
A Comprehensive Analysis of Hungarian MODY Patients—Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases

 , , ,
, , ,
Abstract
1. Introduction
1.1. GCK-MODY (MODY2)
1.2. MODY Prevalence
2. Materials and Methods
2.1. Patients
2.2. Methods
2.3. Variant Confirmation
2.4. Variant Filtering and Interpretation
2.5. Clinical Data Collection
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on Mutations in Glucokinase (GCK), Which Cause Maturity-Onset Diabetes of the Young, Permanent Neonatal Diabetes, and Hyperinsulinemic Hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Iynedjian, P.B. Mammalian Glucokinase and Its Gene. Biochem. J. 1993, 293, 1–13. [Google Scholar] [CrossRef]
- Magnuson, M.A. Glucokinase Gene Structure: Functional Implications of Molecular Genetic Studies. Diabetes 1990, 39, 523–527. [Google Scholar] [CrossRef]
- Capuano, M.; Garcia-Herrero, C.M.; Tinto, N.; Carluccio, C.; Capobianco, V.; Coto, I.; Cola, A.; Iafusco, D.; Franzese, A.; Zagari, A.; et al. Glucokinase (GCK) Mutations and Their Characterization in MODY2 Children of Southern Italy. PLoS ONE 2012, 7, e38906. [Google Scholar] [CrossRef] [PubMed]
- Negahdar, M.; Aukrust, I.; Molnes, J.; Solheim, M.H.; Johansson, B.B.; Sagen, J.V.; Dahl-Jørgensen, K.; Kulkarni, R.N.; Søvik, O.; Flatmark, T.; et al. GCK-MODY Diabetes as a Protein Misfolding Disease: The Mutation R275C Promotes Protein Misfolding, Self-Association and Cellular Degradation. Mol. Cell. Endocrinol. 2014, 382, 55–65. [Google Scholar] [CrossRef]
- Jang, K.M. Maturity-Onset Diabetes of the Young: Update and Perspectives on Diagnosis and Treatment. Yeungnam Univ. J. Med. 2020, 37, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Wensley, K.J.; Ellard, S.; Murphy, R.; Shepherd, M.; Colclough, K.; Hattersley, A.T.; Shields, B.M. Use of HbA1c in the Identification of Patients with Hyperglycaemia Caused by a Glucokinase Mutation: Observational Case Control Studies. PLoS ONE 2013, 8, e65326. [Google Scholar] [CrossRef] [PubMed]
- Karaoglan, M.; Nacarkahya, G. Clinical and Laboratory Clues of Maturity-Onset Diabetes of the Young and Determination of Association with Molecular Diagnosis. J. Diabetes 2021, 13, 154–163. [Google Scholar] [CrossRef]
- McDonald, T.J.; Ellard, S. Maturity Onset Diabetes of the Young: Identification and Diagnosis. Ann. Clin. Biochem. 2013, 50, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Valentínová, L.; Beer, N.L.; Staník, J.; Tribble, N.D.; van de Bunt, M.; Hučková, M.; Barrett, A.; Klimeš, I.; Gašperíková, D.; Gloyn, A.L. Identification and Functional Characterisation of Novel Glucokinase Mutations Causing Maturity-Onset Diabetes of the Young in Slovakia. PLoS ONE 2012, 7, e34541. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, Z.; Sun, X.; Wang, Y.; Xiao, Y.; Luo, S.; Huang, G.; Li, X.; Xia, Y.; Zhou, Z. A New Screening Strategy and Whole-Exome Sequencing for the Early Diagnosis of Maturity-Onset Diabetes of the Young. Diabetes Metab. Res. Rev. 2021, 37, e3381. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.C.; Neves, J.S.; Pérez, A.; Carvalho, D. Maturity-Onset Diabetes of the Young: From a Molecular Basis Perspective toward the Clinical Phenotype and Proper Management. Endocrinol. Diabetes Nutr. 2020, 67, 137–147. [Google Scholar] [CrossRef]
- Shields, B.M.; Shepherd, M.; Hudson, M.; McDonald, T.J.; Colclough, K.; Peters, J.; Knight, B.; Hyde, C.; Ellard, S.; Pearson, E.R.; et al. Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care 2017, 40, 1017–1025. [Google Scholar] [CrossRef]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Bellanné-Chantelot, C.; Hattersley, A.T. European Molecular Genetics Quality Network (EMQN) MODY group Best Practice Guidelines for the Molecular Genetic Diagnosis of Maturity-Onset Diabetes of the Young. Diabetologia 2008, 51, 546–553. [Google Scholar] [CrossRef]
- Nkonge, K.M.; Nkonge, D.K.; Nkonge, T.N. The Epidemiology, Molecular Pathogenesis, Diagnosis, and Treatment of Maturity-Onset Diabetes of the Young (MODY). Clin. Diabetes Endocrinol. 2020, 6, 20. [Google Scholar] [CrossRef]
- Eide, S.A.; Raeder, H.; Johansson, S.; Midthjell, K.; Søvik, O.; Njølstad, P.R.; Molven, A. Prevalence of HNF1A (MODY3) Mutations in a Norwegian Population (the HUNT2 Study). Diabet. Med. J. Br. Diabet. Assoc. 2008, 25, 775–781. [Google Scholar] [CrossRef]
- Johansson, B.B.; Irgens, K.U.; Molnes, J.; Sztromwasser, P.; Aukrust, I.; Juliusson, P.B.; Søvik, O.; Levy, S.; Skrivarhaug, T.; Joner, G.; et al. Targeted Next-Generation Sequencing Reveals MODY in up to 6.5% of Antibody-Negative Diabetes Cases Listed in the Norwegian Childhood Diabetes Registry. Diabetologia 2016, 60, 625–635. [Google Scholar] [CrossRef]
- Fendler, W.; Borowiec, M.; Baranowska-Jazwiecka, A.; Szadkowska, A.; Skala-Zamorowska, E.; Deja, G.; Jarosz-Chobot, P.; Techmanska, I.; Bautembach-Minkowska, J.; Mysliwiec, M.; et al. Prevalence of Monogenic Diabetes amongst Polish Children after a Nationwide Genetic Screening Campaign. Diabetologia 2012, 55, 2631–2635. [Google Scholar] [CrossRef] [PubMed]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovina, S.M.; et al. Prevalence, Characteristics and Clinical Diagnosis of Maturity Onset Diabetes of the Young Due to Mutations in HNF1A, HNF4A, and Glucokinase: Results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee ACMG Recommendations for Standards for Interpretation and Reporting of Sequence Variations: Revisions 2007. Genet. Med. Off. J. Am. Coll. Med. Genet. 2008, 10, 294–300. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, Y.; Einhorn, M.; Kamshov, A.; Lev, O.; Trabelsi, A.; Paz-Yaacov, N.; Gross, S.J. Gene-Specific Artificial Intelligence-Based Variant Classification Engine: Results of a Time-Capsule Experiment. Preprint 2019. [Google Scholar] [CrossRef]
- Lukásová, P.; Vcelák, J.; Vanková, M.; Vejrazková, D.; Andelová, K.; Bendlová, B. Screening of Mutations and Polymorphisms in the Glucokinase Gene in Czech Diabetic and Healthy Control Populations. Physiol. Res. 2008, 57 (Suppl. 1), S99–S108. [Google Scholar] [CrossRef]
- Hager, J.; Blanché, H.; Sun, F.; Vaxillaire, N.V.; Poller, W.; Cohen, D.; Czernichow, P.; Velho, G.; Robert, J.J.; Cohen, N. Six Mutations in the Glucokinase Gene Identified in MODY by Using a Nonradioactive Sensitive Screening Technique. Diabetes 1994, 43, 730–733. [Google Scholar] [CrossRef]
- Prisco, F.; Iafusco, D.; Franzese, A.; Sulli, N.; Barbetti, F. MODY 2 Presenting as Neonatal Hyperglycaemia: A Need to Reshape the Definition of “Neonatal Diabetes”? Diabetologia 2000, 43, 1331–1332. [Google Scholar] [CrossRef][Green Version]
- Gragnoli, C.; Cockburn, B.N.; Chiaramonte, F.; Gorini, A.; Marietti, G.; Marozzi, G.; Signorini, A.M. Early-Onset Type II Diabetes Mellitus in Italian Families Due to Mutations in the Genes Encoding Hepatic Nuclear Factor 1 Alpha and Glucokinase. Diabetologia 2001, 44, 1326–1329. [Google Scholar] [CrossRef][Green Version]
- Froguel, P.; Zouali, H.; Vionnet, N.; Velho, G.; Vaxillaire, M.; Sun, F.; Lesage, S.; Stoffel, M.; Takeda, J.; Passa, P. Familial Hyperglycemia Due to Mutations in Glucokinase. Definition of a Subtype of Diabetes Mellitus. N. Engl. J. Med. 1993, 328, 697–702. [Google Scholar] [CrossRef]
- Thomson, K.L.; Gloyn, A.L.; Colclough, K.; Batten, M.; Allen, L.I.S.; Beards, F.; Hattersley, A.T.; Ellard, S. Identification of 21 Novel Glucokinase (GCK) Mutations in UK and European Caucasians with Maturity-Onset Diabetes of the Young (MODY). Hum. Mutat. 2003, 22, 417. [Google Scholar] [CrossRef]
- Ma, Y.; Han, X.; Zhou, X.; Li, Y.; Gong, S.; Zhang, S.; Cai, X.; Zhou, L.; Luo, Y.; Li, M.; et al. A New Clinical Screening Strategy and Prevalence Estimation for Glucokinase Variant-Induced Diabetes in an Adult Chinese Population. Genet. Med. 2019, 21, 939–947. [Google Scholar] [CrossRef]
- Sagen, J.V.; Odili, S.; Bjørkhaug, L.; Zelent, D.; Buettger, C.; Kwagh, J.; Stanley, C.; Dahl-Jørgensen, K.; de Beaufort, C.; Bell, G.I.; et al. From Clinicogenetic Studies of Maturity-Onset Diabetes of the Young to Unraveling Complex Mechanisms of Glucokinase Regulation. Diabetes 2006, 55, 1713–1722. [Google Scholar] [CrossRef]
- Delvecchio, M.; Mozzillo, E.; Salzano, G.; Iafusco, D.; Frontino, G.; Patera, P.I.; Rabbone, I.; Cherubini, V.; Grasso, V.; Tinto, N.; et al. Monogenic Diabetes Accounts for 6.3% of Cases Referred to 15 Italian Pediatric Diabetes Centers During 2007 to 2012. J. Clin. Endocrinol. Metab. 2017, 102, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Alvelos, M.I.; Gonçalves, C.I.; Coutinho, E.; Almeida, J.T.; Bastos, M.; Sampaio, M.L.; Melo, M.; Martins, S.; Dinis, I.; Mirante, A.; et al. Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. J. Clin. Med. 2020, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Shimada, F.; Makino, H.; Hashimoto, N.; Taira, M.; Seino, S.; Bell, G.I.; Kanatsuka, A.; Yoshida, S. Type 2 (Non-Insulin-Dependent) Diabetes Mellitus Associated with a Mutation of the Glucokinase Gene in a Japanese Family. Diabetologia 1993, 36, 433–437. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Massa, O.; Meschi, F.; Cuesta-Munoz, A.; Caumo, A.; Cerutti, F.; Toni, S.; Cherubini, V.; Guazzarotti, L.; Sulli, N.; Matschinsky, F.M.; et al. High Prevalence of Glucokinase Mutations in Italian Children with MODY. Influence on Glucose Tolerance, First-Phase Insulin Response, Insulin Sensitivity and BMI. Diabetologia 2001, 44, 898–905. [Google Scholar] [CrossRef][Green Version]
- Steele, A.M.; Shields, B.M.; Wensley, K.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Prevalence of Vascular Complications among Patients with Glucokinase Mutations and Prolonged, Mild Hyperglycemia. JAMA 2014, 311, 279–286. [Google Scholar] [CrossRef]
- Beer, N.L.; Osbak, K.K.; van de Bunt, M.; Tribble, N.D.; Steele, A.M.; Wensley, K.J.; Edghill, E.L.; Colcough, K.; Barrett, A.; Valentínová, L.; et al. Insights into the Pathogenicity of Rare Missense GCK Variants from the Identification and Functional Characterization of Compound Heterozygous and Double Mutations Inherited in Cis. Diabetes Care 2012, 35, 1482–1484. [Google Scholar] [CrossRef]
- Ziemssen, F.; Bellanné-Chantelot, C.; Osterhoff, M.; Schatz, H.; Pfeiffer, A.F.H. To: Lindner T, Cockburn BN, Bell GI (1999). Molecular Genetics of MODY in Germany. Diabetologia 42: 121–123. Diabetologia 2002, 45, 286–287. [Google Scholar] [CrossRef]
- Pruhova, S.; Dusatkova, P.; Sumnik, Z.; Kolouskova, S.; Pedersen, O.; Hansen, T.; Cinek, O.; Lebl, J. Glucokinase Diabetes in 103 Families from a Country-Based Study in the Czech Republic: Geographically Restricted Distribution of Two Prevalent GCK Mutations. Pediatr. Diabetes 2010, 11, 529–535. [Google Scholar] [CrossRef]
- Stoffel, M.; Froguel, P.; Takeda, J.; Zouali, H.; Vionnet, N.; Nishi, S.; Weber, I.T.; Harrison, R.W.; Pilkis, S.J.; Lesage, S. Human Glucokinase Gene: Isolation, Characterization, and Identification of Two Missense Mutations Linked to Early-Onset Non-Insulin-Dependent (Type 2) Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 1992, 89, 7698–7702. [Google Scholar] [CrossRef]
- Estalella, I.; Rica, I.; Perez de Nanclares, G.; Bilbao, J.R.; Vazquez, J.A.; San Pedro, J.I.; Busturia, M.A.; Castaño, L. Spanish MODY Group Mutations in GCK and HNF-1alpha Explain the Majority of Cases with Clinical Diagnosis of MODY in Spain. Clin. Endocrinol. 2007, 67, 538–546. [Google Scholar] [CrossRef]
- Gloyn, A.L. Glucokinase (GCK) Mutations in Hyper- and Hypoglycemia: Maturity-Onset Diabetes of the Young, Permanent Neonatal Diabetes, and Hyperinsulinemia of Infancy. Hum. Mutat. 2003, 22, 353–362. [Google Scholar] [CrossRef]
- Caswell, R.C.; Snowsill, T.; Houghton, J.A.L.; Chakera, A.J.; Shepherd, M.H.; Laver, T.W.; Knight, B.A.; Wright, D.; Hattersley, A.T.; Ellard, S. Noninvasive Fetal Genotyping by Droplet Digital PCR to Identify Maternally Inherited Monogenic Diabetes Variants. Clin. Chem. 2020, 66, 958–965. [Google Scholar] [CrossRef]
- Wang, Z.; Diao, C.; Liu, Y.; Li, M.; Zheng, J.; Zhang, Q.; Yu, M.; Zhang, H.; Ping, F.; Li, M.; et al. Identification and Functional Analysis of GCK Gene Mutations in 12 Chinese Families with Hyperglycemia. J. Diabetes Investig. 2019, 10, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Bell, K.L.; Blackburn, C.L.; Powell, K.L.; Seo, T.S.; Takeda, J.; Vionnet, N.; Xiang, K.S.; Gidh-Jain, M.; Pilkis, S.J. Identification of Glucokinase Mutations in Subjects with Gestational Diabetes Mellitus. Diabetes 1993, 42, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Sanyoura, M.; Letourneau, L.; Knight Johnson, A.E.; del Gaudio, D.; Greeley, S.A.W.; Philipson, L.H.; Naylor, R.N. GCK-MODY in the US Monogenic Diabetes Registry: Description of 27 Unpublished Variants. Diabetes Res. Clin. Pract. 2019, 151, 231–236. [Google Scholar] [CrossRef]
- Pruhova, S.; Ek, J.; Lebl, J.; Sumnik, Z.; Saudek, F.; Andel, M.; Pedersen, O.; Hansen, T. Genetic Epidemiology of MODY in the Czech Republic: New Mutations in the MODY Genes HNF-4alpha, GCK and HNF-1alpha. Diabetologia 2003, 46, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Bonfig, W.; Hermanns, S.; Warncke, K.; Eder, G.; Engelsberger, I.; Burdach, S.; Ziegler, A.G.; Lohse, P. GCK-MODY (MODY 2) Caused by a Novel p.Phe330Ser Mutation. ISRN Pediatr. 2011, 2011, 676549. [Google Scholar] [CrossRef]
- Furuzawa, G.K.; Giuffrida, F.M.A.; Oliveira, C.S.V.; Chacra, A.R.; Dib, S.A.; Reis, A.F. Low Prevalence of MODY2 and MODY3 Mutations in Brazilian Individuals with Clinical MODY Phenotype. Diabetes Res. Clin. Pract. 2008, 81, e12–e14. [Google Scholar] [CrossRef]
- Johansen, A.; Ek, J.; Mortensen, H.B.; Pedersen, O.; Hansen, T. Half of Clinically Defined Maturity-Onset Diabetes of the Young Patients in Denmark Do Not Have Mutations in HNF4A, GCK, and TCF1. J. Clin. Endocrinol. Metab. 2005, 90, 4607–4614. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.T.; Vasta, V.; Zhang, M.; Narayanan, J.; Gerrits, P.; Hahn, S.H. Molecular Genetic Testing of Patients with Monogenic Diabetes and Hyperinsulinism. Mol. Genet. Metab. 2015, 114, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Shammas, C.; Neocleous, V.; Phelan, M.M.; Lian, L.-Y.; Skordis, N.; Phylactou, L.A. A Report of 2 New Cases of MODY2 and Review of the Literature: Implications in the Search for Type 2 Diabetes Drugs. Metabolism 2013, 62, 1535–1542. [Google Scholar] [CrossRef] [PubMed]


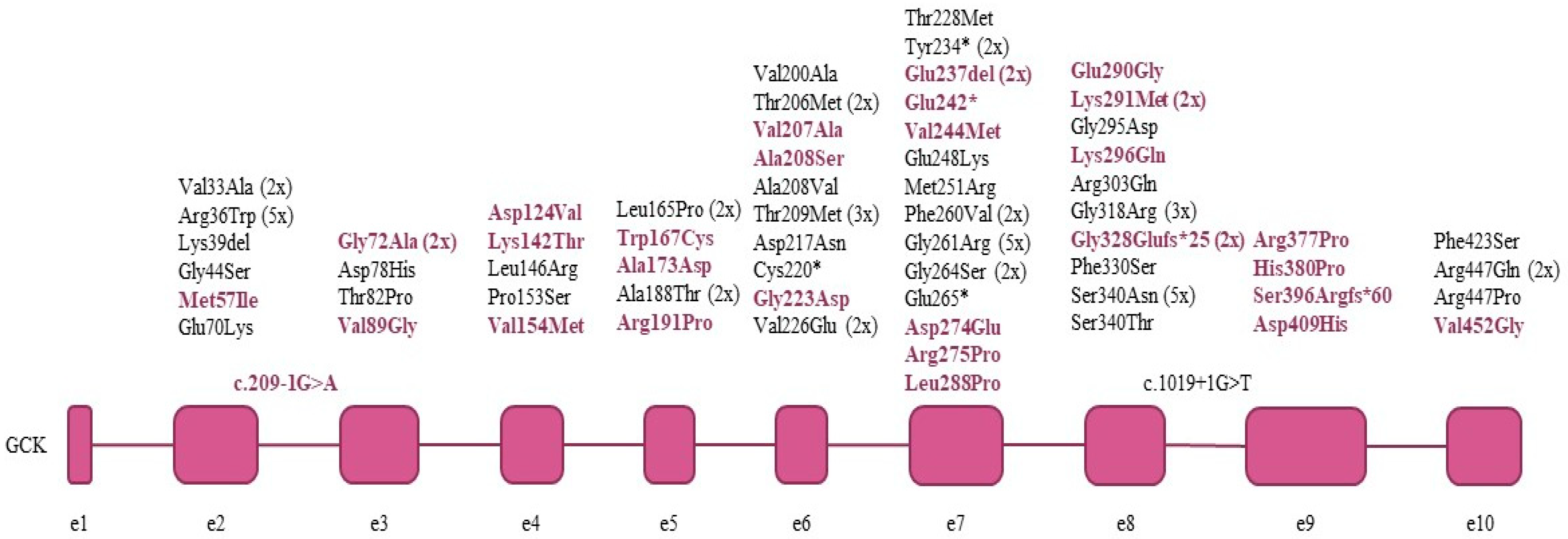{kind=link}
| Gene | No. of Index Patients with ‘P’/’LP’ Mutations | No. of Index Patients and Their Family Members with ‘P’/’LP’ Mutations |
|---|---|---|
| GCK | 91 (68.9%) | 163 (71.8%) |
| HNF1A | 30 (22.7%) | 48 (21.1%) |
| other MODY-causing gene 1 | 11 (8.3%) | 16 (7.0%) |
| Total | 132 (100.0%) | 227 (100.0%) |
| Nucleotide Change | Protein Change | Exon/Intron | Function | ACMG | ACMG Evidence | ClinVar | gnomAD Alleles (MAF) | Pr/FM | Family ID | Novel/Known | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c.98T > C | p.Val33Ala | exon 2 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (1) | N/A | 2/0 | F306, F454 | known | [24] |
| c.106C > T | p.Arg36Trp | exon 2 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (2); PP2 (1); PP3 (1); PP5 (3) | Pathogenic/ Likely pathogenic (4) | 4 (0.00001414) | 5/4 | F028, F105, F310, F433, F434 | known | [25] |
| c.115_117delAAG | p.Lys39del | exon 2 | In-Frame | Pathogenic | PM1 (2); PM2 (2); PM4 (2); PP1 (3) | N/A | N/A | 1/2 | F044 | known | [26] |
| c.130G > A | p.Gly44Ser | exon 2 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (3); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (1) | N/A | 1/3 | F133 | known | [27] |
| c.171G > T | p.Met57Ile | exon 2 | Missense | Pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (3); PS1 (1) | Pathogenic (1) | N/A | 1/0 | F463 | novel | |
| c.208G > A | p.Glu70Lys | exon 2 | Missense/Splicing | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (1) | N/A | N/A | 1/0 | F389 | known | [28] |
| c.209-1G > A | Splice | intron 2 | Splicing | Likely pathogenic | PM2 (2); PVS1 (4) | N/A | 1 (0.00003186) | 1/0 | F499 | novel | |
| c.215G > C | p.Gly72Ala | exon 3 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP1 (2); PP2 (1); PP3 (1) | N/A | N/A | 2/3 | F069, F274 | novel | |
| c.232G > C | p.Asp78His | exon 3 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/3 | F006 | known | [29] |
| c.244A > C | p.Thr82Pro | exon 3 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F472 | known | [30] |
| c.266T > G | p.Val89Gly | exon 3 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F213 | novel | |
| c.371A > T | p.Asp124Val | exon 4 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (1); PP2 (1); PP3 (1); PP5 (2) | Likely pathogenic (1) | N/A | 1/1 | F083 | novel | |
| c.425A > C | p.Lys142Thr | exon 4 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F250 | novel | |
| c.437T > G | p.Leu146Arg | exon 4 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F018 | known | [31] |
| c.457C > T | p.Pro153Ser | exon 4 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1) | Uncertain significance (2) | N/A | 1/1 | F103 | known | [32] |
| c.460G > A | p.Val154Met | exon 4 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F381 | novel | |
| c.494T > C | p.Leu165Pro | exon 5 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 2/0 | F092, F291 | known | [33] |
| c.501G > C | p.Trp167Cys | exon 5 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F349 | novel | |
| c.518C > A | p.Ala173Asp | exon 5 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F002 | novel | |
| c.562G > A | p.Ala188Thr | exon 5 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (2); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (2) | 1 (0.000003982) | 2/2 | F101, F173 | known | [34] |
| c.572G > C | p.Arg191Pro | exon 5 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F374 | novel | |
| c.599T > C | p.Val200Ala | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F094 | known | [10] |
| c.617C > T | p.Thr206Met | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP1 (2); PP2 (1); PP3 (1); PP5 (1) | N/A | 1 (0.000003977) | 2/2 | F031, F116 | known | [35] |
| c.620T > C | p.Val207Ala | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F316 | novel | |
| c.622G > T | p.Ala208Ser | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F205 | novel | |
| c.623C > T | p.Ala208Val | exon 6 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (3); PP2 (1); PP3 (1) | Uncertain significance (1) | 1 (0.000003977) | 1/2 | F227 | known | [36] |
| c.626C > T | p.Thr209Met | exon 6 | Missense | Pathogenic | PM1 (2); PM2 (2); PP1 (2); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (1) | N/A | 3/1 | F041, F042, F153 | known | [25] |
| c.649G > A | p.Asp217Asn | exon 6 | Missense | Likely pathogenic | BS4 (1); PM1 (2); PM2 (2); PP2 (1); PP3 (1) | Uncertain significance (4) | 1 | 1/0 | F150 | known | [37] |
| c.660C > A | p.Cys220* | exon 6 | Nonsense | Likely pathogenic | PM2 (2); PVS1 (4) | N/A | N/A | 1/0 | F165 | known | [38] |
| c.668G > A | p.Gly223Asp | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/2 | F411 | novel | |
| c.677T > A | p.Val226Glu | exon 6 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP1 (2); PP2 (1); PP3 (1) | N/A | N/A | 2/2 | F101, F173 | known | [39] |
| c.683C > T | p.Thr228Met | exon 7 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (5) | 1 (0.000003999) | 1/0 | F065 | known | [40] |
| c.702C > A | p.Tyr234* | exon 7 | Nonsense | Pathogenic | PM2 (2); PP1 (2); PVS1 (4) | N/A | N/A | 2/2 | F011, F244 | known | [41] |
| c.709_711delGAG | p.Glu237del | exon 7 | In-frame | Likely pathogenic | PM1 (2); PM2 (2); PM4 (2); PP1 (2) | N/A | N/A | 2/1 | F070, F246 | novel | |
| c.724G > T | p.Glu242* | exon 7 | Nonsense | Pathogenic | PM2 (2); PP1 (3); PVS1 (4) | N/A | N/A | 1/3 | F382 | novel | |
| c.730G > A | p.Val244Met | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (1) | Conflicting (1 LP, 1 VUS) | N/A | 1/0 | F400 | novel | |
| c.742G > A | p.Glu248Lys | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (1) | N/A | 1 (0.000003982) | 1/0 | F219 | known | [42] |
| c.752T > G | p.Met251Arg | exon 7 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (3); PP2 (1); PP3 (1) | N/A | N/A | 1/4 | F145 | known | [10] |
| c.778T > G | p.Phe260Val | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | N/A | N/A | 2/2 | F408, F455 | known | [43] |
| c.781G > A | p.Gly261Arg | exon 7 | Missense | Pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1); PP5 (3); PS1 (3) | Pathogenic (4) | 1 (0.000003983) | 4/1 | F126, F191, F202, F272 | known | [40] |
| c.781G > C | p.Gly261Arg | exon 7 | Missense | Pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (3); PS1 (3) | Pathogenic (1) | N/A | 1/0 | F108 | known | [44] |
| c.790G > A | p.Gly264Ser | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1); PP5 (1) | Pathogenic (1) | N/A | 2/0 | F080, F216 | known | [35] |
| c.793G > T | p.Glu265* | exon 7 | Nonsense | Pathogenic | PM2 (2); PP1 (1); PP5 (3); PVS1 (4) | Pathogenic (2) | N/A | 1/1 | F035 | known | [45] |
| c.822C > A | p.Asp274Glu | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | 1 (0.000003992) | 1/0 | F280 | novel | |
| c.824G > C | p.Arg275Pro | exon 7 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F107 | novel | |
| c.863T > C | p.Leu288Pro | exon 7 | Missense/splicing | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F403 | novel | |
| c.869A > G | p.Glu290Gly | exon 8 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F375 | novel | |
| c.872A > T | p.Lys291Met | exon 8 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (3); PP2 (1); PP3 (1) | N/A | N/A | 2/6 | F043, F046 | novel | |
| c.884G > A | p.Gly295Asp | exon 8 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F038 | known | [39] |
| c.886A > C | p.Lys296Gln | exon 8 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | 2 (0.000008008) | 1/0 | F296 | novel | |
| c.908G > A | p.Arg303Gln | exon 8 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1); PP5 (2) | Likely pathogenic (1) | N/A | 1/1 | F167 | known | [46] |
| c.952G > A | p.Gly318Arg | exon 8 | Missense | Pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (3); PP2 (1); PP3 (1); PP5 (3) | Pathogenic (1) | N/A | 3/5 | F016, F209, F353 | known | [47] |
| c.982delG | p.Gly328Glufs*25 | exon 8 | Frameshift | Pathogenic | PM2 (2); PP1 (3); PVS1 (4) | N/A | N/A | 2/5 | F085, F162 | novel | |
| c.989T > C | p.Phe330Ser | exon 8 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1) | Uncertain significance (1) | N/A | 1/1 | F122 | known | [48] |
| c.1019G > A | p.Ser340Asn | exon 8 | Missense/ splicing | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (2); PP2 (1); PP3 (1) | N/A | N/A | 5/4 | F027, F062, F163, F187, F197 | known | [32] |
| c.1019G > C | p.Ser340Thr | exon 8 | Missense/ splicing | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F435 | known | [49] |
| c.1019 + 1G > T | Splice | intron 8 | Splicing | Likely pathogenic | PM2 (2); PVS1 (3) | N/A | N/A | 1/0 | F131 | known | [50] |
| c.1130G > C | p.Arg377Pro | exon 9 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F482 | novel | |
| c.1139A > C | p.His380Pro | exon 9 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F200 | novel | |
| c.1186_1193delAGCCGCAG | p.Ser396Argfs*60 | exon 9 | Frameshift | Likely pathogenic | PM2 (2); PVS1 (4) | N/A | N/A | 1/0 | F314 | novel | |
| c.1225G > C | p.Asp409His | exon 9 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP1 (1); PP2 (1); PP3 (1) | N/A | N/A | 1/1 | F113 | novel | |
| c.1268T > C | p.Phe423Ser | exon 10 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | Uncertain significance (1) | N/A | 1/0 | F275 | known | [51] |
| c.1340G > A | p.Arg447Gln | exon 10 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP1 (2); PP2 (1); PP3 (1); PP5 (2) | Likely pathogenic (1) | N/A | 2/2 | F201, F373 | known | [29,42] |
| c.1340G > C | p.Arg447Pro | exon 10 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PM5 (1); PP2 (1); PP3 (1) | Uncertain significance (1) | N/A | 1/0 | F273 | known | [52] |
| c.1355T > G | p.Val452Gly | exon 10 | Missense | Likely pathogenic | PM1 (2); PM2 (2); PP2 (1); PP3 (1) | N/A | N/A | 1/0 | F263 | novel |
| Consequence | No. of Mutations |
|---|---|
| Missense | 51 (78.5%) |
| Missense and/or splicing | 4 (6.2%) |
| Splicing | 21 (3.1%) |
| Nonsense | 4 (6.2%) |
| Frameshift | 2 (3.1%) |
| In-frame | 2 (3.1%) |
| Family ID | Sample ID | Age at Diagnosis of Diabetes | Age at Receiving Genetic Dg | BMI * | Obesity | Complications | Therapy BEFORE Genetic Diagnosis | FPG (0′) (mmol/L) | PPG (120′) (mmol/L) | HbA1c % (mmol/mol) | MODY Calculator (%) | Family Screening |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F002 | P002 | 32 | 47 | 26.2 | no | none | OAD—metformin | 7.3 | N/A | 6.3 (45.4) | 15.1 | no family members tested |
| F006 | P015 | 31 | N/A | 23 | no | none | insulin | 6.8 | 19.0 | 6.7 (49.7) | 12.6 | multiple generations affected |
| F006 | P016 | 46 | N/A | 33 | yes | IHD, PAD | insulin | 7.0 | 12.0 | 8.2 (66.1) | N/A | multiple generations affected |
| F006 | P017 | 3 | 4 | 15.6 | no | none | diet | 6.0 | 9.0 | N/A | N/A | multiple generations affected |
| F006 | P018 | no diabetes | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F011 | P028 | 17 | 27 | 19.8 | no | none | OAD—acarbose | 7.0 | 6.0 | 6.2 (44.3) | 75.5 | multiple generations affected |
| F011 | P029 | 42 | 53 | 24.7 | no | none | OAD— sulphonylurea | 9.2 | 5.2 | 6.5 (47.5) | N/A | multiple generations affected |
| F011 | P030 | 2 | 3 | 15.4 | no | none | diet | 6.7 | N/A | N/A | N/A | multiple generations affected |
| F016 | P037 | childhood | 30 | 25 | no | none | OAD—metformin | 5.8 | 6.2 | 6.6 (48.6) | N/A | multiple generations affected |
| F016 | P038 | no diabetes | 3 | N/A | N/A | none | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F016 | P040 | 15 | 33 | N/A | N/A | none | diet | 7.6 | N/A | N/A | N/A | multiple generations affected |
| F016 | P041 | N/A | 5 | N/A | N/A | none | diet | 5.8 | 8.1 | 6.1 (43.2) | N/A | multiple generations affected |
| F016 | P042 | N/A | 55 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F016 | P044 | N/A | 14 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F018 | P047 | 10 | 14 | N/A | N/A | none | diet | 6.5 | N/A | 6.3 (45.4) | N/A | no family members tested |
| F027 | P063 | N/A | 25 | N/A | N/A | none | diet | 6.1 | 8.8 | 6.2 (44.3) | N/A | no family members tested |
| F028 | P064 | 10 | 12 | 19.4 | no | none | N/A | 7.1 | 8.5 | N/A | N/A | multiple generations affected |
| F028 | P065 | N/A | 11 | 23.2 | no | none | OAD—metformin | 6.2 | 7.1 | 6.5 (47.5) | N/A | multiple generations affected |
| F028 | P066 | N/A | 41 | N/A | N/A | none | none | 6.7 | 6.7 | N/A | N/A | multiple generations affected |
| F031 | P069 | 8 | 11 | 16.2 | no | none | OAD—metformin | 7.6 | 7.9 | 6.7 (49.7) | 75.5 | multiple generations affected |
| F031 | P070 | 27 | 40 | N/A | N/A | N/A | diet | N/A | N/A | N/A | N/A | multiple generations affected |
| F035 | P074 | 8 | 11 | 19.1 | no | none | diet | 6.0 | 15.2 | 6.2 (43.2) | 75.5 | multiple generations affected |
| F035 | P075 | 26 | 40 | 23.2 | no | N/A | OAD—metformin | N/A | N/A | N/A | N/A | multiple generations affected |
| F038 | P078 | N/A | 36 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F038 | P079 | 7 | 12 | 15.2 | no | none | none | 6.5 | 8.6 | N/A | N/A | multiple generations affected |
| F041 | P082 | 8 | 10 | 14.31 | no | none | diet | 8.1 | 11.6 | 6.2 (44.3) | 75.5 | de novo |
| F042 | P085 | 4 | 7 | 15.7 | no | none | diet | 6.0 | 8.5 | 6.4 (46.4) | 75.5 | siblings positive, parents not tested |
| F042 | P086 | 1 | 1 | N/A | N/A | N/A | N/A | N/A | N/A | 4.8 (29.0) | N/A | siblings positive, parents not tested |
| F043 | P088 | 20 | 34 | 21.5 | no | none | diet | 7.9 | N/A | 6.4 (46.4) | 75.5 | no family members tested |
| F044 | P089 | 14 | 17 | 24 | no | none | insulin | 5.1 | N/A | 6.8 (50.8) | 49.4 | multiple generations affected |
| F044 | P090 | N/A | 48 | 29.5 | no | renal cysts | N/A | 6.9 | N/A | 6.7 (49.7) | N/A | multiple generations affected |
| F044 | P091 | 46 | 69 | 21 | no | TIA, glaucoma, osteoporosis | OAD—metformin | 5.1 | N/A | 6.6 (48.6) | N/A | multiple generations affected |
| F046 | P104 | 18 | 45 | 22.0 | no | none | none | N/A | N/A | 5.5 (36.6) | 75.5 | multiple generations affected |
| F046 | P105 | 13 | 18 | 23.7 | no | none | insulin | 7.3 | 11.2 | 6.7 (49.7) | 49.4 | multiple generations affected |
| F046 | P106 | 14 | 24 | 18.5 | no | none | insulin | 6.0 | 9.5 | 6.2 (44.3) | 75.5 | multiple generations affected |
| F046 | P108 | 15 | 20 | 19.4 | no | none | insulin | 7.7 | 8.7 | 7.0 (53.0) | 8.2 | multiple generations affected |
| F046 | P113 | N/A | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F046 | P738 | N/A | 3 | 15.5 | no | none | none | 5.7 | N/A | 6.0 (42.1) | N/A | multiple generations affected |
| F046 | P739 | N/A | 6 | 15.7 | no | none | none | 5.8 | N/A | 6.1 (43.2) | N/A | multiple generations affected |
| F062 | P128 | 3 | 3 | N/A | N/A | none | none | 6.0 | 5.4 | N/A | N/A | multiple generations affected |
| F062 | P129 | no diabetes | 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F062 | P130 | no diabetes | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F065 | P133 | 11 | 23 | 30 | yes | N/A | insulin | N/A | N/A | 6.6 (48.6) | 12.6 | no family members tested |
| F069 | P141 | 16 | 18 | 18.7 | no | none | OAD—metformin | 6.8 | N/A | 5.7 (38.8) | 75.5 | multiple generations affected |
| F069 | P142 | N/A | 51 | N/A | N/A | N/A | N/A | 6.9 | N/A | 6.4 (46.4) | N/A | multiple generations affected |
| F069 | P143 | N/A | 64 | N/A | N/A | N/A | N/A | 7.1 | N/A | 6.4 (46.4) | N/A | multiple generations affected |
| F070 | P144 | N/A | 23 | 18.7 | no | ligament tear | OAD—metformin | 7.1 | N/A | 6.0 (42.1) | N/A | no family members tested |
| F080 | P138 | N/A | 8 | N/A | N/A | N/A | N/A | 5–7 | N/A | N/A | N/A | no family members tested |
| F083 | P157 | N/A | 24 | N/A | N/A | PCOS | none | 6.9 | 7.9 | N/A | N/A | multiple generations affected |
| F083 | P158 | 15 | 50 | N/A | N/A | N/A | OAD—metformin | 7.78 | N/A | 6.3 (45.4) | N/A | multiple generations affected |
| F085 | P160 | 16 | 18 | 20.1 | no | none | diet | 7.1 | 7.9 | 6.8 (50.8) | 75.5 | multiple generations affected |
| F085 | P161 | 15 | 32 | 17.8 | no | none | diet | 6 | N/A | 6.1 (43.2) | 75.5 | multiple generations affected |
| F085 | P162 | 2 | 2 | N/A | N/A | granuloma annulare | diet | 6.1 | N/A | 6.1 (43.2) | N/A | multiple generations affected |
| F092 | P170 | N/A | 15 | N/A | N/A | none | diet | 6.6 | 9.4 | 6.5 (47.5) | N/A | no family members tested |
| F094 | P172 | 10 | 15 | 19.4 | no | none | diet | 7.2 | 9.6 | 6.4 (46.4) | 75.5 | parents not tested |
| F101 | P192 | 6 | 7 | 13.4 | no | none | none | 6.0 | 10.0 | 6.0 (42.1) | 75.5 | multiple generations affected |
| F101 | P193 | no diabetes | 46 | normal | no | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F103 | P196 | 5 | 6 | 17 | no | none | diet | 6.2 | 5.8 | 6.3 (45.4) | 75.5 | multiple generations affected |
| F103 | P197 | 36 | 42 | 24 | no | none | insulin | 6.9 | 13.6 | 6.0 (42.1) | N/A | multiple generations affected |
| F105 | P199 | 9 | 14 | 20.9 | no | N/A | diet | 7.3 | N/A | N/A | N/A | multiple generations affected |
| F105 | P200 | 25 | 37 | 30.1 | yes | none | none | N/A | N/A | N/A | N/A | multiple generations affected |
| F105 | P202 | N/A | 6 | 15.1 | no | none | none | N/A | N/A | N/A | N/A | multiple generations affected |
| F107 | P204 | 16 | 17 | 19 | no | none | diet | 7.9 | 8.8 | 7.0 (53.0) | 75.5 | no family members tested |
| F108 | P205 | 21 | 30 | 15.1 | no | none | insulin | 6.2 | 10.4 | 6.0 (42.1) | 75.5 | no family members tested |
| F113 | P210 | 12 | 17 | 17 | no | none | insulin | N/A | N/A | 6.3 (45.4) | 12.6 | multiple generations affected |
| F113 | P211 | 43 | 45 | 22.2 | no | none | diet | 6.9 | 9.2 | 6.3 (45.4) | N/A | multiple generations affected |
| F116 | P214 | 7 | 19 | 19.8 | no | headache, elevated RR | OAD—metformin | 5.8 | 14 | 6.8 (50.8) | 75.5 | cousin positive, parents not tested |
| F116 | P215 | 9 | 15 | 21.1 | no | none | diet | 5.8 | 6.4 | 6.5 (47.5) | 75.5 | cousin positive, parents not tested |
| F122 | P222 | 14 | 20 | 25 | no | N/A | diet | 6.5 | N/A | 6.2 (44.3) | 75.5 | siblings positive, parents not tested |
| F122 | P223 | 14 | 24 | 24 | no | none | diet | 6.3 | N/A | N/A | N/A | siblings positive, parents not tested |
| F126 | P227 | 14 | 36 | 22.5 | no | PCOS | OAD—metformin | 5.9 | >11 | 5.8 (39.9) | 75.5 | no family members tested |
| F131 | P232 | 19 | 28 | 19.4 | no | none | none | 8.4 | N/A | 6.8 (50.8) | 75.5 | no family members tested |
| F133 | P236 | 13 | 14 | 19 | no | none | diet | 6.4 | 9.4 | 6.4 (46.4) | 75.5 | multiple generations affected |
| F133 | P237 | 11 | 18 | 19 | no | none | insulin | N/A | N/A | 6.4 (46.4) | 8.2 | multiple generations affected |
| F133 | P240 | 31 | 42 | 21 | no | none | OAD | 7.8 | 10.00 | 6.4 (46.4) | 58 | multiple generations affected |
| F133 | P241 | 57 | 63 | 38 | yes | IHD | OAD | 8.7 | 10 | N/A | N/A | multiple generations affected |
| F145 | P257 | 6 | 12 | 15.2 | no | none | insulin | N/A | N/A | 7.1 (54.1) | 12.6 | multiple generations affected |
| F145 | P258 | 37 | 38 | N/A | N/A | N/A | N/A | 7.3 | 7.8 | 5.8 (39.9) | N/A | multiple generations affected |
| F145 | P259 | no diabetes | 2016 | N/A | N/A | none | N/A | 7.0 | 5.9 | N/A | N/A | multiple generations affected |
| F145 | P260 | 4 | 5 | N/A | N/A | none | none | N/A | 9.7 | 6.8 (50.8) | N/A | multiple generations affected |
| F145 | P261 | 39 | 40 | 24.3 | no | N/A | N/A | 7.2 | 5.9 | 6.3 (45.4) | N/A | multiple generations affected |
| F150 | P266 | 35 | 59 | 21.6 | no | none | OAD—metformin | 10.1 | N/A | 7.4 (57.4) | 15.1 | parents not tested |
| F153 | P271 | 9 | 16 | 23.6 | no | none | diet | 6.9 | 12.0 | 6.2 (44.3) | 75.5 | no family members tested |
| F162 | P280 | 9 | 12 | 32.4 | yes | acanthosis nigricans | OAD—metformin | N/A | N/A | 6.5 (47.5) | 75.5 | multiple generations affected |
| F162 | P281 | 3 | 4 | 14.1 | no | N/A | diet | 4.7 | 5.7 | 6.4 (46.4) | 75.5 | multiple generations affected |
| F162 | P282 | N/A | 2 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F162 | P283 | no diabetes | 21 | N/A | N/A | N/A | none | 5.4 | 6.2 | N/A | N/A | multiple generations affected |
| F163 | P284 | 7 | 11 | 29.0 | yes | none | none | 10.0 | 6.3 | 6.2 (44.3) | 75.5 | multiple generations affected |
| F163 | P285 | 9 | 13 | 23.5 | no | none | none | 6.6 | 10.9 | 5.9 (41.0) | 75.5 | multiple generations affected |
| F163 | P286 | N/A | 43 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F165 | P288 | 10 | 11 | 18.0 | no | N/A | diet | 7 | 10.8 | 5.9 (41.0) | 75.5 | no family members tested |
| F167 | P290 | 13 | 13 | 19.2 | no | none | diet | 5.2 | 10.7 | 5.7 (38.8) | 75.5 | multiple generations affected |
| F167 | P292 | no diabetes | 48 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F173 | P298 | 1 | 2 | 15 | no | N/A | diet | 5.6 | 9.9 | 6.4 (46.4) | 75.5 | multiple generations affected |
| F173 | P299 | 16 | 33 | N/A | N/A | N/A | insulin | N/A | N/A | N/A | N/A | multiple generations affected |
| F187 | P319 | 1 | 4 | 16.5 | no | none | none | 6.3—7 | 5.8—7 | 6.5 (47.5) | 75.5 | no family members tested |
| F191 | P323 | 1 | 12 | 16.8 | no | none | diet | 6.6 | 7.3 | 6.6 (48.6) | 75.5 | no family members tested |
| F197 | P335 | 13 | 27 | 21.9 | no | none | diet | 6.9 | 8.5 | 6.5 (47.5) | 75.5 | no family members tested |
| F200 | P338 | 5 | 18 | 17.7 | no | none | diet | 6.17 | 8.3 | 6.8 (50.8) | 75.5 | multiple generations affected |
| F200 | P339 | 47 | 51 | 23.9 | no | none | OAD—metformin + sulphonylurea | N/A | N/A | N/A | N/A | multiple generations affected |
| F201 | P340 | 23 | 31 | 19.7 | no | none | OAD— sulphonylurea | 6.6 | 11.2 | 6.1 (43.2) | 75.5 | multiple generations affected |
| F201 | P341 | 57 | 60 | 30.8 | yes | N/A | diet | 6.9 | 14.5 | 6.3 (45.4) | N/A | multiple generations affected |
| F201 | P342 | 22 | 35 | 23.1 | no | none | diet | 7.3 | 9.8 | 5.8 (39.9) | 75.5 | multiple generations affected |
| F202 | P343 | 5 | 6 | 17.0 | no | none | none | 6.2 | 8.2 | 6.4 (46.4) | 75.5 | siblings positive, parents not tested |
| F202 | P344 | 9 | 10 | 15.4 | no | none | none | 6.5 | 6.9 | 6.9 (51.9) | 75.5 | siblings positive, parents not tested |
| F205 | P349 | 4 | 10 | 16.5 | no | none | diet | 5.8 | 8.5 | 5.7 (38.8) | 75.5 | no family members tested |
| F209 | P353 | 9 | 43 | 29 | no | none | OAD—metformin | 7.2 | 9.5 | 7.0 (53.0) | 75.5 | no family members tested |
| F213 | P357 | N/A | 12 | 17.8 | no | none | diet | 7.1 | 8.6 | 6.2 (44.3) | N/A | multiple generations affected |
| F213 | P358 | N/A | 34 | N/A | N/A | N/A | diet | N/A | N/A | N/A | N/A | multiple generations affected |
| F216 | P361 | N/A | 14 | 16.2 | no | none | diet | 5.6 | 5.9 | 6.3 (45.4) | N/A | no family members tested |
| F219 | P364 | 32 | 39 | 25.2 | no | none | diet | 6.9 | 8.7 | 5.5 (36.6) | 62.4 | parents not tested |
| F227 | P373 | 7 | 21 | 25.7 | no | none | insulin | 7.0 | 14.7 | 6.1 (43.2) | 75.5 | multiple generations affected |
| F227 | P374 | N/A | 15 | 15.5 | no | N/A | N/A | 6.8 | N/A | N/A | N/A | multiple generations affected |
| F227 | P377 | 45 | 51 | 33 | yes | none | OAD—metformin | 7.8 | 8.9 | N/A | N/A | multiple generations affected |
| F244 | P394 | 15 | 16 | 17.6 | no | none | diet | 6.5 | 8.8 | 6.2 (44.3) | 75.5 | no family members tested |
| F246 | P396 | 6 | 7 | 19.9 | yes | none | none | 6.7 | 9.4 | 6.3 (45.4) | 75.5 | multiple generations affected |
| F246 | P398 | no diabetes | 43 | N/A | N/A | N/A | N/A | 7.5 | N/A | 7.0 (53.0) | N/A | multiple generations affected |
| F250 | P403 | N/A | 35 | N/A | N/A | cardiac | N/A | N/A | N/A | N/A | N/A | no family members tested |
| F263 | P426 | 18 | 31 | 21.5 | no | N/A | OAD—metformin | 6.9 | 8.5 | 6.4 (46.4) | 75.5 | no family members tested |
| F272 | P435 | 20 | 22 | 17.4 | no | none | none | 6.7 | 22.9 | 6.0 (42.1) | 75.5 | no family members tested |
| F273 | P436 | N/A | 18 | 19.5 | no | N/A | N/A | 5.8 | N/A | N/A | N/A | no family members tested |
| F274 | P437 | 5 | 16 | 32.7 | yes | hypertension, obesity | OAD—metformin | 6.5 | 9.4 | 6.5 (47.5) | 75.5 | siblings positive, parents not tested |
| F274 | P438 | 2 | 7 | 16.7 | no | none | diet | 6.7 | N/A | 6.4 (46.4) | 75.5 | siblings positive, parents not tested |
| F275 | P439 | 8 | 10 | 16.3 | no | none | diet | 6.1 | 8.3 | 6.1 (43.2) | 75.5 | no family members tested |
| F280 | P444 | N/A | 10 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | no family members tested |
| F291 | P455 | 10 | 16 | 19.2 | no | none | insulin | 6.5 | 11.7 | 6.8 (50.8) | 4 | no family members tested |
| F296 | P460 | 13 | 18 | N/A | N/A | none | insulin | N/A | N/A | N/A | N/A | no family members tested |
| F306 | P470 | 7 | 8 | 14.6 | no | repeated acute laryngitis | none | 6.10 | N/A | 6.0 (42.0) | 75.5 | no family members tested |
| F310 | P477 | 14 | 15 | 22.2 | no | N/A | N/A | N/A | N/A | 6.4 (46.4) | 75.5 | mother no carrier, father not tested |
| F314 | P482 | 4 | 6 | 14.9 | no | none | diet | N/A | N/A | 6.0 (42.1) | 75.5 | no family members tested |
| F316 | P488 | 30 | 37 | 20.6 | no | none | OAD—metformin | 6.3 | 6.9 | 6.3 (45.4) | 35.8 | no family members tested |
| F349 | P532 | 17 | 39 | 20.6 | no | none | OAD—metformin | 5.7 | N/A | 6.4 (46.4) | 75.5 | no family members tested |
| F353 | P536 | 14 | 23 | 26.7 | no | none | diet | 6.9 | 9.6 | 6.5 (47.5) | 75.5 | no family members tested |
| F373 | P557 | 8 | 18 | 19.2 | no | N/A | diet | 5.7 | 9 | 5.6 (37.7) | 75.5 | no family members tested |
| F374 | P558 | 2 | 19 | 20 | no | N/A | diet | 6.0 | 9.4 | 6.7 (49.7) | 75.5 | no family members tested |
| F375 | P559 | 8 | 10 | normal | no | none | diet | 7.9 | 9.5 | 6.7 (49.7) | N/A | de novo |
| F381 | P570 | 12 | 12 | 21.2 | no | none | diet | 5.3 | 8.8 | 6.8 (50.8) | 75.5 | multiple generations affected |
| F381 | P572 | 2 | 41 | 24 | no | N/A | diet | N/A | N/A | N/A | N/A | multiple generations affected |
| F382 | P574 | 13 | 13 | 22 | no | none | diet | 7.7 | 9.8 | 6.3 (45.4) | 75.5 | multiple generations affected |
| F382 | P575 | N/A | 41 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F382 | P577 | N/A | 61 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F382 | P578 | N/A | 29 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F389 | P586 | 28 | 31 | 20 | no | none | diet | 7.2 | 9.5 | 6.3 (45.4) | 62.4 | no family members tested |
| F400 | P598 | 14 | 18 | 21 | no | none | none | 6.7 | 8.9 | 5.7 (38.8) | 75.5 | no family members tested |
| F403 | P601 | 10 | 11 | 15.1 | no | none | insulin | 6.0 | 11.0 | 6.3 (45.4) | 12.6 | mother no carrier, father not tested |
| F403 | P604 | no diabetes | 7 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | mother no carrier, father not tested |
| F408 | P610 | 5 | 18 | 18.7 | no | none | insulin | 7.3 | N/A | 6.4 (46.4) | 1.9 | siblings positive, parents not tested |
| F408 | P611 | 14 | 14 | 17.9 | no | none | diet | 7.2 | 8.8 | 7.1 (54.1) | 75.5 | siblings positive, parents not tested |
| F411 | P614 | 12 | 12 | 14.7 | no | none | diet | 5.9 | 9.3 | 7.0 (53.0) | 75.5 | multiple generations affected |
| F411 | P616 | no diabetes | 11 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F411 | P617 | no diabetes | 42 | 26 | no | none | diet | 7.1 | 9.1 | 6.2 (44.3) | N/A | multiple generations affected |
| F433 | P639 | N/A | 13 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | no family members tested |
| F434 | P640 | N/A | 10 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | no family members tested |
| F435 | P641 | 7 | 13 | 18 | no | none | insulin | N/A | N/A | 7.1 (54.1) | 8.2 | multiple generations affected |
| F435 | P642 | 24 | 37 | 24 | no | none | none | N/A | N/A | 7.2 (55.2) | 62.4 | multiple generations affected |
| F454 | P665 | 10 | 11 | 17.4 | no | possible coeliac disease | none | 6.2 | 5.9 | N/A | N/A | no family members tested |
| F455 | P666 | 3 | 4 | N/A | N/A | N/A | diet | 4.9 | 12.7 | 6.1 (43.2) | N/A | multiple generations affected |
| F455 | P667 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F463 | P675 | 20 | 27 | 18.9 | no | none | OAD—metformin + sulphonylurea | 7.2 | N/A | 6.6 (48.6) | 75.5 | no family members tested |
| F472 | P684 | 1 | 2 | N/A | N/A | N/A | N/A | 6.4 | 8.6 | 6.9 (51.9) | N/A | multiple generations affected |
| F472 | P733 | N/A | 50 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | multiple generations affected |
| F482 | P696 | 6 | 6 | 28 | yes | N/A | diet | 6.4 | 11.0 | 6.2 (44.3) | 75.5 | no family members tested |
| F499 | P713 | N/A | 21 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | no family members tested |
| Therapy | Number of Patients |
|---|---|
| insulin | 19 (11.7%) |
| OAD—sulphonylurea | 2 (1.2%) |
| OAD—metformin | 19 (11.7%) |
| OAD—metformin and sulphonylurea | 2 (1.2%) |
| OAD—acarbose | 1 (0.6%) |
| other OAD | 2 (1.2%) |
| diet | 56 (34.4%) |
| no treatment | 24 (14.7%) |
| N/A | 38 (23.3%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaál, Z.; Szűcs, Z.; Kántor, I.; Luczay, A.; Tóth-Heyn, P.; Benn, O.; Felszeghy, E.; Karádi, Z.; Madar, L.; Balogh, I. A Comprehensive Analysis of Hungarian MODY Patients—Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases. Life 2021, 11, 771. https://doi.org/10.3390/life11080771
Gaál Z, Szűcs Z, Kántor I, Luczay A, Tóth-Heyn P, Benn O, Felszeghy E, Karádi Z, Madar L, Balogh I. A Comprehensive Analysis of Hungarian MODY Patients—Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases. Life. 2021; 11(8):771. https://doi.org/10.3390/life11080771
Chicago/Turabian StyleGaál, Zsolt, Zsuzsanna Szűcs, Irén Kántor, Andrea Luczay, Péter Tóth-Heyn, Orsolya Benn, Enikő Felszeghy, Zsuzsanna Karádi, László Madar, and István Balogh. 2021. "A Comprehensive Analysis of Hungarian MODY Patients—Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases" Life 11, no. 8: 771. https://doi.org/10.3390/life11080771
APA StyleGaál, Z., Szűcs, Z., Kántor, I., Luczay, A., Tóth-Heyn, P., Benn, O., Felszeghy, E., Karádi, Z., Madar, L., & Balogh, I. (2021). A Comprehensive Analysis of Hungarian MODY Patients—Part II: Glucokinase MODY Is the Most Prevalent Subtype Responsible for about 70% of Confirmed Cases. Life, 11(8), 771. https://doi.org/10.3390/life11080771