 16, 4006–4013. [Google Scholar] [CrossRef]
- Khabiri, M.; Freddolino, P.L. Deficiencies in Molecular Dynamics Simulation-Based Prediction of Protein–DNA Binding Free Energy Landscapes. J. Phys. Chem. B 2017, 121, 5151–5161. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.A.; García, A.E. High-resolution reversible folding of hyperstable RNA tetraloops using molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825. [Google Scholar] [CrossRef] [Green Version]
- Perez, A.; Morrone, J.A.; Brini, E.; MacCallum, J.L.; Dill, K. Blind protein structure prediction using accelerated free-energy simulations. Sci. Adv. 2016, 2, e1601274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, G.R.; Ensign, D.L.; Pande, V.S. Enhanced Modeling via Network Theory: Adaptive Sampling of Markov State Models. J. Chem. Theory Comput. 2010, 6, 787–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scale | Solvation | Solvent | State | Temperature (K) | Time (s) | Replicates |
|---|---|---|---|---|---|---|
| All-Atom | explicit | OPC | bound | 298 | 1 | 3 |
| 368 | 1 | 3 | ||||
| unbound | 298 | 1 | 3 | |||
| 368 | 1 | 3 | ||||
| bound | 298 | 1 | 3 | |||
| 368 | 1 | 3 | ||||
| unbound | 298 | 1 | 3 | |||
| 368 | 1 | 3 | ||||
| implicit | GBneck2 | bound | 300–500 | 1 | 1 | |
| 1 | 50 replicas | |||||
| unbound | 300–500 | 1 | 1 | |||
| 1 | 50 replicas | |||||
| Coarse Grained | explicit | WT4 | bound | 298 | 30 | 3 |
| 368 | 30 | 3 | ||||
| unbound | 298 | 30 | 3 | |||
| 368 | 30 | 3 | ||||
| implicit | HCT | bound | 298 | 50 | 3 | |
| 368 | 50 | 3 | ||||
| unbound | 298 | 50 | 3 | |||
| 368 | 50 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esmaeeli, R.; Andal, B.; Perez, A. Searching for Low Probability Opening Events in a DNA Sliding Clamp. Life 2022, 12, 261. https://doi.org/10.3390/life12020261
Esmaeeli R, Andal B, Perez A. Searching for Low Probability Opening Events in a DNA Sliding Clamp. Life. 2022; 12(2):261. https://doi.org/10.3390/life12020261
Chicago/Turabian StyleEsmaeeli, Reza, Benedict Andal, and Alberto Perez. 2022. "Searching for Low Probability Opening Events in a DNA Sliding Clamp" Life 12, no. 2: 261. https://doi.org/10.3390/life12020261
APA StyleEsmaeeli, R., Andal, B., & Perez, A. (2022). Searching for Low Probability Opening Events in a DNA Sliding Clamp. Life, 12(2), 261. https://doi.org/10.3390/life12020261