
Prokineticin-Receptor Network: Mechanisms of Regulation


Abstract
1. Introduction
2. Prokineticin Receptors
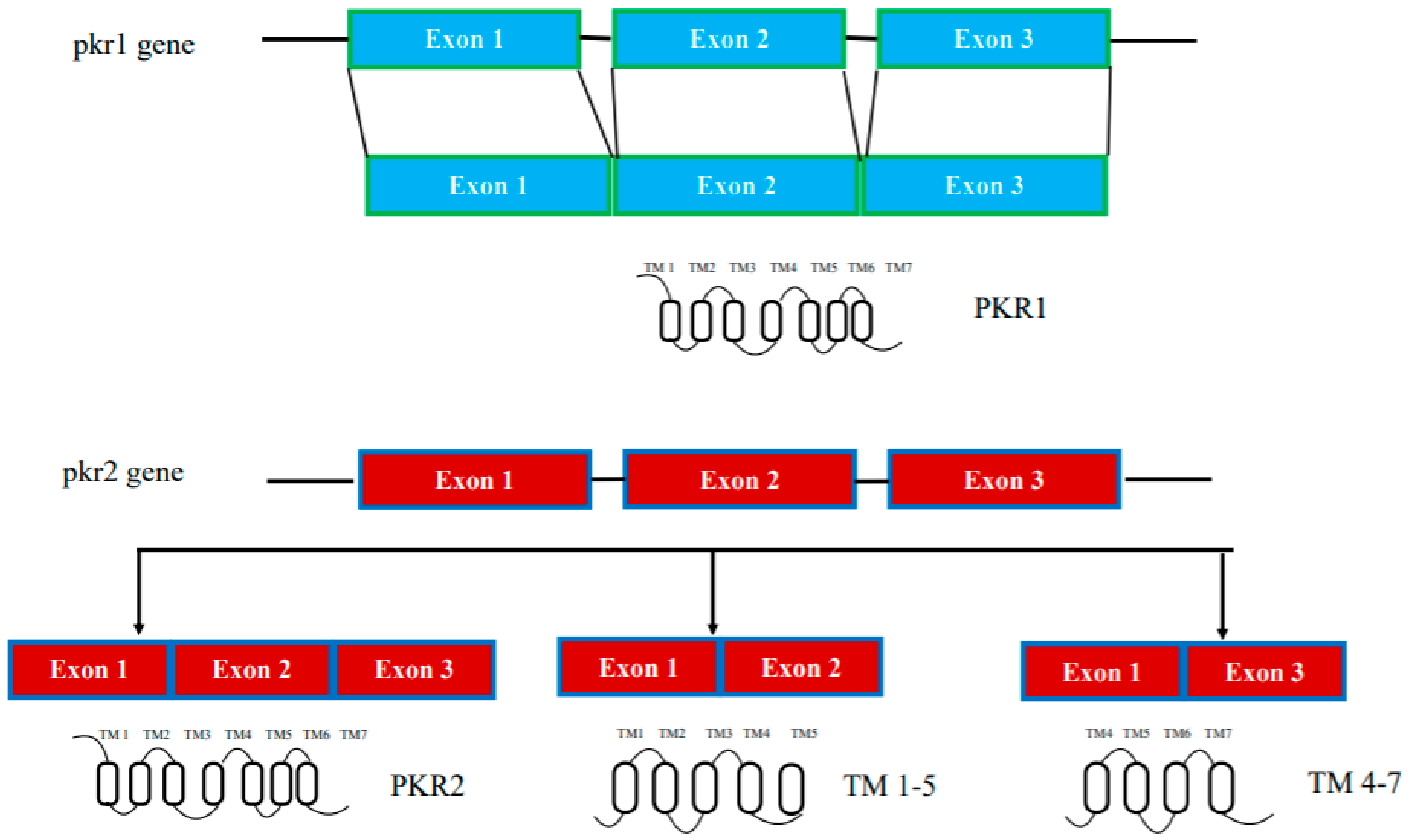
2.1. Genetic and Splicing Variants
2.2. Transcriptional Regulation
2.3. Structural Elements Underlying Receptors Function
2.3.1. Receptor-Prokineticin Interactions
2.3.2. Receptor-G-Protein Interactions
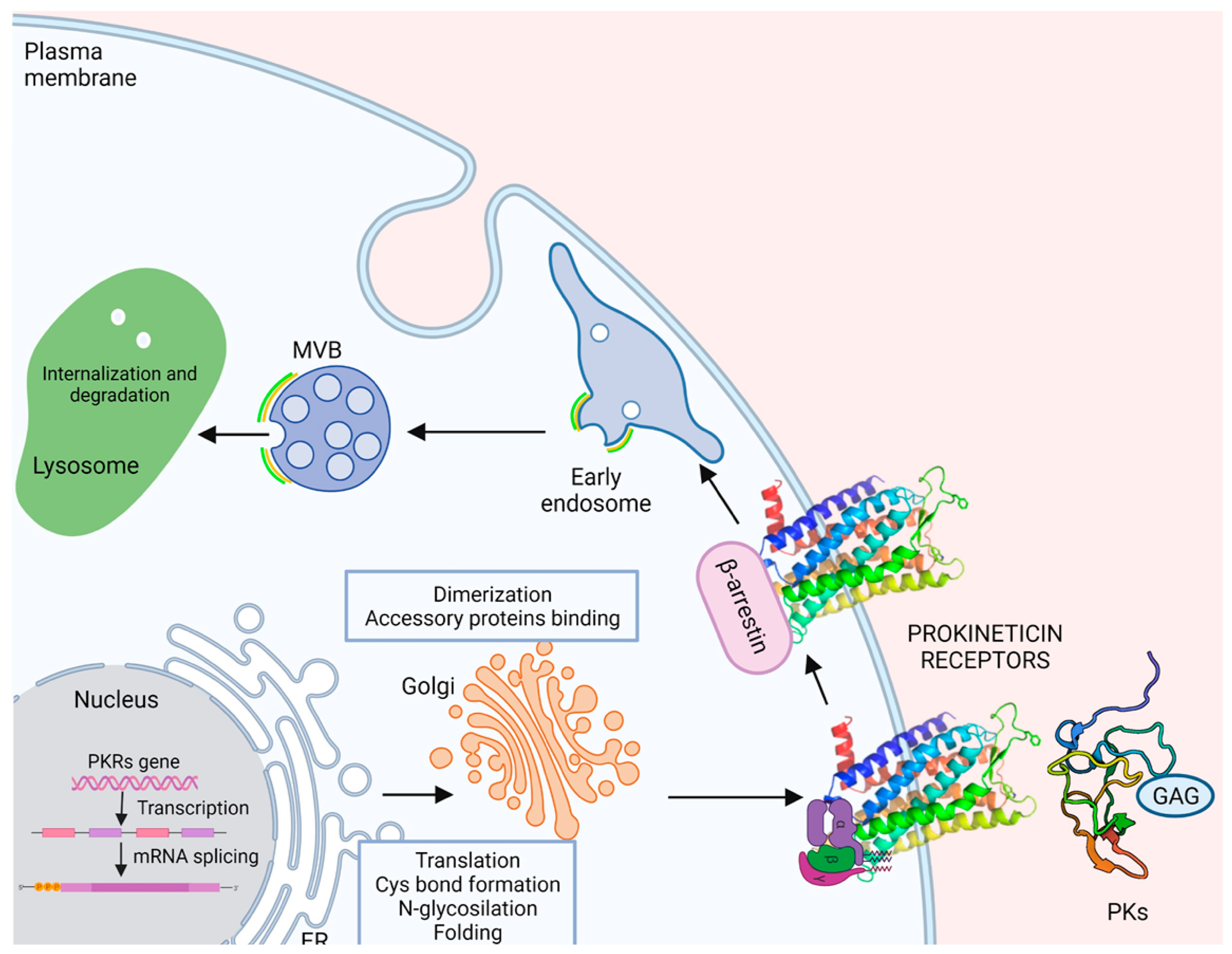
2.4. Maturation, Folding, and Localization
2.4.1. Post-Translation Modifications: N-Glycosylation
2.4.2. Dimerization
2.4.3. Binding to Accessory Proteins
2.5. Internalization Recycling, or Degradation
3. Prokineticins
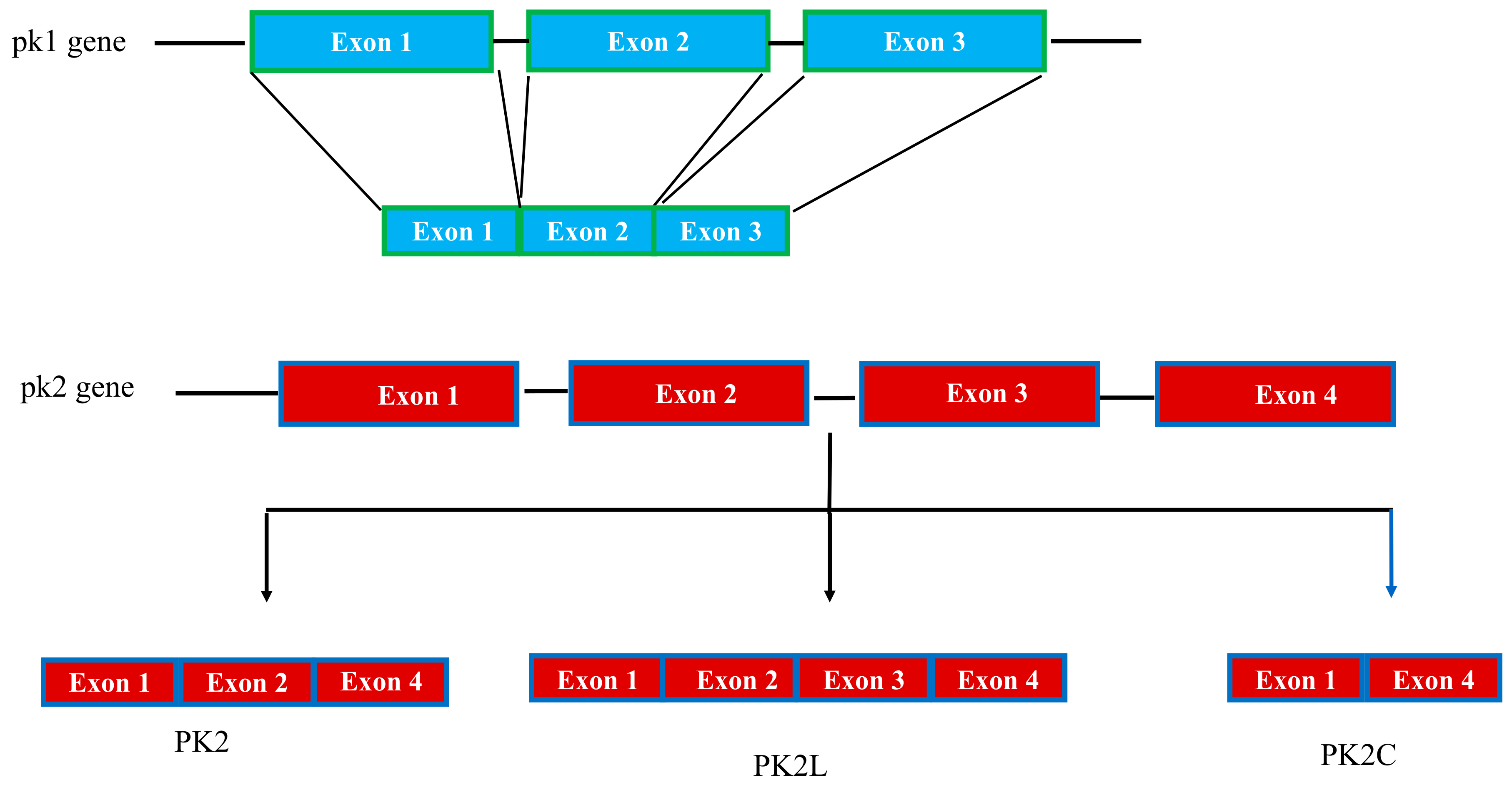
3.1. Genetic and mRNA Splice Variants
3.2. Transcriptional and Post-Transcriptional Regulation
3.3. Structural Elements Underlying Prokineticin-Receptor Interactions
3.4. Post-Translational Modifications
4. Natural and Pharmacological Inhibitors
4.1. Parasites
4.2. Antagonists
5. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Monnier, J.; Samson, M. Cytokine properties of prokineticins. FEBS J. 2008, 275, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Winklmayr, M.; Lepperdinger, G.; Kreil, G. The AVIT protein family. Secreted cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. 2003, 4, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Miele, R.; Lattanzi, R.; di Patti, M.C.B.; Paiardini, A.; Negri, L.; Barra, D. Expression of Bv8 in Pichia pastoris to identify structural features for receptor binding. Protein Expr. Purif. 2010, 73, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Sacerdote, P.; Franchi, S.; Canestrelli, M.; Miele, R.; Barra, D.; Visentin, S.; De Nuccio, C.; Porreca, F.; De Felice, M.; et al. Pharmacological activity of a Bv8 analogue modified in position 24. Br. J. Pharmacol. 2012, 166, 950–963. [Google Scholar] [CrossRef]
- Désaubry, L.; Kanthasamy, A.G.; Nebigil, C.G. Prokineticin signaling in heart-brain developmental axis: Therapeutic options for heart and brain injuries. Pharm. Res. 2020, 160, 105190. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Leslie, F.M.; Zhou, Q.Y. Expression of prokineticins and their receptors in the adult mouse brain. J. Comp. Neurol. 2006, 498, 796–809. [Google Scholar] [CrossRef]
- Negri, L.; Ferrara, N. The Prokineticins: Neuromodulators and Mediators of Inflammation and Myeloid Cell-Dependent Angiogenesis. Physiol. Rev. 2018, 98, 1055–1082. [Google Scholar] [CrossRef]
- Lattanzi, R.; Miele, R. Versatile Role of Prokineticins and Prokineticin Receptors in Neuroinflammation. Biomedicines 2021, 9, 1648. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Trypanosoma cruzi trans-sialidase induces STAT3 and ERK activation by prokineticin receptor 2 binding. Cell Biochem. Funct. 2020, 39, 326–334. [Google Scholar] [CrossRef]
- Dodé, C.; Rondard, P. PROK2/PROKR2 Signaling and Kallmann Syndrome. Front. Endocrinol. 2013, 4, 19. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Jia, H.; Wang, X.; Zheng, R.; Jiang, F.; Chen, D.; Chen, Z.; Li, J. PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: Selective disruption of the binding to a Gα-protein leads to biased signaling. FASEB J. 2019, 33, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Bullock, C.M.; Ehlert, F.J.; Chen, J.L.; Tian, H.; Zhou, Q.Y. Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J. Biol. Chem. 2002, 277, 19276–19280. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Takatsu, Y.; Terao, Y.; Kumano, S.; Ishibashi, Y.; Suenaga, M.; Abe, M.; Fukusumi, S.; Watanabe, T.; Shintani, Y.; et al. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem. Biophys. Res. Commun. 2002, 293, 396–402. [Google Scholar] [CrossRef]
- Soga, T.; Matsumoto, S.; Oda, T.; Saito, T.; Hiyama, H.; Takasaki, J.; Kamohara, M.; Ohishi, T.; Matsushime, H.; Furuichi, K. Molecular cloning and characterization of prokineticin receptors. Biochim Biophys. Acta 2002, 1579, 173–179. [Google Scholar] [CrossRef]
- Parker, R.; Liu, M.; Eyre, H.J.; Copeland, N.G.; Gilbert, D.J.; Crawford, J.; Sutherland, G.R.; Jenkins, N.A.; Herzog, H. Y-receptor-like genes GPR72 and GPR73: Molecular cloning, genomic organisation and assignment to human chromosome 11q21.1 and 2p14 and mouse chromosome 9 and 6. Biochim. Biophys. Acta 2000, 1491, 369–375. [Google Scholar] [CrossRef]
- Ngan, E.S.W.; Tam, P.H.K. Prokineticin-signaling pathway. Int. J. Biochem. Cell Biol. 2008, 4, 1679–1684. [Google Scholar] [CrossRef]
- Yin, W.; Liu, H.; Peng, Z.; Che, D.; Li, J.; Li, J.D. Mechanisms that underlie the internalization and extracellular signal regulated kinase 1/2 activation by PKR2 receptor. Cell. Signal. 2014, 26, 1118–1124. [Google Scholar] [CrossRef]
- Su, M.T.; Lin, S.H.; Lee, I.W. Polymorphisms of endocrine gland-derived vascular endothelial growth factor gene and its receptor genes are associated with recurrent pregnancy loss. Hum. Reprod. 2010, 25, 2923–2930. [Google Scholar] [CrossRef]
- Cao, Y.L.; Zhang, Z.F.; Wang, J.; Miao, M.H.; Xu, J.H.; Shen, Y.P.; Chen, A.M.; Du, J.; Yuan, W.J. Zhejiang Association between polymorphisms of prokineticin receptor (PKR1 rs4627609 and PKR2 rs6053283) and recurrent pregnancy loss. Univ. Sci. B. 2016, 17, 218–224. [Google Scholar] [CrossRef]
- Kishi, T.; Kitajima, T.; Tsunoka, T.; Okumura, T.; Ikeda, M.; Okochi, T.; Kinoshita, Y.; Kawashima, K.; Yamanouchi, Y.; Ozaki, N.; et al. Possible association of prokineticin 2 receptor gene (PROKR2) with mood disorders in the Japanese population. Neuromol. Med. 2009, 11, 114–122. [Google Scholar] [CrossRef]
- Aiello, F.; Cirillo, G.; Cassio, A.; Di Mase, R.; Tornese, G.; Umano, G.R.; del Giudice, E.M.; Grandone, A. Molecular screening of PROKR2 gene in girls with idiopathic central precocious puberty. Ital. J. Pediatr. 2021, 47, 5. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ferrer, M.; Torroglosa, A.; Nunez-Torres, R.; de Agustin, J.C.; Antinolo, G.; Borrego, S. Expression of PROKR1 and PROKR2 in human enteric neural precursor cells and identification of sequence variants suggest a role in HSC. PLoS ONE 2011, 6, e23475. [Google Scholar]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Identification and characterization of Prokineticin receptor 2 splicing variant and its modulation in an animal model of Alzheimer’s disease. Neuropeptides 2019, 73, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Sposini, S.; Caltabiano, G.; Hanyaloglu, A.C.; Miele, R. Identification of transmembrane domains that regulate spatial arrangements and activity of prokineticin receptor 2 dimers. Mol. Cell. Endocrinol. 2015, 399, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Fukami, M.; Suzuki, E.; Izumi, Y.; Torii, T.; Narumi, S.; Igarashi, M.; Miyado, M.; Katsumi, M.; Fujisawa, Y.; Nakabayashi, K.; et al. Paradoxical gain-of-function mutant of the G-protein-coupled receptor PROKR2 promotes early puberty. J. Cell. Mol. Med. 2017, 21, 2623–2626. [Google Scholar] [CrossRef] [PubMed]
- Monnier, C.; Dodé, C.; Fabre, L.; Teixeira, L.; Labesse, G.; Pin, J.P.; Hardelin, J.P.; Rondard, P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet. 2009, 18, 75–81. [Google Scholar] [CrossRef]
- Abreu, A.P.; Noel, S.D.; Xu, S.; Carroll, R.S.; Latronico, A.C.; Kaiser, U.B. Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function. Mol. Endocrinol. 2012, 26, 1417–1427. [Google Scholar] [CrossRef]
- Cole, L.W.; Sidis, Y.; Zhang, C.K.; Quinton, R.; Plummer, L.; Pignatelli, D.; Hughes, V.A.; Dwyer, A.A.; Raivio, T.; Hayes, F.J.; et al. Mutations in Prokineticin 2 and Prokineticin receptor 2 genes in Human Gonadotrophin-Releasing Hormone Deficiency: Molecular Genetics and Clinical Spectrum. J. Clin. Endocrinol. Metab. 2008, 93, 3551–3559. [Google Scholar] [CrossRef]
- Abreu, A.P.; Kaiser, U.B.; Latronico, A.C. The role of prokineticins in the pathogenesis of hypogonadotropic hypogonadism. Neuroendocrinology 2010, 91, 283–290. [Google Scholar] [CrossRef]
- Libri, D.V.; Kleinau, G.; Vezzoli, V.; Busnelli, M.; Guizzardi, F.; Sinisi, A.A.; Pincelli, A.I.; Mancini, A.; Russo, G.; Beck-Peccoz, P.; et al. Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differential modulation of distinct intracellular pathways. J. Clin. Endocrin. Metab. 2014, 99, E458–E463. [Google Scholar] [CrossRef]
- Cox, K.H.; Oliveira, L.M.B.; Plummer, L.; Corbin, B.; Gardella, T.; Balasubramanian, R.; Crowley, W.F. Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum. Mol. Genet. 2018, 27, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.N.; Ma, Y.T.; Liu, H.; Zhou, Q.Y.; Li, J.D. Functional rescue of Kallmann syndrome-associated prokineticin receptor 2 (PKR2) mutants deficient in trafficking. J. Biol. Chem. 2014, 289, 15518–15526. [Google Scholar] [CrossRef] [PubMed]
- Dodé, C.; Teixeira, L.; Levilliers, J.; Fouveaut, C.; Bouchard, P.; Kottler, M.-L.; Lespinasse, J.; Lienhardt-Roussie, A.; Mathieu, M.; Moerman, A.; et al. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006, 2, e175. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Feige, J.J.; Alfaidy, N. Expression and oxygen regulation of endocrine gland-derived vascular endothelial growth factor/prokineticin-1 and its receptors in human placenta during early pregnancy. Endocrinology 2016, 147, 1675–1684. [Google Scholar] [CrossRef]
- Meidan, R.; Klipper, E.; Zalman, Y.; Ronit, Y. The role of hypoxia-induced genes in ovarian angiogenesis. Reprod. Fertil. Dev. 2012, 25, 343–350. [Google Scholar] [CrossRef]
- Licht, P.; Russu, V.; Wildt, L. On the role of human chorionic gonadotropin (hCG) in the embryo-endometrial microenvironment: Implications for differentiation and implantation. Semin. Reprod. Med. 2001, 19, 37–47. [Google Scholar] [CrossRef]
- Hoffmann, P.; Saoudi, Y.; Benharouga, M.; Graham, C.H.; Schaal, J.P.; Mazouni, C.; Feige, J.J.; Alfaidy, N. Role of EG-VEGF in human placentation: Physiological and pathological implications. J. Cell. Mol. Med. 2009, 13, 2224–2235. [Google Scholar] [CrossRef]
- Ragancokova, D.; Rocca, E.; Oonk, A.M.; Schulz, H.; Rohde, E.; Bednarsch, J.; Feenstra, I.; Pennings, R.J.; Wende, H.; Garratt, A.N. TSHZ1-dependent gene regulation is essential for olfactory bulb development and olfaction. J. Clin. Investig. 2014, 124, 1214–1227. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef]
- Fullone, M.R.; Lattanzi, R.; Maftei, D.; Bonaccorsi, M.C.; Miele, R. Analysis of role of aromatic residues in extracellular loop 2 of Prokineticin receptor 2 in ligand binding probed with genetically encoded photo-crosslinkers. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183549. [Google Scholar] [CrossRef] [PubMed]
- Levit, A.; Yarnitzky, T.; Wiener, A.; Meidan, R.; Niv, M.Y. Modeling of human prokineticin receptors: Interactions with novel small-molecule binders and potential off-target drugs. PLoS ONE 2011, 6, e27990. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gasser, A.; Brogi, S.; Urayama, K.; Nishi, T.; Kurose, H.; Tafi, A.; Ribeiro, N.; Désaubry, L.; Nebigil, C.G. Discovery and cardioprotective effects of the first non-Peptide agonists of the G protein-coupled prokineticin receptor-1. PLoS ONE 2015, 10, e0121027. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kuei, C.; Sutton, S.; Wilson, S.; Yu, J.; Kamme, F.; Mazur, C.; Lovenberg, T.; Liu, C. Identification and pharmacological characterization of prokineticin 2 beta as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 2005, 67, 2070–2076. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Maftei, D.; Negri, L.; Fusco, I.; Miele, R. PK2β ligand, a splice variant of prokineticin 2, is able to modulate and drive signaling through PKR1 receptor. Neuropeptides 2018, 71, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Tang, Y.; Luo, H.; Jiang, F.; Yang, J.; Sun, L.; Li, J.-D. Disease-causing mutation in PKR2 receptor reveals a critical role of positive charges in the second intracellular loop for G-protein coupling and receptor trafficking. J. Biol. Chem. 2011, 286, 16615–16622. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, A.; Cheng, N.; Trejo, J.N. Post-Translational Modifications of G Protein-Coupled Receptors Control Cellular Signaling Dynamics in Space and Time. Pharmacol. Rev. 2021, 3, 120–151. [Google Scholar] [CrossRef]
- Verdinez, J.A.; Sebag, J.A. Role of N-Linked Glycosylation in PKR2 Trafficking and Signaling. Front. Neurosci. 2021, 15, 730417. [Google Scholar] [CrossRef]
- Ferre, S.; Casado, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef]
- Marsango, S.; di Patti, M.C.B.; Barra, D.; Miele, R. Evidence that prokineticin receptor 2 exists as a dimer in vivo. Cell. Mol. Life Sci. 2011, 68, 2919–2929. [Google Scholar] [CrossRef]
- Berruien, N.N.A.; Smith, C.L. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene 2020, 757, 144949. [Google Scholar] [CrossRef] [PubMed]
- Chaly, A.L.; Srisai, D.; Gardner, E.E.; Sebag, J.A. The Melanocortin Receptor Accessory Protein 2 promotes food intake through inhibition of the Prokineticin Receptor-1. eLife 2016, 5, e12397. [Google Scholar] [CrossRef] [PubMed]
- Rouault, A.A.J.; Lee, A.A.; Sebag, J.A. Regions of MRAP2 required for the inhibition of orexin and prokineticin receptor signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2322–2329. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Morishima, S.; Tanaka, T.; Muramatsu, I. Snapin, a New Regulator of Receptor Signaling, Augments α1A-Adrenoceptor-operated Calcium Influx through TRPC6. J. Biol. Chem. 2007, 282, 29563–29573. [Google Scholar] [CrossRef]
- Song, J.; Li, J.; Liu, H.; Liu, W.; Feng, Y.; Zhou, X.T.; Li, Y.D. Snapin interacts with G-protein coupled receptor PKR2. Biochem. Biophys. Res. Commun. 2016, 469, 501–516. [Google Scholar] [CrossRef]
- Mollay, C.; Wechselberger, C.; Mignogna, G.; Negri, L.; Melchiorri, P.; Barra, D.; Kreil, G. Bv8, a small protein from frog skin and its homologue from snake venom induce hyperalgesia in rats. Eur. J. Pharmacol. 1999, 374, 189–196. [Google Scholar] [CrossRef]
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachi-asmatic nucleus. Nature 2002, 417, 405–410. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by b-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Casella, I.; Ambrosio, C. Prokineticin receptors interact unselectively with several G protein subtypes but bind selectively to β-arrestin 2. Cell. Signal. 2021, 83, 110000. [Google Scholar] [CrossRef]
- Chen, T.; Xue, Y.; Zhou, M.; Shaw, C. Molecular cloning of mRNA from toad granular gland secretion and lyophilized skin: Identification of Bo8—A novel prokineticin from Bombina orientalis. Peptides 2005, 26, 377–383. [Google Scholar] [CrossRef]
- Schweitz, H.; Pacaud, P.; Diochot, S.; Moinier, D.; Lazdunski, M. MIT (1), a black mamba toxin with a new and highly potent activity on intestinal contraction. FEBS Lett. 1999, 461, 183–188. [Google Scholar] [CrossRef]
- Li, M.; Bullock, C.M.; Knauer, D.J.; Ehlert, F.J.; Zhou, Q.Y. Identification of two prokineticin cDNAs: Recombinant proteins potently contract gastrointestinal smooth muscle. Mol. Pharmacol. 2001, 59, 692–698. [Google Scholar] [CrossRef] [PubMed]
- LeCouter, J.; Kowalski, J.; Foster, J.; Hass, P.; Zhang, Z.; Dillard-Telm, L.; Frantz, G.; Rangell, L.; DeGuzman, L.; Keller, G.A.; et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature 2001, 412, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Jilek, A.; Engel, E.; Beier, D.; Lepperdinger, G. Murine Bv8 gene maps near a synteny breakpoint of mouse chromosome 6 and human 3p21. Gene 2000, 256, 189–195. [Google Scholar] [CrossRef]
- Maftei, D.; Lattanzi, R.; Vincenzi, M.; Squillace, S.; Fullone, M.R.; Miele, R. The balance of concentration between Prokineticin 2β and Prokineticin 2 modulates the food intake by STAT3 signaling. BBA Adv. 2021, 1, 100028. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Vincenzi, M.; Fullone, M.R.; Miele, R. Identification and characterization of a new splicing variant of Prokineticin 2. Life 2021, 1443, 400–406. [Google Scholar]
- Mortreux, M.; Foppen, E.; Denis, R.G.; Montaner, M.; Kassis, N.; Denom, J.; Vincent, M.; Fumeron, F.; Kujawski-Lafourcade, M.; AndreÃÅelli, F.; et al. New roles for prokineticin 2 in feeding behavior, insulin resistance and type 2 diabetes: Studies in mice and humans. Mol. Metab. 2019, 29, 182–196. [Google Scholar] [CrossRef]
- LeCouter, J.; Lin, R.; Frantz, G.; Zhang, Z.; Hillan, K.; Ferrara, N. Mouse endocrine gland-derived vascular endothelial growth factor: A distinct expression pattern from its human ortholog suggests different roles as a regulator of organ-specific angiogenesis. Endocrinology 2003, 144, 2606–2616. [Google Scholar] [CrossRef]
- Brouillet, S.; Hoffmann, P.; Chauvet, S.; Salomon, A.; Chamboredon, S.; Sergent, F.; Benharouga, M.; Feige, J.-J.; Alfaidy, N. Revisiting the role of hCG: New regulation of the angiogenic factor EG-VEGF and its receptors. Cell. Mol. Life Sci. 2012, 69, 1537–1550. [Google Scholar] [CrossRef]
- Traboulsi, W.; Brouillet, S.; Sergent, F.; Boufettal, H.; Samouh, N.; Aboussaouira, T.; Hoffmann, P.; Feige, J.J.; Benharouga, M.; Alfaidy, N. Prokineticins in central and peripheral control of human reproduction. Horm. Mol. Biol. Clin. Investig. 2015, 24, 73–81. [Google Scholar] [CrossRef]
- Ujvari, D.; Jakson, I.; Oldmark, C.; Attarha, S.; Alkasalias, T.; Salamon, D.; Gidlof, S.; Hirschberg, A.L. Prokineticin 1 is up-regulated by insulin in decidualizing human endometrial stromal cells. J. Cell. Mol. Med. 2018, 22, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ujvari, D.; Graells Brugalla, C.; Hirschberg, A.L. Dihydrotestosterone potentiates insulin to up-regulate prokineticin-1 in decidualizing human endometrial stromal cells. J. Cell. Mol. Med. 2020, 24, 3242–3245. [Google Scholar] [CrossRef] [PubMed]
- Garnier, V.; Traboulsi, W.; Salomon, A.; Brouillet, S.; Fournier, T.; Winkler, C.; Desvergne, B.; Hoffmann, P.; Zhou, Q.-Y.; Congiu, C.; et al. PPARgamma controls pregnancy outcome through activation of EG-VEGF: New insights into the mechanism of placental development. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E357–E369. [Google Scholar] [CrossRef] [PubMed]
- Marsango, S.; di Patti, M.C.B.; Barra, D.; Miele, R. The Bv8 gene from Bombina orientalis: Molecular cloning, genomic organization and functional characterization of the promoter. Peptides 2009, 30, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Bittman, E.L.; Hattar, S.; Zhou, Q.Y. Regulation of prokineticin 2 expression by light and the circadian clock. BMC Neurosci. 2005, 6, 17. [Google Scholar] [CrossRef]
- Zhang, C.; Ng, K.L.; Li, J.D.; He, F.; Anderson, D.J.; Sun, Y.E.; Zhou, Q.Y. Prokineticin 2 is a target gene of proneural basic helix-loop-helix factors for olfactory bulb neurogenesis. J. Biol. Chem. 2007, 282, 6917–6921. [Google Scholar] [CrossRef]
- Meng, S.; Gu, Q.; Yang, X.; Lv, J.; Owusu, I.; Matrone, G.; Chen, K.; Cooke, J.P.; Fang, L. TBX20 Regulates Angiogenesis through the Prokineticin 2-Prokineticin Receptor 1 Pathway. Circulation 2018, 138, 913–928. [Google Scholar] [CrossRef]
- Xin, H.; Lu, R.; Lee, H.; Zhang, W.; Zhang, C.; Deng, J.; Liu, Y.; Shen, S.; Wagner, K.U.; Forman, S.; et al. G-protein-coupled receptor agonist BV8/prokineticin-2 and STAT3 protein form a feed-forward loop in both normal and malignant myeloid cells. J. Biol. Chem. 2013, 288, 13842–13849. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef]
- Meng, L.; Yang, H.; Jin, C.; Quan, S. miR-28-5p suppresses cell proliferation and weakens the progression of polycystic ovary syndrome by targeting prokineticin-1. Mol. Med. Rep. 2019, 20, 2468–2475. [Google Scholar] [CrossRef]
- Su, M.T.; Tsai, P.Y.; Tsai, H.L.; Chen, Y.C.; Kuo, P.L. miR-346 and miR-582-3p-regulated EG-VEGF expression and trophoblast invasion via matrix metalloproteinases 2 and 9. Biofactors 2017, 43, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Tsai, P.Y.; Chen, T.Y.; Tsai, H.L.; Kuo, P.L.; Su, M.T. Elevated miR-200a and miR-141 inhibit endocrine gland-derived vascular endothelial growth factor expression and ciliogenesis in preeclampsia. J. Physiol. 2019, 597, 3069–3083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Li, P. The novel function of miR-3195 for mutant PROK2 (c.223-4C>A) degradation. Cell Biol. Int. 2021, 45, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Boisbouvier, J.; Albrand, J.P.; Blackledge, M.; Jaquinod, M.; Schweitz, H.; Lazdunski, M.; Marion, D. A structural homologue of colipase in black mamba venom revealed by NMR floating disulphide bridge analysis. J. Mol. Biol. 1998, 283, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.A.; Daly, N.L.; Vetter, I.; Mobli, M.; Napier, I.A.; Craik, D.J.; Lewis, R.J.; Christie, M.J.; King, G.F.; Alewood, P.F.; et al. Chemical synthesis and structure of the prokineticin Bv8. ChemBioChem 2010, 11, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Van Tilbeurgh, H.; Bezzine, S.; Cambillau, C.; Verger, R.; Carriere, F. Colipase: Structure and interaction with pancreatic lipase. Biochim. Biophys. Acta 1999, 1441, 173–184. [Google Scholar] [CrossRef]
- Niehrs, C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Bullock, C.M.; Li, J.D.; Zhou, Q.Y. Structural Determinants Required for the Bioactivities of Prokineticins and Identification of Prokineticin Receptor Antagonists. Mol. Pharmacol. 2004, 65, 582–588. [Google Scholar] [CrossRef]
- Negri, L.; Lattanzi, R.; Giannini, E.; Colucci, M.; Grohovaz, F.; Codazzi, F.; Mignogna, G.; Barra, D.; Kaiser, A.; Kreil, G.; et al. Biological activity of Bv8 analogues. Br. J. Pharmacol. 2005, 146, 625–632. [Google Scholar] [CrossRef]
- Giannini, E.; Lattanzi, R.; Nicotra, A.; Campese, A.F.; Grazioli, P.; Screpanti, I.; Balboni, G.; Severo, S.; Sacerdote, P.; Negri, L. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc. Natl. Acad. Sci. USA 2009, 106, 14646–14651. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Khusal, K.G.; Tonelli, R.R.; Mattos, E.C.; Soares, C.-O.; Di Genova, B.M.; Juliano, M.A.; Urias, U.; Colli, W.; Alves, M.J.M. Prokineticin receptor identified by phage display is an entry receptor for Trypanosoma cruzi into mammalian cells. Parasitol. Res. 2015, 114, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Ferrara, N. Role of the microenvironment in tumor growth and in refract toriness/resistance to anti-angiogenic therapies. Drug Resist. Updat. 2008, 11, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Monnier, J.; Samson, M. Prokineticins in angiogenesis and cancer. Cancer Lett. 2010, 296, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Congiu, C.; Onnis, V.; Deplano, A.; Salvadori, S.; Marconi, V.; Maftei, D.; Negri, L.; Lattanzi, R.; Balboni, G. A new convenient synthetic method and preliminary pharmacological characterization of triazinediones as prokineticin receptor antagonists. Eur. J. Med. Chem. 2014, 81, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Congiu, C.; Onnis, V.; Deplano, A.; Salvadori, S.; Marconi, V.; Maftei, D.; Francioso, A.; Ambrosio, C.; Casella, I.; et al. Halogenated triazinediones behave as antagonists of PKR1: In vitro and in vivo pharmacological characterization. Int. J. Pharm. Sci. Res. 2015, 10, 1033–1042. [Google Scholar] [CrossRef]
- Maftei, D.; Marconi, V.; Florenzano, F.; Giancotti, L.A.; Castelli, M.; Moretti, S.; Borsani, E.; Rodella, L.F.; Balboni, G.; Luongo, L.; et al. Controlling the activation of the Bv8/prokineticin system reduces neuroinflammation and abolishes thermal and tactile hyperalgesia in neuropathic animals. Br. J. Pharmacol. 2014, 171, 4850–4865. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Marconi, V.; Florenzano, F.; Franchi, S.; Borsani, E.; Rodella, F.L.; Balboni, G.; Salvadori, S.; Sacerdote, P.; et al. Prokineticin 2 upregulation in the peripheral nervous system has a major role in triggering and maintaining neuropathic pain in the chronic constriction injury model. BioMed Res. Int. 2015, 2015, 301292. [Google Scholar] [CrossRef]
- Guida, F.; Lattanzi, R.; Boccella, S.; Maftei, D.; Romano, R.; Marconi, V.; Balboni, G.; Salvadori, S.; Scafuro, M.A.; De Novellis, V.; et al. PC1, a non-peptide PKR1-preferring antagonist, reduces pain behavior and spinal neuronal sensitization in neuropathic mice. Pharmacol. Res. 2015, 91, 36–46. [Google Scholar] [CrossRef]
- Castelli, M.; Amodeo, G.; Negri, L.; Lattanzi, R.; Maftei, D.; Gotti, C.; Pistillo, F.; Onnis, V.; Congiu, C.; Panerai, A.E.; et al. Antagonism of the Prokineticin System Prevents and Reverses Allodynia and Inflammation in a Mouse Model of Diabetes. PLoS ONE 2016, 11, e0146259. [Google Scholar] [CrossRef]
- Moschetti, G.; Amodeo, G.; Maftei, D.; Lattanzi, R.; Procacci, P.; Sartori, P.; Balboni, G.; Onnis, V.; Conte, V.; Panerai, A.; et al. Targeting prokineticin system counteracts hypersensitivity, neuroinflammation, and tissue damage in a mouse model of bortezomib-induced peripheral neuropathy. J. Neuroinflamm. 2019, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, G.; Amodeo, G.; Paladini, M.S.; Molteni, R.; Balboni, G.; Panerai, A.; Sacerdote, P.; Franchi, S. Prokineticin 2 promotes and sustains neuroinflammation in vincristine treated mice: Focus on pain and emotional like behavior. Brain Behav. Immun. 2019, 82, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, G.; Kalpachidou, T.; Amodeo, G.; Lattanzi, R.; Sacerdote, P.; Kress, M.; Franchi, S. Prokineticin Receptor Inhibition with PC1 protects Mouse Primary Sensory Neurons from Neurotoxic Effects of Chemotherapeutic Drugs In Vitro. Front. Immunol. 2020, 11, 2119. [Google Scholar] [CrossRef] [PubMed]
- Maftei, D.; Ratano, P.; Fusco, I.; Marconi, V.; Squillace, S.; Negri, L.; Severini, C.; Balboni, G.; Steardo, L.; Bronzuoli, M.R.; et al. The prokineticin receptor antagonist PC1 rescues memory impairment induced by β Amyloid administration through the modulation of prokineticin system. Neuropharmacology 2019, 158, 107739. [Google Scholar] [CrossRef] [PubMed]
- Abou-hamdan, M.; Costanza, M.; Fontana, E.; Di Dario, M.; Musio, S.; Congiu, C.; Onnis, V.; Lattanzi, R.; Radaelli, M.; Martinelli, V.; et al. Critical role for prokineticin 2 in central nervous system autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e95. [Google Scholar] [CrossRef]
- Goryszewska-Szczurek, E.; Baryla, M.; Kaczynski, P.; Waclawik, A. Prokineticin 1–prokineticin receptor 1 signaling in trophoblast promotes embryo implantation and placenta development. Sci. Rep. 2019, 11, 13715. [Google Scholar] [CrossRef]
- Curtis, V.F.; Wang, H.; Yang, P.; McLendon, R.E.; Li, X.; Zhou, Q.Y.; Wang, X.F. A PK2/Bv8/PROK2 Antagonist Suppresses Tumorigenic Processes by Inhibiting Angiogenesis in Glioma and Blocking Myeloid Cell Infiltration in Pancreatic Cancer. PLoS ONE 2013, 8, e54916. [Google Scholar] [CrossRef]
- Ito, H.; Noda, K.; Yoshida, K.; Otani, K.; Yoshiga, M.; Oto, Y.; Saito, S.; Kurosaka, D. Prokineticin 2 antagonist, PKRA7 suppresses arthritis in mice with collagen-induced arthritis. BMC Musculoskelet. Disord. 2016, 17, 387. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Su, Y.; Zhou, T.; Hu, Z.; Wei, J.; Wang, W.; Liu, C.; Zhang, H.; Zhao, K. Activation of the NLRP3 Inflammasome Pathway by Prokineticin 2 in Testicular Macrophages of Uropathogenic Escherichia coli—Induced Orchitis. Front. Immunol. 2019, 14, 1872. [Google Scholar] [CrossRef]
- Negri, L.; Lattanzi, R.; Giannini, E.; Melchiorri, P. Bv8/Prokineticin proteins and their receptors. Life Sci. 2007, 81, 1103–1116. [Google Scholar] [CrossRef]
- Balboni, G.; Lazzari, I.; Trapella, C.; Negri, L.; Lattanzi, R.; Giannini, E.; Nicotra, A.; Melchiorri, P.; Visentin, S.; De Nuccio, C.; et al. Triazine Compounds as Antagonists at Bv8-Prokineticin Receptors. J. Med. Chem. 2008, 51, 7635–7639. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Polymorphisms | Phatology | Mechanism | References |
|---|---|---|---|
| rs6053283 | PL, DD | Alteration of the exonic splicing | Su et al., 2010 [18] Cao et al., 2016 [19] |
| Splice variants | |||
| TM 4-7 | AD | Impaired dimerization with PKR2 | Lattanzi et al., 2019 [23] |
| TM 1-5 | PP | Increase of Gα coupling capacity | Sposini et al., 2015 [24] Fukami et al., 2017 [25] |
| Mutations | |||
| R80C | KS, IHH | Impaired PKRs N-glycosylation | Monnier et al., 2009 [26] Abreu et al., 2012 [27] |
| R85H | IHH, HSCR | Impaired PKRs N-glycosylation | Monnier et al., 2009 [26] Ruiz Ferrer et al., 2011 [22] Abreu et al., 2012 [27] |
| R85C | IHH, HSCR | Impaired PKRs N-glycosylation | Cole et al., 2008 [28] Ruiz Ferrer et al., 2011 [22] Abreu et al., 2012 [27] |
| R85G | IHH | Impaired Gα coupling | Cole et al., 2008 [28] Monnier et al., 2009 [26] |
| Y113H | KS, IHH | Impaired Gα coupling | Cole et al., 2008 [28] Zhao et al., 2019 [11] |
| V115M | KS | Impaired Gα coupling | Cole et al., 2008 [28] |
| L218P | IHH | Impaired Gα coupling | Zhao et al., 2019 [11] |
| R164Q | KS | Impaired Gα coupling | Cole et al., 2008 [28] Monnier et al., 2009 [26] |
| L173R | KS, IHH | Inability to reach the cell surface | Cole et al., 2008 [28] Monnier et al., 2009 [26] Abreu et al., 2010 [29] Libri et al., 2014 [30] Cox et al., 2018 [31] |
| W178S | KS, IHH | Inability to reach the cell surface | Cole et al., 2008 [28] Monnier et al., 2009 [26] Chen et al., 2014 [32] Zhao et al., 2019 [11] |
| S188L | KS | Impaired Gα coupling | Cole et al., 2008 [28] |
| Q210R | KS, IHH | Impaired ligand binding | Dodé et al., 2006 [33] Monnier et al., 2009 [26] |
| L218P | IHH | Impaired Gα coupling | Zhao et al., 2019 [11] |
| G229R | IHH | Inability to reach the cell surface | Zhao et al., 2019 [11] |
| E231K | IHH | Inability to reach the cell surface | Zhao et al., 2019 [11] |
| G234D | KS, IHH | Impaired dimerization with PKR2 | Chen et al., 2014 [32] Cox et al., 2018 [31] |
| R248Q | KS | Impaired Gα coupling | Cole et al., 2008 [29] |
| T260M | IHH | Impaired Gα coupling | Monnier et al., 2009 [26] Libri et al., 2014 [30] |
| R268C | IHH, HSCR | Impaired Gα coupling | Libri et al., 2014 [30] Ruiz Ferrer et al., 2011 [22] Cox et al., 2018 [31] |
| R270H | IHH | Impaired Gα coupling | Zhao et al., 2019 [11] |
| V274D | KS, IHH | Inability to reach the cell surface | Libri et al., 2014 [30] |
| P290S | KS, IHH, HSCR | Inability to reach the cell surface | Monnier et al., 2009 [26] Ruiz Ferrer et al., 2011 [22] Chen et al., 2014 [32] Cox et al., 2018 [31] |
| V331M | KS, IHH | Impaired Gα coupling | Cole et al., 2008 [29] Monnier et al., 2009 [26] Libri et al., 2014 [30] |
| V334M | IHH | Increased ability to reach cell surface | Libri et al., 2014 [30] |
| R353H | IHH | Impaired Gα coupling | Zhao et al., 2019 [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattanzi, R.; Miele, R. Prokineticin-Receptor Network: Mechanisms of Regulation. Life 2022, 12, 172. https://doi.org/10.3390/life12020172
Lattanzi R, Miele R. Prokineticin-Receptor Network: Mechanisms of Regulation. Life. 2022; 12(2):172. https://doi.org/10.3390/life12020172
Chicago/Turabian StyleLattanzi, Roberta, and Rossella Miele. 2022. "Prokineticin-Receptor Network: Mechanisms of Regulation" Life 12, no. 2: 172. https://doi.org/10.3390/life12020172
APA StyleLattanzi, R., & Miele, R. (2022). Prokineticin-Receptor Network: Mechanisms of Regulation. Life, 12(2), 172. https://doi.org/10.3390/life12020172