
Multiprotein GLI Transcriptional Complexes as Therapeutic Targets in Cancer


Abstract
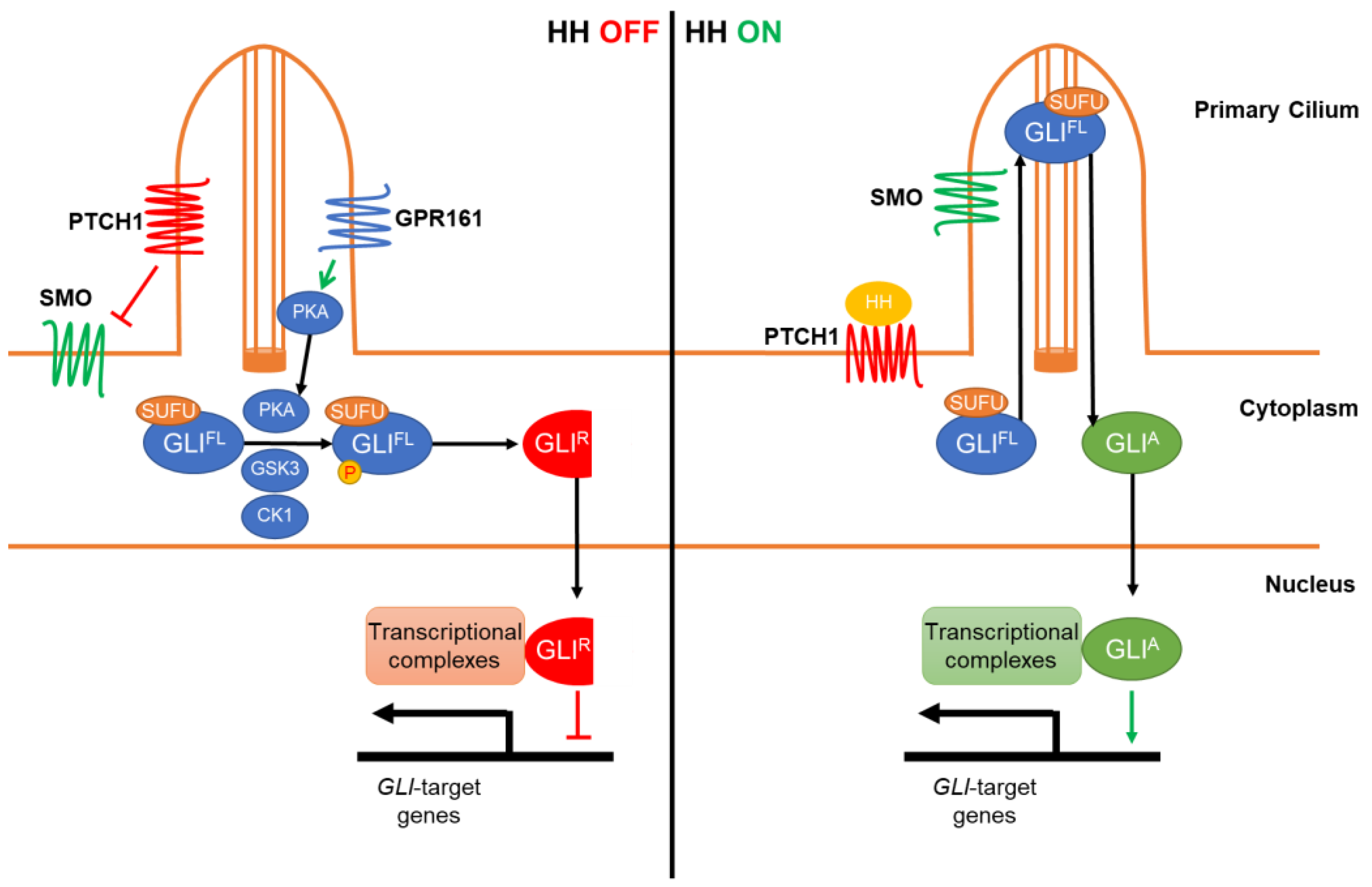
:1. The Hedgehog Signaling and GLI Regulation
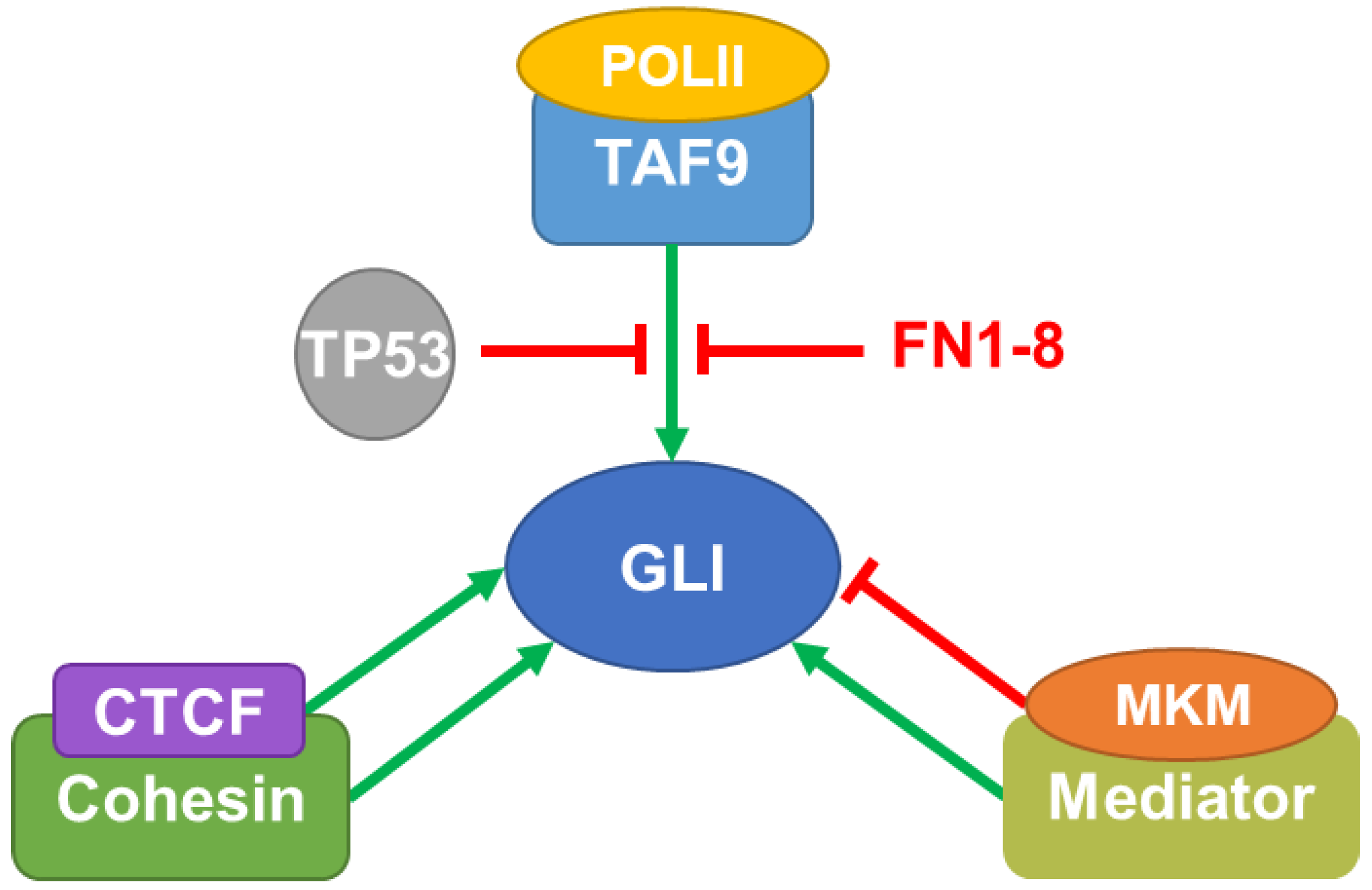
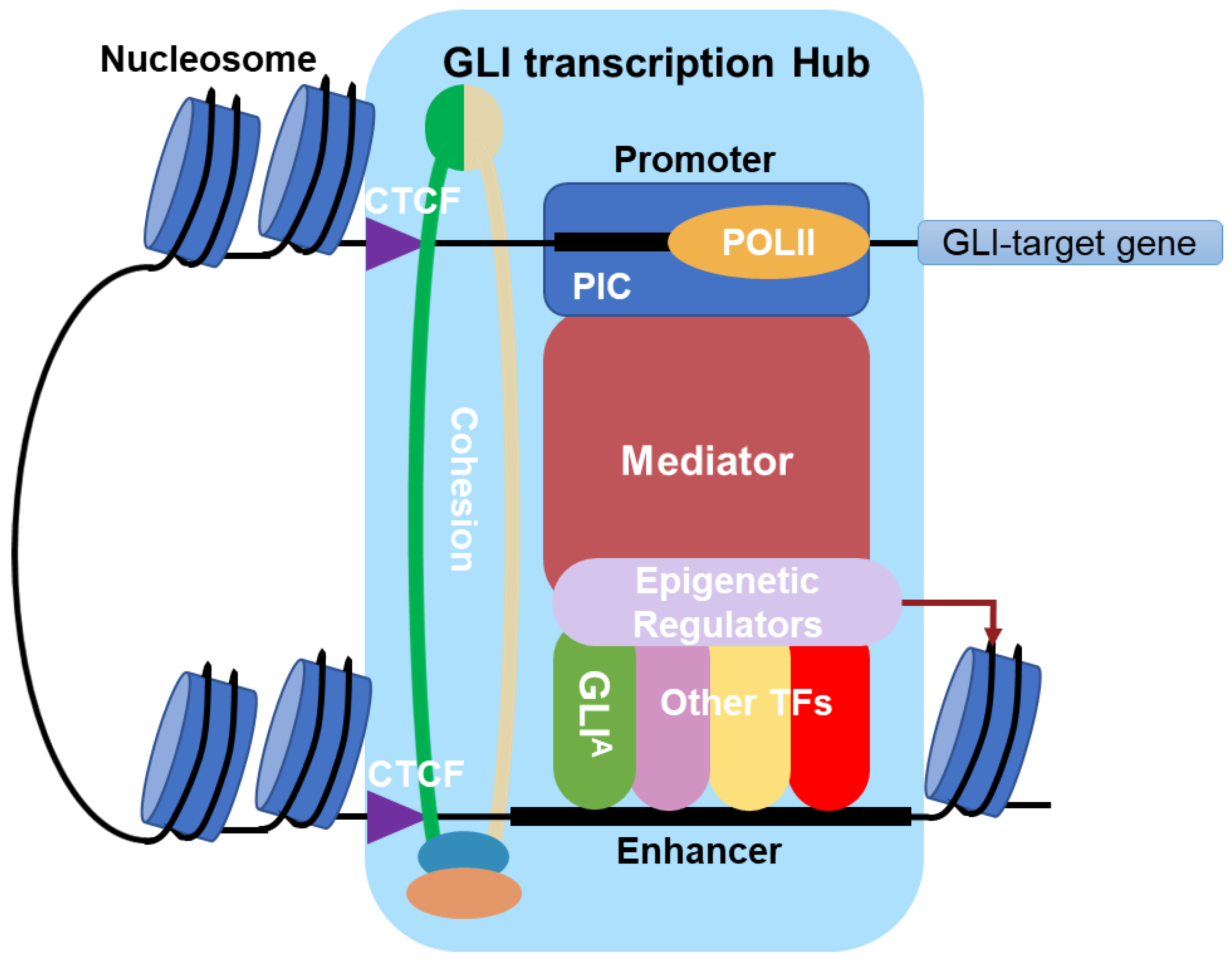
2. Multi-Protein GLI Transcriptional Complexes
2.1. General Transcription Machinery
2.1.1. Transcription Factor II D Complex and TP53
2.1.2. Mediator Complex
2.1.3. Cohesin-CCCTC-Binding Factor Complex
2.2. Epigenetic Regulators
2.2.1. Bromodomain-Containing 4 Complexes and SRY-Box Transcription Factor 2
2.2.2. Histone/GLI-Dual Regulators
2.2.3. Polycomb Repressive Complex 2
2.2.4. Switch/Sucrose Non-Fermentable Complex
2.2.5. Ubiquitin Like with PHD and Ring Finger Domains 1-DNA Methyltransferase 1 Complex
2.3. Transcription Factors
2.3.1. Serum Response Factor-Megakaryoblastic Leukemia 1 Complex
2.3.2. Activator Protein 1 Complex
2.3.3. SMAD Complex
2.3.4. GATA Binding Protein-Friend of GATA Complex
2.3.5. SUFU-Containing Complexes
2.3.6. Potential GLI Complexes
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, J. Hedgehog signaling mechanism and role in cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; E Hooper, J. The Drosophila smoothened Gene Encodes a Seven-Pass Membrane Protein, a Putative Receptor for the Hedgehog Signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.-L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.S.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M. US Food and Drug Administration approval: Vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Regl, G.; Frischauf, A.-M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [CrossRef] [Green Version]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Speer, R.M.; Volk, L.; Hudson, L.G.; Liu, K.J. Arsenic co-carcinogenesis: Inhibition of DNA repair and interaction with zinc finger proteins. Semin. Cancer Biol. 2021, 76, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderburgh, J.P.; Kwakwa, K.A.; Werfel, T.A.; Merkel, A.; Gupta, M.; Johnson, R.W.; Guelcher, S.A.; Duvall, C.L.; Rhoades, J.A. Systemic delivery of a Gli inhibitor via polymeric nanocarriers inhibits tumor-induced bone disease. J. Control. Release 2019, 311–312, 257–272. [Google Scholar] [CrossRef]
- Calcaterra, A.; Iovine, V.; Botta, B.; Quaglio, D.; D’Acquarica, I.; Ciogli, A.; Iazzetti, A.; Alfonsi, R.; Lospinoso Severini, L.; Infante, P.; et al. Chemical, computational and functional insights into the chemical stability of the Hedgehog pathway inhibitor GANT61. J. Enzym. Inhib. Med. Chem. 2018, 33, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Zhang, Y.; Fang, M.; Jehan, S.; Zhou, W. Current Advances of Nanomedicines Delivering Arsenic Trioxide for Enhanced Tumor Therapy. Pharmaceutics 2022, 14, 743. [Google Scholar] [CrossRef]
- Ingallina, C.; Costa, P.M.; Ghirga, F.; Klippstein, R.; Wang, J.T.; Berardozzi, S.; Hodgins, N.; Infante, P.; Pollard, S.M.; Botta, B.; et al. Polymeric glabrescione B nanocapsules for passive targeting of Hedgehog-dependent tumor therapy in vitro. Nanomedicine 2017, 12, 711–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.; Malfanti, A.; Quaglio, D.; Balducci, S.; De Martin, S.; Bufalieri, F.; Mastrotto, F.; Basili, I.; Garofalo, M.; Severini, L.L.; et al. Glabrescione B delivery by self-assembling micelles efficiently inhibits tumor growth in preclinical models of Hedgehog-dependent medulloblastoma. Cancer Lett. 2020, 499, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Chenna, V.; Hu, C.; Pramanik, D.; Aftab, B.T.; Karikari, C.; Campbell, N.R.; Hong, S.-M.; Zhao, M.; Rudek, M.A.; Khan, S.R.; et al. A Polymeric Nanoparticle Encapsulated Small-Molecule Inhibitor of Hedgehog Signaling (NanoHHI) Bypasses Secondary Mutational Resistance to Smoothened Antagonists. Mol. Cancer Ther. 2012, 11, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Struhl, G. Dual Roles for Patched in Sequestering and Transducing Hedgehog. Cell 1996, 87, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Ingham, P.W.; Taylor, A.M.; Nakano, Y. Role of the Drosophila patched gene in positional signalling. Nature 1991, 353, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Rohatgi, R.; Siebold, C. Cholesterol access in cellular membranes controls Hedgehog signaling. Nat. Chem. Biol. 2020, 16, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Wen, X.; Ratti, N.; Loktev, A.; Rangell, L.; Scales, S.J.; Jackson, P.K. The Ciliary G-Protein-Coupled Receptor Gpr161 Negatively Regulates the Sonic Hedgehog Pathway via cAMP Signaling. Cell 2013, 152, 210–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [CrossRef]
- Aza-Blanc, P.; Ramírez-Weber, F.-A.; Laget, M.-P.; Schwartz, C.; Kornberg, T.B. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 1997, 89, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Fallon, J.F.; A Beachy, P. Hedgehog-Regulated Processing of Gli3 Produces an Anterior/Posterior Repressor Gradient in the Developing Vertebrate Limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog Signaling Effector Cubitus interruptus Requires Phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that Patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Charron, F.; Morin, S.; Shin, D.S.; Wong, K.; Fabre, P.J.; Tessier-Lavigne, M.; McConnell, S.K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature 2006, 444, 369–373. [Google Scholar] [CrossRef]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Pal, K.; Hwang, S.-H.; Somatilaka, B.; Badgandi, H.; Jackson, P.K.; DeFea, K.; Mukhopadhyay, S. Smoothened determines β-arrestin–mediated removal of the G protein–coupled receptor Gpr161 from the primary cilium. J. Cell Biol. 2016, 212, 861–875. [Google Scholar] [CrossRef] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef]
- Ding, Q.; Fukami, S.-I.; Meng, X.; Nishizaki, Y.; Zhang, X.; Sasaki, H.; Dlugosz, A.; Nakafuku, M.; Hui, C.-C. Mouse Suppressor of fused is a negative regulator of Sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr. Biol. 1999, 9, 1119–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.R.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P. Mammalian Suppressor-of-Fused modulates nuclear–cytoplasmic shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Barnfield, P.C.; Zhang, X.; Thanabalasingham, V.; Yoshida, M.; Hui, C.-C. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation 2005, 73, 397–405. [Google Scholar] [CrossRef]
- Ingham, P.W.; Nakano, Y.; Seger, C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 2011, 12, 393–406. [Google Scholar] [CrossRef]
- Bai, C.; Stephen, D.; Joyner, A.L. All Mouse Ventral Spinal Cord Patterning by Hedgehog Is Gli Dependent and Involves an Activator Function of Gli3. Dev. Cell 2004, 6, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Motoyama, J.; Gasca, S.; Mo, R.; Sasaki, H.; Rossant, J.; Hui, C. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 1998, 125, 2533–2543. [Google Scholar] [CrossRef]
- Motoyama, J.; Liu, J.; Mo, R.; Ding, Q.; Post, M.; Hui, C.-C. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 1998, 20, 54–57. [Google Scholar] [CrossRef]
- Park, H.; Bai, C.; Platt, K.; Matise, M.; Beeghly, A.; Hui, C.; Nakashima, M.; Joyner, A. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2016, 9, a022137. [Google Scholar] [CrossRef] [PubMed]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [Green Version]
- Kern, D.; Regl, G.; Hofbauer, S.W.; Altenhofer, P.; Achatz, G.; Dlugosz, A.; Schnidar, H.; Greil, R.; Hartmann, T.N.; Aberger, F. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene 2015, 34, 5341–5351. [Google Scholar] [CrossRef] [Green Version]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with Resistance to Smoothened Antagonists by Inhibition of the PI3K Pathway in Medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [Green Version]
- Kebenko, M.; Drenckhan, A.; Gros, S.J.; Jücker, M.; Grabinski, N.; Ewald, F.; Grottke, A.; Schultze, A.; Izbicki, J.R.; Bokemeyer, C.; et al. ErbB2 signaling activates the Hedgehog pathway via PI3K–Akt in human esophageal adenocarcinoma: Identification of novel targets for concerted therapy concepts. Cell. Signal. 2015, 27, 373–381. [Google Scholar] [CrossRef]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF–MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Bosco-Clément, G.; Zhang, F.; Chen, Z.; Zhou, H.-M.; Li, H.; Mikami, I.; Hirata, T.; Yagui-Beltran, A.; Lui, N.; Do, H.T.; et al. Targeting Gli transcription activation by small molecule suppresses tumor growth. Oncogene 2013, 33, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.W.; Lamm, M.; Iannaccone, S.; Higashiyama, N.; Leong, K.F.; Iannaccone, P.; Walterhouse, D. p53 modulates the activity of the GLI1 oncogene through interactions with the shared coactivator TAF9. DNA Repair 2015, 34, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Kim, S.; Ishii, S.; Boyer, T.G. Mediator Modulates Gli3-Dependent Sonic Hedgehog Signaling. Mol. Cell. Biol. 2006, 26, 8667–8682. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Spaeth, J.M.; Kim, N.H.; Xu, X.; Friez, M.J.; Schwartz, C.E.; Boyer, T.G. MED12 mutations link intellectual disability syndromes with dysregulated GLI3-dependent Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 19763–19768. [Google Scholar] [CrossRef] [Green Version]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet. 2018, 50, 460–471. [Google Scholar] [CrossRef]
- Hallson, G.; Syrzycka, M.; Beck, S.A.; Kennison, J.A.; Dorsett, D.; Page, S.L.; Hunter, S.M.; Keall, R.; Warren, W.D.; Brock, H.W. The Drosophila cohesin subunit Rad21 is a trithorax group (trxG) protein. Proc. Natl. Acad. Sci. USA 2008, 105, 12405–12410. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Niu, B.; Cheah, K.S.E.; Alman, B. Unique and overlapping GLI1 and GLI2 transcriptional targets in neoplastic chondrocytes. PLoS ONE 2019, 14, e0211333. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI Underlies a Role for BET Bromodomains in Pancreatic Cancer Growth and the Tumor Microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gaudio, E.; Gagliardi, S.; Zitani, M.; Carrassa, L.; Migliorini, F.; Petricci, E.; Manetti, F.; Makukhin, N.; Bond, A.G.; et al. Targeting non-canonical activation of GLI1 by the SOX2-BRD4 transcriptional complex improves the efficacy of HEDGEHOG pathway inhibition in melanoma. Oncogene 2021, 40, 3799–3814. [Google Scholar] [CrossRef]
- Tang, Y.; Gholamin, S.; Schubert, S.; I Willardson, M.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2019, 27, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Swiderska-Syn, M.; Mir-Pedrol, J.; Oles, A.; Schleuger, O.; Salvador, A.D.; Greiner, S.M.; Seward, C.; Yang, F.; Babcock, B.R.; Shen, C.; et al. Noncanonical activation of GLI signaling in SOX2+ cells drives medulloblastoma relapse. Sci. Adv. 2022, 8, eabj9138. [Google Scholar] [CrossRef]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced Activation of the Gli1Promoter Is Mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone Acetyltransferase PCAF Is Required for Hedgehog–Gli-Dependent Transcription and Cancer Cell ProliferationPCAF Is Required for GLI-Dependent Transcription. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Shinagawa, T.; Nomura, T.; Harada, J.; Kaul, S.C.; Wadhwa, R.; Khan, M.; Akimaru, H.; Sasaki, H.; Colmenares, C.; et al. Ski is involved in transcriptional regulation by the repressor and full-length forms of Gli3. Genes Dev. 2002, 16, 2843–2848. [Google Scholar] [CrossRef] [Green Version]
- Lex, R.K.; Ji, Z.; Falkenstein, K.N.; Zhou, W.; Henry, J.L.; Ji, H.; A Vokes, S. GLI transcriptional repression regulates tissue-specific enhancer activity in response to Hedgehog signaling. eLife 2020, 9, e50670. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, S.; Wang, D.; Chen, X.; Kalvakolanu, D.V.; Zheng, H.; Wei, X.; Wen, N.; Liang, H.; Guo, B. A Combination of BRD4 and HDAC3 Inhibitors Synergistically Suppresses Glioma Stem Cell Growth by Blocking GLI1/IL6/STAT3 Signaling AxisGlioma Stem Cell Suppression by BRD4 and HDAC3 Inhibitors. Mol. Cancer Ther. 2020, 19, 2542–2553. [Google Scholar] [CrossRef]
- Gurung, B.; Feng, Z.; Iwamoto, D.V.; Thiel, A.; Jin, G.; Fan, C.-M.; Ng, J.M.; Curran, T.; Hua, X. Menin Epigenetically Represses Hedgehog Signaling in MEN1 Tumor Syndrome. Cancer Res. 2013, 73, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- Gurung, B.; Feng, Z.; Hua, X. Menin Directly Represses Gli1 Expression Independent of Canonical Hedgehog SignalingMenin/PRMT5 Directly Suppresses Gli1 Expression. Mol. Cancer Res. 2013, 11, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhang, J.; Tian, T.; Fu, X.; Wang, W.; Li, S.; Shi, T.; Suo, A.; Ruan, Z.; Guo, H. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumor Biol. 2016, 37, 9311–9322. [Google Scholar] [CrossRef]
- Jeon, S.; Seong, R.H. Anteroposterior Limb Skeletal Patterning Requires the Bifunctional Action of SWI/SNF Chromatin Remodeling Complex in Hedgehog Pathway. PLoS Genet. 2016, 12, e1005915. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Zhang, Z.; Zhan, X.; Cao, M.; Satoh, T.; Akira, S.; Shpargel, K.; Magnuson, T.; Li, Q.; Wang, R.; et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat. Commun. 2014, 5, 5425. [Google Scholar] [CrossRef]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Shi, X.; Zhang, Z.; Chen, Y.; Wu, J.I. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 2011, 108, 12758–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safgren, S.L.; Olson, R.L.; Vrabel, A.M.; Almada, L.L.; Marks, D.L.; Hernandez-Alvarado, N.; Gaspar-Maia, A.; Fernandez-Zapico, M.E. The transcription factor GLI1 cooperates with the chromatin remodeler SMARCA2 to regulate chromatin accessibility at distal DNA regulatory elements. J. Biol. Chem. 2020, 295, 8725–8735. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Wu, J.I.; Ranish, J.A.; Wan, M.; Winslow, M.M.; Staahl, B.T.; Wu, H.; Aebersold, R.; Graef, I.A.; Crabtree, G.R. An Essential Switch in Subunit Composition of a Chromatin Remodeling Complex during Neural Development. Neuron 2007, 55, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Rodriguez-Blanco, J.; Long, J.; Swiderska-Syn, M.; Wynn, D.T.; Li, B.; Shen, C.; Nayak, A.; Ban, Y.; Sun, X. A druggable UHRF1/DNMT1/GLI complex regulates Sonic hedgehog dependent tumor growth. Mol. Cancer Res. 2022, 20, 1598–1610. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Anichini, G.; Sala, C.; Manetti, F.; Almada, L.L.; Pepe, S.; Carr, R.M.; Paradise, B.D.; Sarkaria, J.N.; Davila, J.I.; et al. ST3GAL1 is a target of the SOX2-GLI1 transcriptional complex and promotes melanoma metastasis through AXL. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Peterson, K.A.; Nishi, Y.; Ma, W.; Vedenko, A.; Shokri, L.; Zhang, X.; McFarlane, M.; Baizabal, J.-M.; Junker, J.P.; van Oudenaarden, A.; et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev. 2012, 26, 2802–2816. [Google Scholar] [CrossRef] [Green Version]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Selvadurai, H.J.; Luis, E.; Desai, K.; Lan, X.; Vladoiu, M.C.; Whitley, O.; Galvin, C.; Vanner, R.J.; Lee, L.; Whetstone, H.; et al. Medulloblastoma Arises from the Persistence of a Rare and Transient Sox2+ Granule Neuron Precursor. Cell Rep. 2020, 31, 107511. [Google Scholar] [CrossRef]
- Laner-Plamberger, S.; Kaser, A.; Paulischta, M.; Hauser-Kronberger, C.; Eichberger, T.; Frischauf, A.M. Cooperation between GLI and JUN enhances transcription of JUN and selected GLI target genes. Oncogene 2009, 28, 1639–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.D.; Haensel, D.; Gaddam, S.; Patel, T.; Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; McKellar, S.; Shankar, G.; Aasi, S.; et al. AP-1 and TGFß cooperativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nye, M.D.; Almada, L.L.; Fernandez-Barrena, M.G.; Marks, D.L.; Elsawa, S.F.; Vrabel, A.; Tolosa, E.J.; Ellenrieder, V.; Fernandez-Zapico, M.E. The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J. Biol. Chem. 2014, 289, 15495–15506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, I.; Alvarez-Rodríguez, R.; Martí, E.; Pons, S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development 2004, 131, 3159–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Massagué, J. Carboxy-terminally truncated Gli3 proteins associate with Smads. Nat. Genet. 1998, 20, 325–326. [Google Scholar] [CrossRef]
- Kozhemyakina, E.; Ionescu, A.; Lassar, A.B. GATA6 Is a Crucial Regulator of Shh in the Limb Bud. PLoS Genet. 2014, 10, e1004072. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Akiyama, R.; Wong, J.; Tahara, N.; Kawakami, H.; Kawakami, Y. Gata6-Dependent GLI3 Repressor Function is Essential in Anterior Limb Progenitor Cells for Proper Limb Development. PLoS Genet. 2016, 12, e1006138. [Google Scholar] [CrossRef] [Green Version]
- Daoud, G.; Kempf, H.; Kumar, D.; Kozhemyakina, E.; Holowacz, T.; Kim, D.-W.; Ionescu, A.; Lassar, A.B. BMP-mediated induction of GATA4/5/6 blocks somitic responsiveness to SHH. Development 2014, 141, 3978–3987. [Google Scholar] [CrossRef] [Green Version]
- Stone, D.; Murone, M.; Luoh, S.; Ye, W.; Armanini, M.; Gurney, A.; Phillips, H.; Brush, J.; Goddard, A.; de Sauvage, F.; et al. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J. Cell Sci. 1999, 112, 4437–4448. [Google Scholar] [CrossRef]
- Dunaeva, M.; Michelson, P.; Kogerman, P.; Toftgard, R. Characterization of the Physical Interaction of Gli Proteins with SUFU Proteins. J. Biol. Chem. 2003, 278, 5116–5122. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5442–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.-C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00421-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Yao, E.; Wang, K.; Nozawa, Y.; Shimizu, H.; Johnson, J.R.; Chen, J.-N.; Krogan, N.J.; Chuang, P.-T. Regulation of Sufu activity by p66β and Mycbp provides new insight into vertebrate Hedgehog signaling. Genes Dev. 2014, 28, 2547–2563. [Google Scholar] [CrossRef] [Green Version]
- Pearse, R.V., 2nd; Collier, L.S.; Scott, M.P.; Tabin, C.J. Vertebrate Homologs of Drosophila Suppressor of Fused Interact with the Gli Family of Transcriptional Regulators. Dev. Biol. 1999, 212, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.-C.; Satkunendran, T.; Mo, R.; Morrissy, S.; Zhang, X.; Huang, E.S.; Uusküla-Reimand, L.; Hou, H.; Son, J.E.; Liu, W.; et al. Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Dev. Cell 2018, 48, 167–183.e5. [Google Scholar] [CrossRef] [Green Version]
- Ayrault, O.; Zhao, H.; Zindy, F.; Qu, C.; Sherr, C.J.; Roussel, M.F. Atoh1 Inhibits Neuronal Differentiation and Collaborates with Gli1 to Generate Medulloblastoma-Initiating CellsAtoh1 in Medulloblastoma. Cancer Res. 2010, 70, 5618–5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Lex, R.K.; Chung, H.; Giovanetti, S.M.; Ji, Z.; Ji, H.; Person, M.D.; Kim, J.; Vokes, S.A. The Pluripotency Factor NANOG Binds to GLI Proteins and Represses Hedgehog-mediated Transcription. J. Biol. Chem. 2016, 291, 7171–7182. [Google Scholar] [CrossRef] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [Green Version]
- Girbig, M.; Misiaszek, A.D.; Müller, C.W. Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases. Nat. Rev. Mol. Cell Biol. 2022, 23, 603–622. [Google Scholar] [CrossRef]
- Farnung, L.; Vos, S.M. Assembly of RNA polymerase II transcription initiation complexes. Curr. Opin. Struct. Biol. 2022, 73, 102335. [Google Scholar] [CrossRef] [PubMed]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Mahindroo, N.; Connelly, M.C.; Punchihewa, C.; Kimura, H.; Smeltzer, M.; Wu, S.; Fujii, N. Structure−Activity Relationships and Cancer-Cell Selective Toxicity of Novel Inhibitors of Glioma-Associated Oncogene Homologue 1 (Gli1) Mediated Transcription. J. Med. Chem. 2009, 52, 4277–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, J.; Behrmann, L.; Muschhammer, J.; Modemann, F.; Khoury, K.; Brauneck, F.; Bokemeyer, C.; Campeau, E.; Fiedler, W. The BET bromodomain inhibitor ZEN-3365 targets the Hedgehog signaling pathway in acute myeloid leukemia. Ann. Hematol. 2021, 100, 2933–2941. [Google Scholar] [CrossRef] [PubMed]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mönkemeyer, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor Effects of a Combined 5-Aza-2′Deoxycytidine and Valproic Acid Treatment on Rhabdomyosarcoma and Medulloblastoma in Ptch Mutant Mice. Cancer Res. 2009, 69, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Richter, W.F.; Nayak, S.; Iwasa, J.; Taatjes, D.J. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2022, 23, 732–749. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2017, 19, 262–274. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 2020, 20, 504–515. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 1–25. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterveen, T.; Kurdija, S.; Alekseenko, Z.; Uhde, C.W.; Bergsland, M.; Sandberg, M.; Andersson, E.; Dias, J.M.; Muhr, J.; Ericson, J. Mechanistic Differences in the Transcriptional Interpretation of Local and Long-Range Shh Morphogen Signaling. Dev. Cell 2012, 23, 1006–1019. [Google Scholar] [CrossRef] [Green Version]
- Gavai, A.V.; Norris, D.; Delucca, G.; Tortolani, D.; Tokarski, J.S.; Dodd, D.; O’Malley, D.; Zhao, Y.; Quesnelle, C.; Gill, P.; et al. Discovery and Preclinical Pharmacology of an Oral Bromodomain and Extra-Terminal (BET) Inhibitor Using Scaffold-Hopping and Structure-Guided Drug Design. J. Med. Chem. 2021, 64, 14247–14265. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Coni, S.; Antonucci, L.; D’Amico, D.; Di Magno, L.; Infante, P.; De Smaele, E.; Giannini, G.; Di Marcotullio, L.; Screpanti, I.; Gulino, A.; et al. Gli2 Acetylation at Lysine 757 Regulates Hedgehog-Dependent Transcriptional Output by Preventing Its Promoter Occupancy. PLoS ONE 2013, 8, e65718. [Google Scholar] [CrossRef] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E. Histone deacetylase and Cullin3–REN KCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132. [Google Scholar] [CrossRef]
- Gai, X.; Tu, K.; Li, C.; Lu, Z.; Roberts, L.; Zheng, X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1712. [Google Scholar] [CrossRef] [Green Version]
- Pak, E.; MacKenzie, E.L.; Zhao, X.; Pazyra-Murphy, M.F.; Park, P.M.C.; Wu, L.; Shaw, D.L.; Addleson, E.C.; Cayer, S.S.; Lopez, B.G.-C.; et al. A large-scale drug screen identifies selective inhibitors of class I HDACs as a potential therapeutic option for SHH medulloblastoma. Neuro-Oncology 2019, 21, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Holz, P.S.; Finkernagel, F.; Fendrich, V.; Lauth, M. Histone Deacetylase 6 Represents a Novel Drug Target in the Oncogenic Hedgehog Signaling PathwayHDAC6 Regulates Hh Signaling. Mol. Cancer Ther. 2015, 14, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Liu, J.; Jiang, H.; Wang, J.; Li, X.; Wang, J.; Zhu, S.; Guo, J.; Li, T.; Zhong, Y.; et al. Proteasome inhibitor induced SIRT1 deacetylates GLI2 to enhance hedgehog signaling activity and drug resistance in multiple myeloma. Oncogene 2019, 39, 922–934. [Google Scholar] [CrossRef]
- Abe, Y.; Suzuki, Y.; Kawamura, K.; Tanaka, N. MEP50/PRMT5-mediated methylation activates GLI1 in Hedgehog signalling through inhibition of ubiquitination by the ITCH/NUMB complex. Commun. Biol. 2019, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Wu, H.; Cheng, S.Y.; Gao, D.; Zhang, L.; Zhao, Y. Set7 mediated Gli3 methylation plays a positive role in the activation of Sonic Hedgehog pathway in mammals. eLife 2016, 5, 15690. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Klose, R.J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 815–833. [Google Scholar] [CrossRef] [PubMed]
- Lex, R.K.; Zhou, W.; Ji, Z.; Falkenstein, K.N.; Schuler, K.E.; Windsor, K.E.; Kim, J.D.; Ji, H.; Vokes, S.A. GLI transcriptional repression is inert prior to Hedgehog pathway activation. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W. The SWI/SNF complex in cancer—biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Silva, C.; Vermeulen, W.; Lans, H. SWI/SNF: Complex complexes in genome stability and cancer. DNA Repair 2019, 77, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.J.; Fernandez-Barrena, M.G.; Iguchi, E.; McCleary-Wheeler, A.L.; Carr, R.M.; Almada, L.L.; Flores, L.F.; Vera, R.E.; Alfonse, G.W.; Marks, D.L. GLI1/GLI2 functional interplay is required to control Hedgehog/GLI targets gene expression. Biochem. J. 2020, 477, 3131–3145. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Alhosin, M.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mousli, M.; Bronner, C. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J. Exp. Clin. Cancer Res. 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hervouet, E.; Vallette, F.M.; Cartron, P.-F. Dnmt1/transcription factor interactions: An alternative mechanism of DNA methylation inheritance. Genes Cancer 2010, 1, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Joppien, S.; Nemeth, A.; Diermeier, S.; Thalhammer, V.; Dobner, T.; Kremmer, E.; Kappler, R.; Längst, G.J.N.A.R. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res. 2011, 39, 8355–8365. [Google Scholar] [CrossRef] [Green Version]
- Mirny, L.A. Nucleosome-mediated cooperativity between transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 22534–22539. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Et Biophys. Acta (BBA)-Rev. Cancer 2019, 1872, 11–23. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Molkentin, J.D. The zinc finger-containing transcription factors GATA-4,-5, and-6: Ubiquitously expressed regulators of tissue-specific gene expression. J. Biol. Chem. 2000, 275, 38949–38952. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.; Sanchez-Ferras, O.; Bouchard, M. GATA transcription factors in development and disease. Development 2018, 145, dev164384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Cai, J.; Liu, C.; Shen, L.; Pu, X.; Yao, Y.; Han, B.; Yu, T.; Cheng, S.Y.; Yue, S. Protein phosphatase 4 promotes Hedgehog signaling through dephosphorylation of Suppressor of fused. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Palacio, M.; Taatjes, D.J. Merging Established Mechanisms with New Insights: Condensates, Hubs, and the Regulation of RNA Polymerase II Transcription. J. Mol. Biol. 2021, 434, 167216. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GLI Transcriptional Co-Factor/Muti-Protein Complex | Role in GLI-Mediated Transcription | References |
|---|---|---|---|
| General transcription machinery | TFIID | activator | [59,60] |
| Mediator | dual-regulator | [61,62,63] | |
| Cohesin-CTCF | activator | [64,65] | |
| Epigenetic regulators | BRD4 | activator | [66,67,68,69,70,71] |
| HATs | activator | [72,73] | |
| HDACs | repressor | [74,75,76] | |
| PRMT5 | repressor | [77,78] | |
| SETD7 | repressor | [79] | |
| PRC2 | repressor | [80,81] | |
| SWI/SNF | dual-regulator | [80,82,83,84,85] | |
| UHRF1-DNMT1 | repressor | [86,87] | |
| Transcription factors | TP53 | repressor | [60] |
| SOX2 | activator | [71,88,89,90,91] | |
| SRF-MKL1 | activator | [58] | |
| AP-1 | activator | [92,93] | |
| SMAD1-5 | dual-regulator | [94,95,96] | |
| GATA4-6 | repressor | [97,98,99] | |
| SUFU | repressor | [100,101,102,103,104,105] | |
| ATOH1 | activator | [106,107] | |
| NANOG | dual-regulator | [108,109] |
| Target | Inhibitor | Preclinical Study | Clinical Study |
|---|---|---|---|
| TAF9 | FN1-8 | lung cancer [59] | n.a. |
| BRDs | JQ1 | MB, BCC, ATRT [69], melanoma [68] | n.a. |
| I-BET151 | MB [67] | NCT02630251 (terminated): advanced or recurrent solid tumors | |
| CPI203 | PDAC [66] | n.a. | |
| BMS-986158 | MB [71] | NCT03936465: Solid tumor, childhood; lymphoma; brain tumor, pediatric | |
| NCT04817007: myelofibrosis | |||
| NCT02419417: advanced tumors | |||
| NCT05372354: multiple myeloma | |||
| MZ1 | melanoma [68] | n.a. | |
| ZEN-3365 | AML [118] | n.a. | |
| DNMT1 | 5-Azacytidine | MB [86,87] | FDA-approved for MDS, AML, JMML |
| 5-aza-2′-deoxycytidine | MB [119] | FDA-approved for MDS | |
| MKL1 | CCG-1423 | BCC [58] | n.a. |
| CCG-203971 | BCC [58] | n.a. | |
| AP-1 | T5224 | BCC [93] | n.a. |
| SR11302 | BCC [93] | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Wynn, D.T.; Shen, C.; Ayad, N.G.; Robbins, D.J. Multiprotein GLI Transcriptional Complexes as Therapeutic Targets in Cancer. Life 2022, 12, 1967. https://doi.org/10.3390/life12121967
Yang F, Wynn DT, Shen C, Ayad NG, Robbins DJ. Multiprotein GLI Transcriptional Complexes as Therapeutic Targets in Cancer. Life. 2022; 12(12):1967. https://doi.org/10.3390/life12121967
Chicago/Turabian StyleYang, Fan, Daniel T. Wynn, Chen Shen, Nagi G. Ayad, and David J. Robbins. 2022. "Multiprotein GLI Transcriptional Complexes as Therapeutic Targets in Cancer" Life 12, no. 12: 1967. https://doi.org/10.3390/life12121967
APA StyleYang, F., Wynn, D. T., Shen, C., Ayad, N. G., & Robbins, D. J. (2022). Multiprotein GLI Transcriptional Complexes as Therapeutic Targets in Cancer. Life, 12(12), 1967. https://doi.org/10.3390/life12121967