

Candidate Multi-Epitope Vaccine against Corona B.1.617 Lineage: In Silico Approach

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of SEQUENCE of Delta Variant
2.2. Prediction of T Cell Epitope
2.3. Structural Modeling of Multi-Epitope Vaccine, Refinement, and Validation
2.4. Prediction of Antigenicity, Allergenicity, Immunogenicity and Physiochemical Features
2.5. Population Coverage
2.6. Molecular Docking
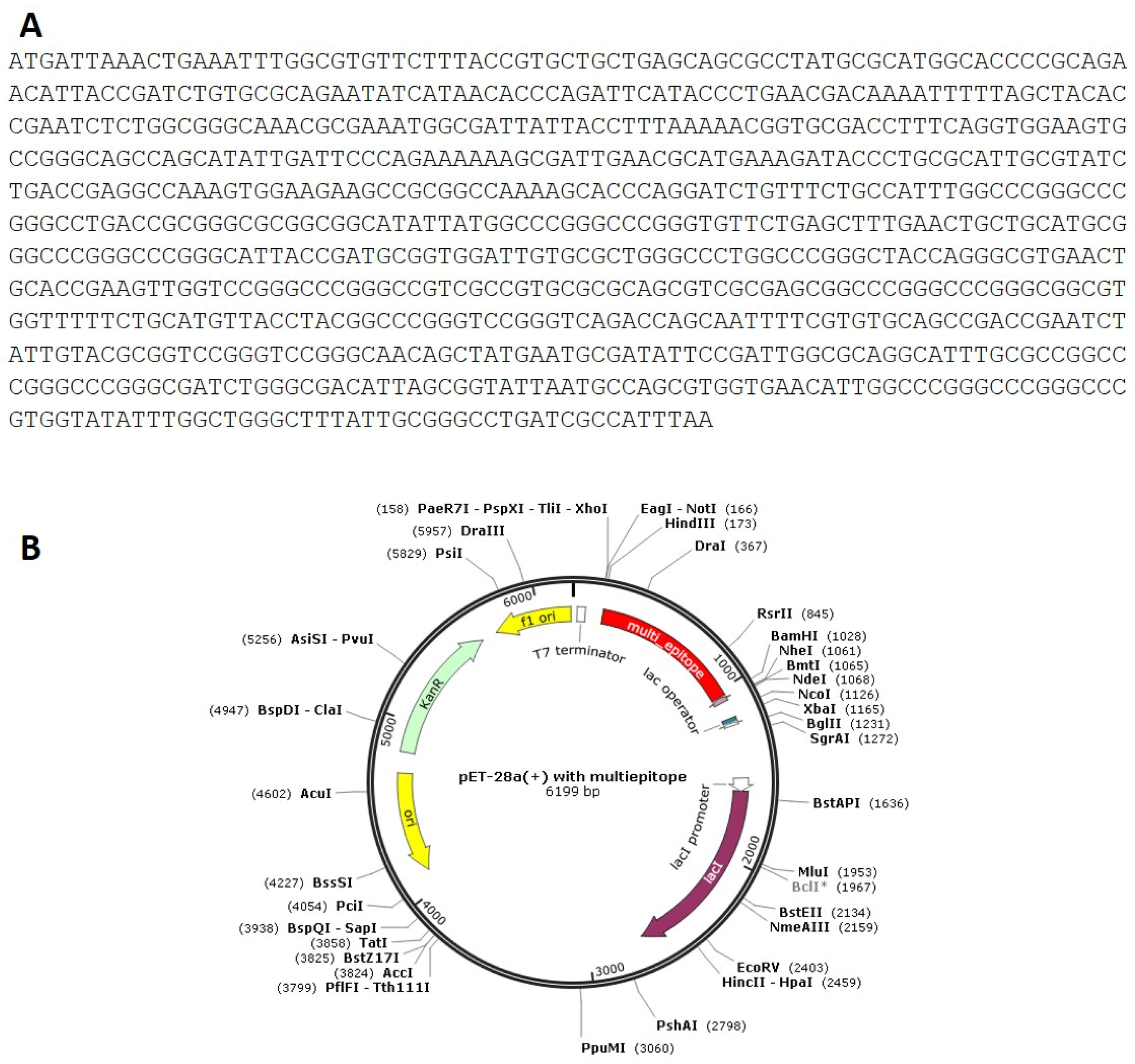
2.7. Codon Adaptation, In Silico Cloning and Testing the Stability of the Secondary Structure of the Produced mRNA
3. Results
3.1. CTL/HTL Epitope Prediction and Assessment
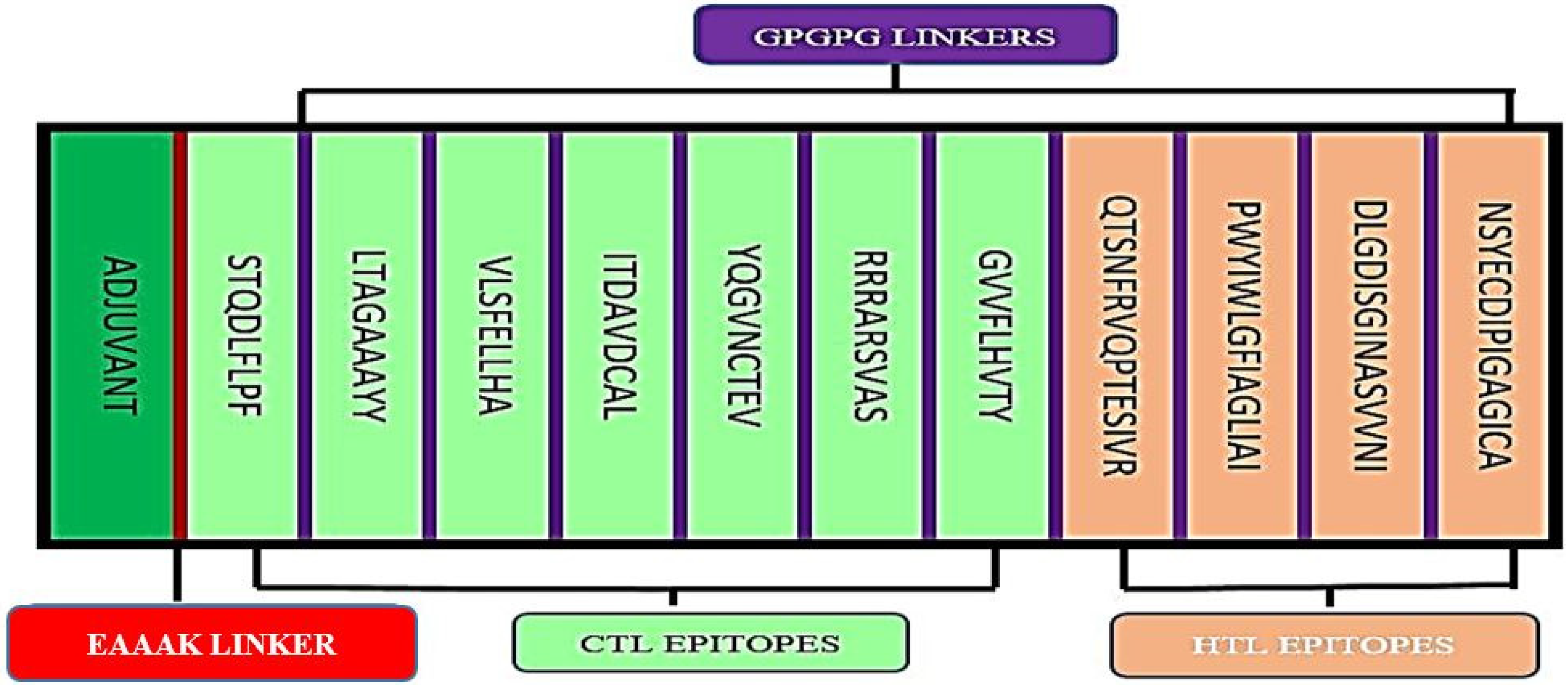
3.2. Multi-Epitope Vaccine Construct Design
3.3. Prediction of Immunogenicity, Antigenicity, Allergenicity, and Physicochemical Parameters
3.4. Population Coverage Study
3.5. D Structure Modeling and Validation of the Multi-Epitope
3.6. Molecular Docking Analyses of Multi-Epitope-Based Vaccine against TLR4
3.7. Molecular Dynamics Simulation of the Vaccine-Receptor Complex
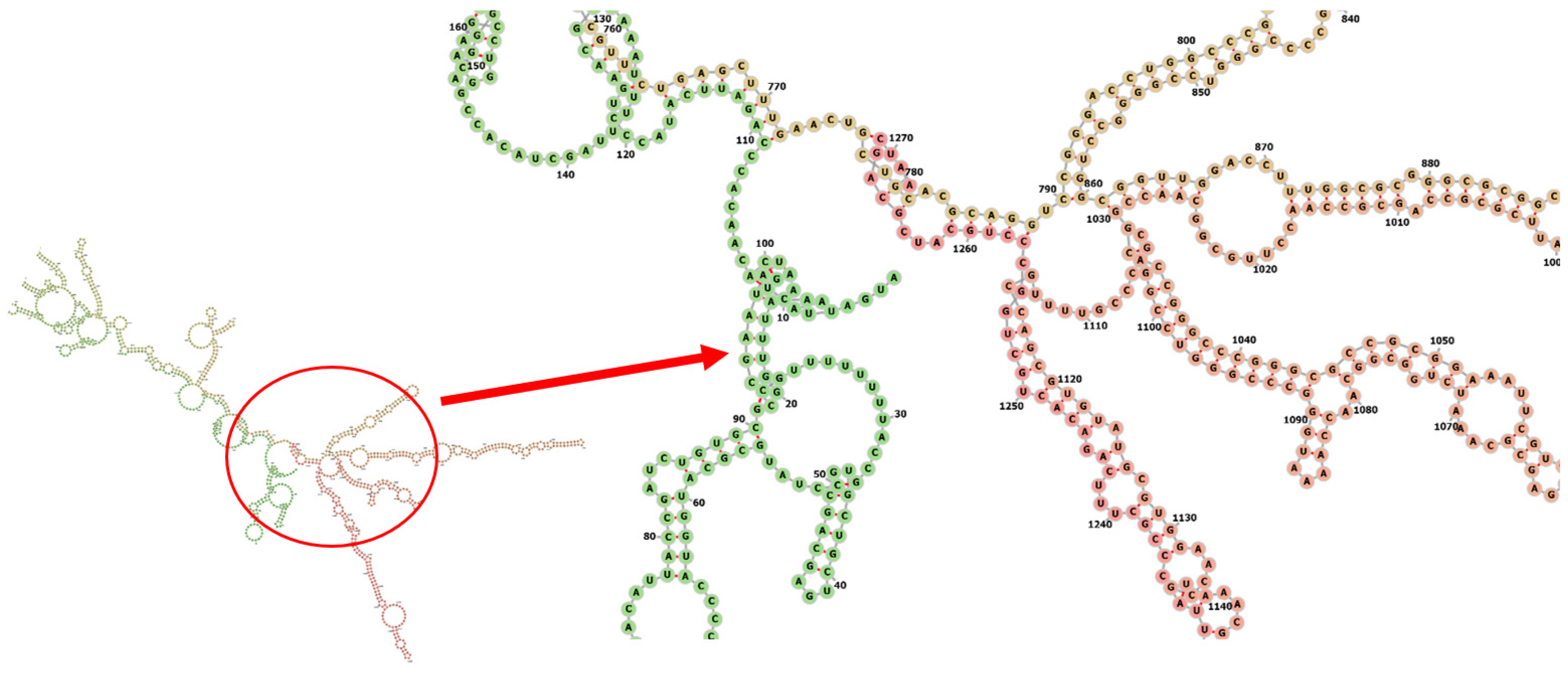
3.8. Codon Adaptation, In Silico Cloning and Testing the Stability of the Secondary Structure of the Produced mRNA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yi, Y.; Lagniton, P.N.; Ye, S.; Li, E.; Xu, R.-H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Bonavia, A.; Zelus, B.D.; Wentworth, D.; Talbot, P.J.; Holmes, K.V. Identification of a Receptor-Binding Domain of the Spike Glycoprotein of Human Coronavirus HCoV-229E. J. Virol. 2003, 77, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Zohar, T.; Alter, G. Dissecting antibody-mediated protection against SARS-CoV-2. Nat. Rev. Immunol. 2020, 20, 392–394. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 30 January 2022).
- Saito, A.; Nasser, H.; Uriu, K.; Kosugi, Y.; Irie, T.; Shirakawa, K. SARS-CoV-2 spike P681R mutation, a hallmark of the Delta variant, enhances viral fusogenicity and pathogenicity. BioRxiv 2021, 17, 448820. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Shi, P.Y. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. BioRxiv 2021, 39, 110829. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, W.; Guo, J.; Zhao, G.; Sun, S.; Yu, H.; Guo, Y.; Li, J.; Jin, X.; Du, L.; et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 2015, 11, 795–805. [Google Scholar] [CrossRef]
- Dar, H.; Zaheer, T.; Rehman, M.T.; Ali, A.; Javed, A.; Khan, G.A.; Babar, M.M.; Waheed, Y. Prediction of promiscuous T-cell epitopes in the Zika virus polyprotein: An in silico approach. Asian Pac. J. Trop. Med. 2016, 9, 844–850. [Google Scholar] [CrossRef]
- Patronov, A.; Doytchinova, I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2013, 3, 120139. [Google Scholar] [CrossRef]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020, 92, 495–500. [Google Scholar] [CrossRef]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef]
- Kar, T.; Narsaria, U.; Basak, S.; Deb, D.; Castiglione, F.; Mueller, D.M.; Srivastava, A.P. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 1–24. [Google Scholar] [CrossRef]
- Mitra, D.; Pandey, J.; Jain, A.; Swaroop, S. In silico design of multi-epitope-based peptide vaccine against SARS-CoV-2 using its spike protein. J. Biomol. Struct. Dyn. 2020, 40, 5189–5202. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.P.H.; Young, B.E. Clinical and Virological Features of SARS-CoV-2 Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta). SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M. (Ed.) The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr. Protoc. Immunol. 2016, 114, 18.19.1–18.19.24. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Orti, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Gruber, A.; Lorenz, R.; Bernhart, S.H.F.; Neuböck, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Testa, J.S.; Philip, R. Role of T-cell epitope-based vaccine in prophylactic and therapeutic applications. Future Virol. 2012, 7, 1077–1088. [Google Scholar] [CrossRef]
- Wilson, C.C.; Palmer, B.; Southwood, S.; Sidney, J.; Higashimoto, Y.; Appella, E.; Chesnut, R.; Sette, A.; Livingston, B.D. Identification and Antigenicity of Broadly Cross-Reactive and Conserved Human Immunodeficiency Virus Type 1-Derived Helper T-Lymphocyte Epitopes. J. Virol. 2001, 75, 4195–4207. [Google Scholar] [CrossRef]
- Carty, M.; Bowie, A.G. Recent insights into the role of Toll-like receptors in viral infection. Clin. Exp. Immunol. 2010, 161, 397–406. [Google Scholar] [CrossRef]
- Hu, W.; Yen, Y.T.; Singh, S.; Kao, C.L.; Wu-Hsieh, B.A. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol. 2012, 25, 277–288. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Lorenzo-Redondo, R.; Nam, H.H.; Roberts, S.C.; Simons, L.M.; Jennings, L.J.; Qi, C.; Achenbach, C.J.; Hauser, A.R.; Ison, M.G.; Hultquist, J.F.; et al. A Unique Clade of SARS-CoV-2 Viruses is Associated with Lower Viral Loads in Patient Upper Airways. MedRxiv 2020. [Google Scholar] [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pr. 2020, 74, e13525. [Google Scholar] [CrossRef]
- Mirza, M. Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016, 6, 37313. [Google Scholar] [CrossRef]
- Sette, A.; Fikes, J. Epitope-based vaccines: An update on epitope identification, vaccine design and delivery. Curr. Opin. Immunol. 2003, 15, 461–470. [Google Scholar] [CrossRef]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef]
- Hou, J.; Liu, Y.; Hsi, J.; Wang, H.; Tao, R.; Shao, Y. Cholera toxin B subunit acts as a potent systemic adjuvant for HIV-1 DNA vaccination intramuscularly in mice. Hum. Vaccines Immunother. 2014, 10, 1274–1283. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.K.; Seo, S.B.; Lee, H.J.; Kim, H.J. Intranasal vaccination with peptides and cholera toxin subunit B as adjuvant to enhance mucosal and systemic immunity to respiratory syncytial virus. Arch. Pharm. Res. 2007, 30, 366–371. [Google Scholar] [CrossRef]
- Tamura, S.I.; Yamanaka, A.; Shimohara, M.; Tomita, T.; Komase, K.; Tsuda, Y.; Suzuki, Y.; Nagamine, T.; Kawahara, K.; Danbara, H.; et al. Synergistic action of cholera toxin B subunit (and Escherichia coli heat-labile toxin B subunit) and a trace amount of cholera whole toxin as an adjuvant for nasal influenza vaccine. Vaccine 1994, 12, 419–426. [Google Scholar] [CrossRef]
- Bazhan, S.I.; Antonets, D.V.; Karpenko, L.I.; Oreshkova, S.F.; Kaplina, O.N.; Starostina, E.V.; Dudko, S.G.; Fedotova, S.A.; Ilyichev, A.A. In silico designed ebola virus T-cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines 2019, 7, 34. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A. Thermostability and Aliphatic Index of Globular Proteins. Commun. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Foroutan, M.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101413. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Xagorari, A.; Chlichlia, K. Toll-Like Receptors and Viruses: Induction of Innate Antiviral Immune Responses. Open Microbiol. J. 2008, 2, 49–59. [Google Scholar] [CrossRef]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-κB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- Manan, A.; Pirzada, R.H.; Haseeb, M.; Choi, S. Toll-like Receptor Mediation in SARS-CoV-2: A Therapeutic Approach. Int. J. Mol. Sci. 2022, 23, 10716. [Google Scholar] [CrossRef]
- Banerjee, S.; Majumder, K.; Gutierrez, G.J.; Gupta, D.; Mittal, B. Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Oladipo, E.K.; Ajayi, A.F.; Onile, O.S.; Ariyo, O.E.; Jimah, E.M.; Ezediuno, L.O.; Adebayo, O.I.; Adebayo, E.T.; Odeyemi, A.N.; Oyeleke, M.O.; et al. Designing a conserved peptide-based subunit vaccine against SARS-CoV-2 using immunoinformatics approach. Silico Pharmacol. 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Bin Sayed, S.; Nain, Z.; Khan, S.A.; Abdulla, F.; Tasmin, R.; Adhikari, U.K. Exploring Lassa Virus Proteome to Design a Multi-epitope Vaccine Through Immunoinformatics and Immune Simulation Analyses. Int. J. Pept. Res. Ther. 2020, 26, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Kamthania, M.; Pandey, R.K.; Saxena, A.K.; Saxena, V.; Singh, S.K.; Sharma, R.K.; Sharma, N. Design of novel multi-epitope vaccines against severe acute respiratory syndrome validated through multistage molecular interaction and dynamics. J. Biomol. Struct. Dyn. 2019, 37, 4345–4360. [Google Scholar] [CrossRef]
- Sallenave, J.-M.; Guillot, L. Innate Immune Signaling and Proteolytic Pathways in the Resolution or Exacerbation of SARS-CoV-2 in Covid-19: Key Therapeutic Targets? Front. Immunol. 2020, 11, 1229. [Google Scholar] [CrossRef]
- Das, S.; Chottopadhyay, B.; Sahoo, S. Comparative Analysis of Predicted Gene Expression among Crenarchaeal Genomes. Genom. Inform. 2017, 15, 38–47. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | ||||
|---|---|---|---|---|
| Supertype | Epitope | Position | Antigenicity | Immunogenicity |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | STQDLFLPF | 50 | 0.6619 | 0.06828 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | LTAGAAAYY | 258 | 0.9269 | 0.15259 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | VLSFELLHA | 510 | 1.0776 | 0.1607 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | ITDAVDCAL | 283 | 0.5260 | 0.08501 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | YQGVNCTEV | 610 | 1.3957 | 0.08675 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | RRRARSVAS | 681 | 1.1645 | 0.00854 |
| A1-A2-A3-A24-A26-B7-B7-B27-B39-B44-B58-B62 | GVVFLHVTY | 1057 | 1.4104 | 0.20837 |
| B | ||||
| Supertype | Epitope | Position | Antigenicity | Immunogenicity |
| HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*07:01, HLA-DRB1*08:01, HLA-DRB1*13:01, HLA-DRB1*15:01 | QTSNFRVQPTESIVR | 312 | 0.4885 | 0.1368 |
| HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*07:01, HLA-DRB1*08:01, HLA-DRB1*13:01, HLA-DRB1*15:01 | PWYIWLGFIAGLIAI | 1211 | 0.6761 | 0.81006 |
| HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*07:01, HLA-DRB1*08:01, HLA-DRB1*13:01, HLA-DRB1*15:01 | DLGDISGINASVVNI | 1163 | 0.8798 | 0.10504 |
| HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*07:01, HLA-DRB1*08:01, HLA-DRB1*13:01, HLA-DRB1*15:01 | NSYECDIPIGAGICA | 656 | 0.5726 | 0.50087 |
| Population/Area | Coverage |
|---|---|
| Brazil | 85.39% |
| East Africa | 82.0% |
| England | 99.82% |
| Europe | 99.3% |
| India | 88.89% |
| North Africa | 88.79% |
| Russia | 95.21% |
| Saudi Arabia | 95.64% |
| United States | 97.93% |
| World | 96.85% |
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored |
|---|---|---|---|---|---|---|
| Initial | 1.0000 | 0.000 | 3.906 | 132.2 | 7.1 | 83.9 |
| MODEL 1 | 0.8963 | 0.550 | 2.031 | 15.2 | 0.5 | 95 |
| MODEL 2 | 0.8901 | 0.540 | 2.031 | 15.2 | 0.5 | 95 |
| MODEL 3 | 0.8972 | 0.554 | 2.078 | 14.7 | 0.9 | 93.9 |
| MODEL 4 | 0.8892 | 0.553 | 2.040 | 14.7 | 0.9 | 94.6 |
| MODEL 5 | 0.8963 | 0.534 | 2.193 | 15.4 | 1.4 | 94.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seadawy, M.G.; Zekri, A.R.N.; Saeed, A.A.; San, E.J.; Ageez, A.M. Candidate Multi-Epitope Vaccine against Corona B.1.617 Lineage: In Silico Approach. Life 2022, 12, 1715. https://doi.org/10.3390/life12111715
Seadawy MG, Zekri ARN, Saeed AA, San EJ, Ageez AM. Candidate Multi-Epitope Vaccine against Corona B.1.617 Lineage: In Silico Approach. Life. 2022; 12(11):1715. https://doi.org/10.3390/life12111715
Chicago/Turabian StyleSeadawy, Mohamed G., Abdel Rahman N. Zekri, Aya A. Saeed, Emmanuel James San, and Amr M. Ageez. 2022. "Candidate Multi-Epitope Vaccine against Corona B.1.617 Lineage: In Silico Approach" Life 12, no. 11: 1715. https://doi.org/10.3390/life12111715
APA StyleSeadawy, M. G., Zekri, A. R. N., Saeed, A. A., San, E. J., & Ageez, A. M. (2022). Candidate Multi-Epitope Vaccine against Corona B.1.617 Lineage: In Silico Approach. Life, 12(11), 1715. https://doi.org/10.3390/life12111715