


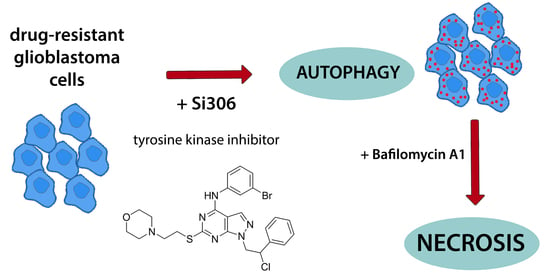
Autophagy Inhibition Enhances Anti-Glioblastoma Effects of Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors

 , , ,
, , , 
 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Reagents
2.3. Cell Cultures
2.4. MTT Assay
2.5. Median Effect Analysis
2.6. Cell Death Detection by Flow Cytometry
2.7. CFSE Cell Proliferation Assay
2.8. Western Blot
2.9. Transfection
2.10. mRFP-LC3 Detection by Flow Cytometry and Fluorescence Microscopy
2.11. Immunocytochemistry
2.12. Acridine Orange Assay
2.13. Statistical Analysis
3. Results
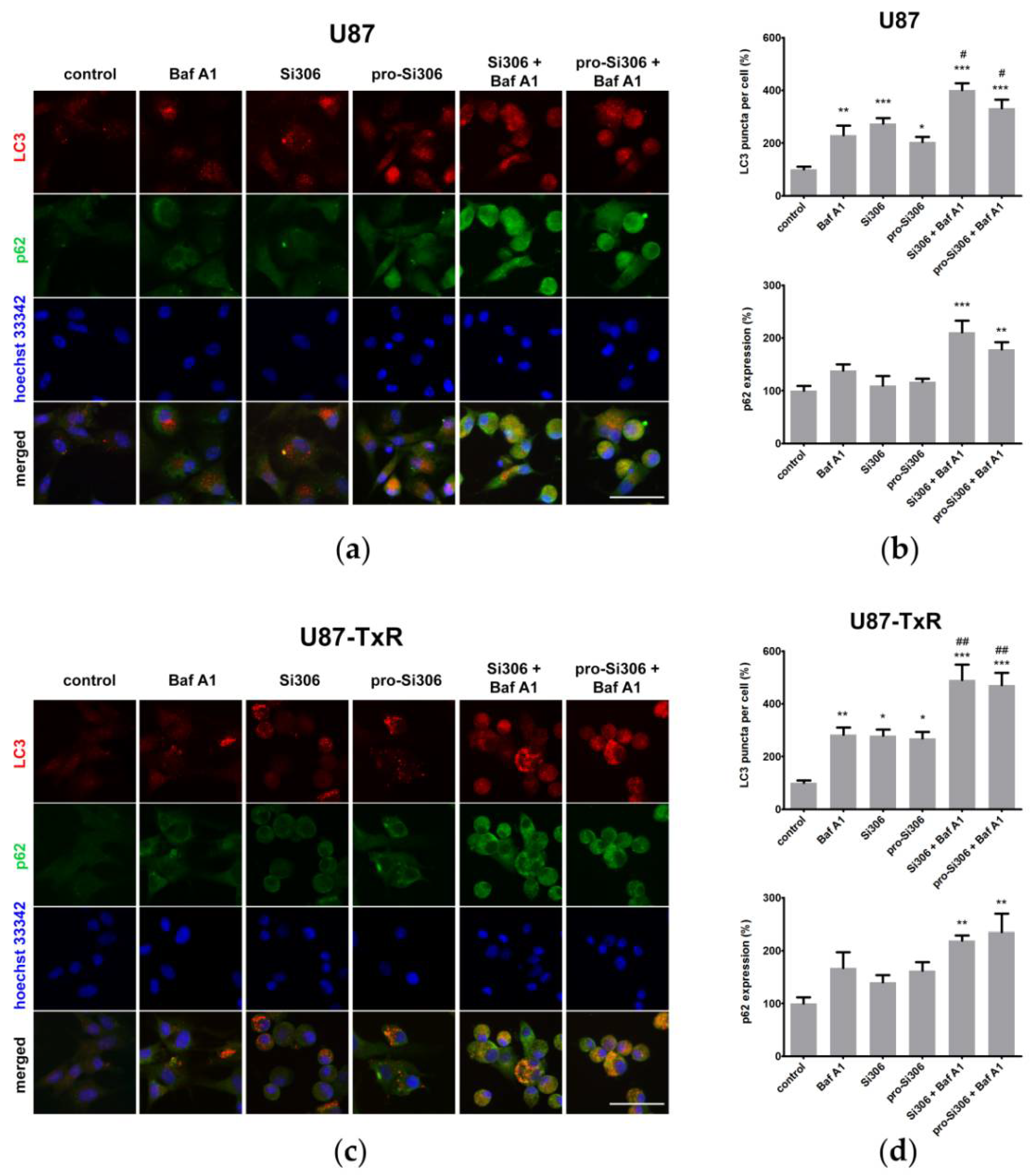
3.1. Si306 and Pro-Si306 Induce Autophagy in Glioblastoma Cells
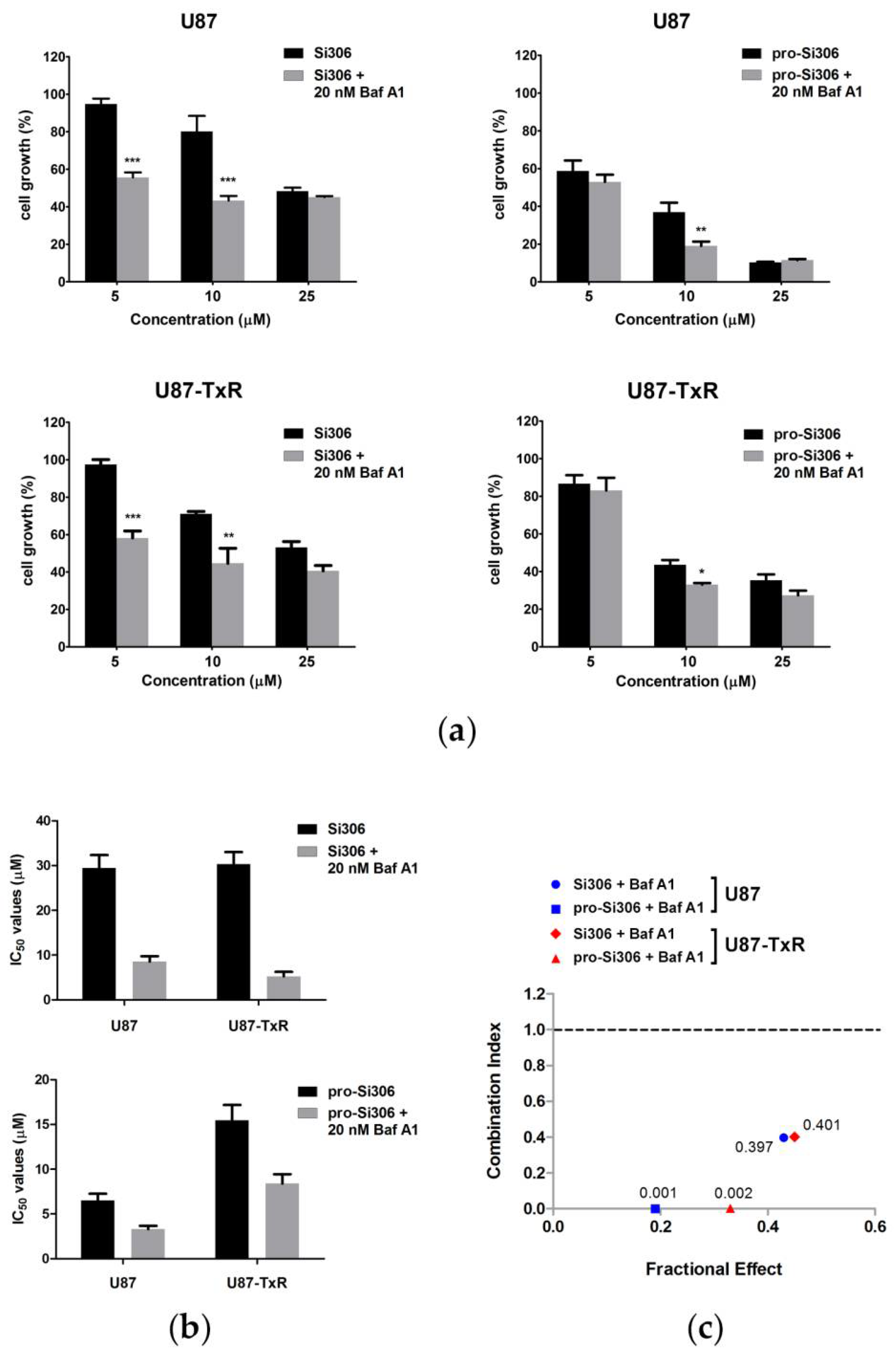
3.2. Si306 and Pro-Si306 Have Synergistic Interaction with Bafilomycin A1 in Glioblastoma Cells
3.3. Si306 and Pro-Si306 Have Prolonged Effects on Autophagy in Glioblastoma Cells
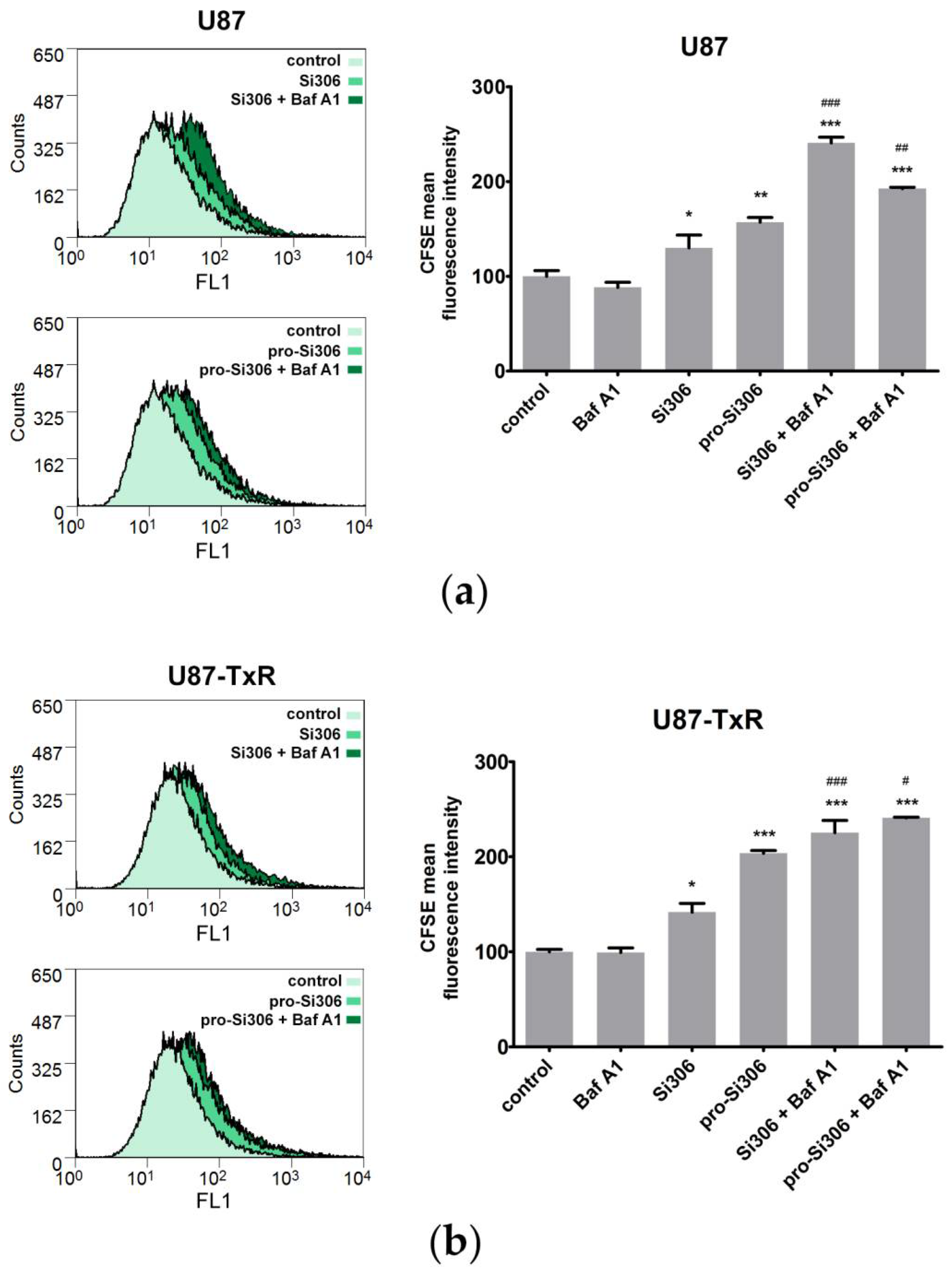
3.4. Bafilomycin A1 Enhances the Antiproliferative Effects of Si306 and Pro-Si306 in Glioblastoma Cells
3.5. Si306 and Pro-Si306 in Combination with Bafilomycin A1 Induce Necrotic Cell Death in Glioblastoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanaka, H.; Hino, H.; Moriya, S.; Kazama, H.; Miyazaki, M.; Takano, N.; Hiramoto, M.; Tsukahara, K.; Miyazawa, K. Comparison of autophagy inducibility in various tyrosine kinase inhibitors and their enhanced cytotoxicity via inhibition of autophagy in cancer cells in combined treatment with azithromycin. Biochem. Biophys. Rep. 2020, 22, 100750. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.R.; Neves, S.P.; Santos, L.S.; Dias, R.B.; Bezerra, D.P. Challenges and Therapeutic Opportunities of Autophagy in Cancer Therapy. Cancers 2020, 12, 3461. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zou, Z. Targeting autophagy to overcome drug resistance: Further developments. J. Hematol. Oncol. 2020, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chang, P.C.; Yang, J.C.; Chu, C.Y.; Wang, L.Y.; Chen, N.T.; Ma, A.H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy Blockade Sensitizes Prostate Cancer Cells towards Src Family Kinase Inhibitors. Genes Cancer 2010, 1, 40–49. [Google Scholar] [CrossRef]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2020, 7, 620677. [Google Scholar] [CrossRef]
- Khan, I.; Baig, M.H.; Mahfooz, S.; Rahim, M.; Karacam, B.; Elbasan, E.B.; Ulasov, I.; Dong, J.J.; Hatiboglu, M.A. Deciphering the Role of Autophagy in Treatment of Resistance Mechanisms in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 1318. [Google Scholar] [CrossRef]
- Ou, A.; Yung, W.K.A.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 22, 351. [Google Scholar] [CrossRef]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Cong, X.; Nomura, N.; Gallagher, M.; Abbasi, T.; et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 2014, 12, 13. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Corallo, D.; Pastorino, F.; Pantile, M.; Mariotto, E.; Caicci, F.; Viola, G.; Ponzoni, M.; Tonini, G.P.; Aveic, S. Autophagic flux inhibition enhances cytotoxicity of the receptor tyrosine kinase inhibitor ponatinib. J. Exp. Clin. Cancer Res. CR 2020, 39, 195. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef]
- Litwack, G. Enzymes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 95–129. [Google Scholar]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Schenone, S.; Radi, M.; Musumeci, F.; Brullo, C.; Botta, M. Biologically driven synthesis of pyrazolo[3,4-d]pyrimidines as protein kinase inhibitors: An old scaffold as a new tool for medicinal chemistry and chemical biology studies. Chem. Rev. 2014, 114, 7189–7238. [Google Scholar] [CrossRef]
- Calgani, A.; Vignaroli, G.; Zamperini, C.; Coniglio, F.; Festuccia, C.; Di Cesare, E.; Gravina, G.L.; Mattei, C.; Vitale, F.; Schenone, S.; et al. Suppression of SRC Signaling Is Effective in Reducing Synergy between Glioblastoma and Stromal Cells. Mol. Cancer Ther. 2016, 15, 1535–1544. [Google Scholar] [CrossRef]
- Kostic, A.; Jovanovic Stojanov, S.; Podolski-Renic, A.; Nesovic, M.; Dragoj, M.; Nikolic, I.; Tasic, G.; Schenone, S.; Pesic, M.; Dinic, J. Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors Induce Oxidative Stress in Patient-Derived Glioblastoma Cells. Brain Sci. 2021, 11, 884. [Google Scholar] [CrossRef]
- Nesovic, M.; Divac Rankov, A.; Podolski-Renic, A.; Nikolic, I.; Tasic, G.; Mancini, A.; Schenone, S.; Pesic, M.; Dinic, J. Src Inhibitors Pyrazolo[3,4-d]pyrimidines, Si306 and Pro-Si306, Inhibit Focal Adhesion Kinase and Suppress Human Glioblastoma Invasion In Vitro and In Vivo. Cancers 2020, 12, 1570. [Google Scholar] [CrossRef]
- Fallacara, A.L.; Zamperini, C.; Podolski-Renic, A.; Dinic, J.; Stankovic, T.; Stepanovic, M.; Mancini, A.; Rango, E.; Iovenitti, G.; Molinari, A.; et al. A New Strategy for Glioblastoma Treatment: In Vitro and In Vivo Preclinical Characterization of Si306, a Pyrazolo[3,4-d]Pyrimidine Dual Src/P-Glycoprotein Inhibitor. Cancers 2019, 11, 848. [Google Scholar] [CrossRef]
- Podolski-Renic, A.; Dinic, J.; Stankovic, T.; Tsakovska, I.; Pajeva, I.; Tuccinardi, T.; Botta, L.; Schenone, S.; Pesic, M. New Therapeutic Strategy for Overcoming Multidrug Resistance in Cancer Cells with Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Cancers 2021, 13, 5308. [Google Scholar] [CrossRef]
- Zawilska, J.B.; Wojcieszak, J.; Olejniczak, A.B. Prodrugs: A challenge for the drug development. Pharmacol. Rep. 2013, 65, 1–14. [Google Scholar] [CrossRef]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef]
- Podolski-Renic, A.; Andelkovic, T.; Bankovic, J.; Tanic, N.; Ruzdijic, S.; Pesic, M. The role of paclitaxel in the development and treatment of multidrug resistant cancer cell lines. Biomed. Pharmacother. 2011, 65, 345–353. [Google Scholar] [CrossRef]
- Mele, L.; Del Vecchio, V.; Liccardo, D.; Prisco, C.; Schwerdtfeger, M.; Robinson, N.; Desiderio, V.; Tirino, V.; Papaccio, G.; La Noce, M. The role of autophagy in resistance to targeted therapies. Cancer Treat. Rev. 2020, 88, 102043. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef]
- Eng, K.E.; Panas, M.D.; Karlsson Hedestam, G.B.; McInerney, G.M. A novel quantitative flow cytometry-based assay for autophagy. Autophagy 2010, 6, 634–641. [Google Scholar] [CrossRef]
- Dacevic, M.; Isakovic, A.; Podolski-Renic, A.; Isakovic, A.M.; Stankovic, T.; Milosevic, Z.; Rakic, L.; Ruzdijic, S.; Pesic, M. Purine nucleoside analog--sulfinosine modulates diverse mechanisms of cancer progression in multi-drug resistant cancer cell lines. PLoS ONE 2013, 8, e54044. [Google Scholar] [CrossRef]
- Podolski-Renic, A.; Bosze, S.; Dinic, J.; Kocsis, L.; Hudecz, F.; Csampai, A.; Pesic, M. Ferrocene-cinchona hybrids with triazolyl-chalcone linkers act as pro-oxidants and sensitize human cancer cell lines to paclitaxel. Metallomics 2017, 9, 1132–1141. [Google Scholar] [CrossRef]
- Podolski-Renic, A.; Jadranin, M.; Stankovic, T.; Bankovic, J.; Stojkovic, S.; Chiourea, M.; Aljancic, I.; Vajs, V.; Tesevic, V.; Ruzdijic, S.; et al. Molecular and cytogenetic changes in multi-drug resistant cancer cells and their influence on new compounds testing. Cancer Chemother. Pharmacol. 2013, 72, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Aveic, S.; Pantile, M.; Polo, P.; Sidarovich, V.; De Mariano, M.; Quattrone, A.; Longo, L.; Tonini, G.P. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s tyrosine kinase inhibitor, exhibits antitumoral activity and induces autophagy in glioblastoma. J. Exp. Clin. Cancer Res. CR 2017, 36, 96. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Palmieri, M.; Chaudhury, A.; Klisch, T.J.; di Ronza, A.; Neilson, J.R.; Rodney, G.G.; Sardiello, M. Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nat. Commun. 2018, 9, 4351. [Google Scholar] [CrossRef]
- Ahn, J.H.; Lee, M. Suppression of autophagy sensitizes multidrug resistant cells towards Src tyrosine kinase specific inhibitor PP2. Cancer Lett. 2011, 310, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef]
- Wang, Z.; Du, T.; Dong, X.; Li, Z.; Wu, G.; Zhang, R. Autophagy inhibition facilitates erlotinib cytotoxicity in lung cancer cells through modulation of endoplasmic reticulum stress. Int. J. Oncol. 2016, 48, 2558–2566. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.; Liu, H.; Zhang, W.; Shen, C.; Cho, K.; Chen, X.; Peng, F.; Bi, Y.; Hou, X.; et al. Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 1303–1316. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Shen, J.; Zheng, H.; Ruan, J.; Fang, W.; Li, A.; Tian, G.; Niu, X.; Luo, S.; Zhao, P. Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. Br. J. Cancer 2013, 109, 164–171. [Google Scholar] [CrossRef]
- Fan, Q.W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, e19671. [Google Scholar] [CrossRef]
- Escamilla-Ramirez, A.; Castillo-Rodriguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseno, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- Kamat, S.; Kumari, M. Repurposing Chloroquine Against Multiple Diseases With Special Attention to SARS-CoV-2 and Associated Toxicity. Front. Pharmacol. 2021, 12, 576093. [Google Scholar] [CrossRef]
- Lu, X.; Chen, L.; Chen, Y.; Shao, Q.; Qin, W. Bafilomycin A1 inhibits the growth and metastatic potential of the BEL-7402 liver cancer and HO-8910 ovarian cancer cell lines and induces alterations in their microRNA expression. Exp. Ther. Med. 2015, 10, 1829–1834. [Google Scholar] [CrossRef]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef]
- Drose, S.; Altendorf, K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997, 200, 1–8. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanović Stojanov, S.; Kostić, A.; Ljujić, M.; Lupšić, E.; Schenone, S.; Pešić, M.; Dinić, J. Autophagy Inhibition Enhances Anti-Glioblastoma Effects of Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Life 2022, 12, 1503. https://doi.org/10.3390/life12101503
Jovanović Stojanov S, Kostić A, Ljujić M, Lupšić E, Schenone S, Pešić M, Dinić J. Autophagy Inhibition Enhances Anti-Glioblastoma Effects of Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Life. 2022; 12(10):1503. https://doi.org/10.3390/life12101503
Chicago/Turabian StyleJovanović Stojanov, Sofija, Ana Kostić, Mila Ljujić, Ema Lupšić, Silvia Schenone, Milica Pešić, and Jelena Dinić. 2022. "Autophagy Inhibition Enhances Anti-Glioblastoma Effects of Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors" Life 12, no. 10: 1503. https://doi.org/10.3390/life12101503
APA StyleJovanović Stojanov, S., Kostić, A., Ljujić, M., Lupšić, E., Schenone, S., Pešić, M., & Dinić, J. (2022). Autophagy Inhibition Enhances Anti-Glioblastoma Effects of Pyrazolo[3,4-d]pyrimidine Tyrosine Kinase Inhibitors. Life, 12(10), 1503. https://doi.org/10.3390/life12101503