
Flavivirus: From Structure to Therapeutics Development

Abstract
1. General Information of Flavivirus and the Infected Diseases
1.1. YFV
1.2. DENV
1.3. JEV
1.4. WNV
1.5. ZIKV
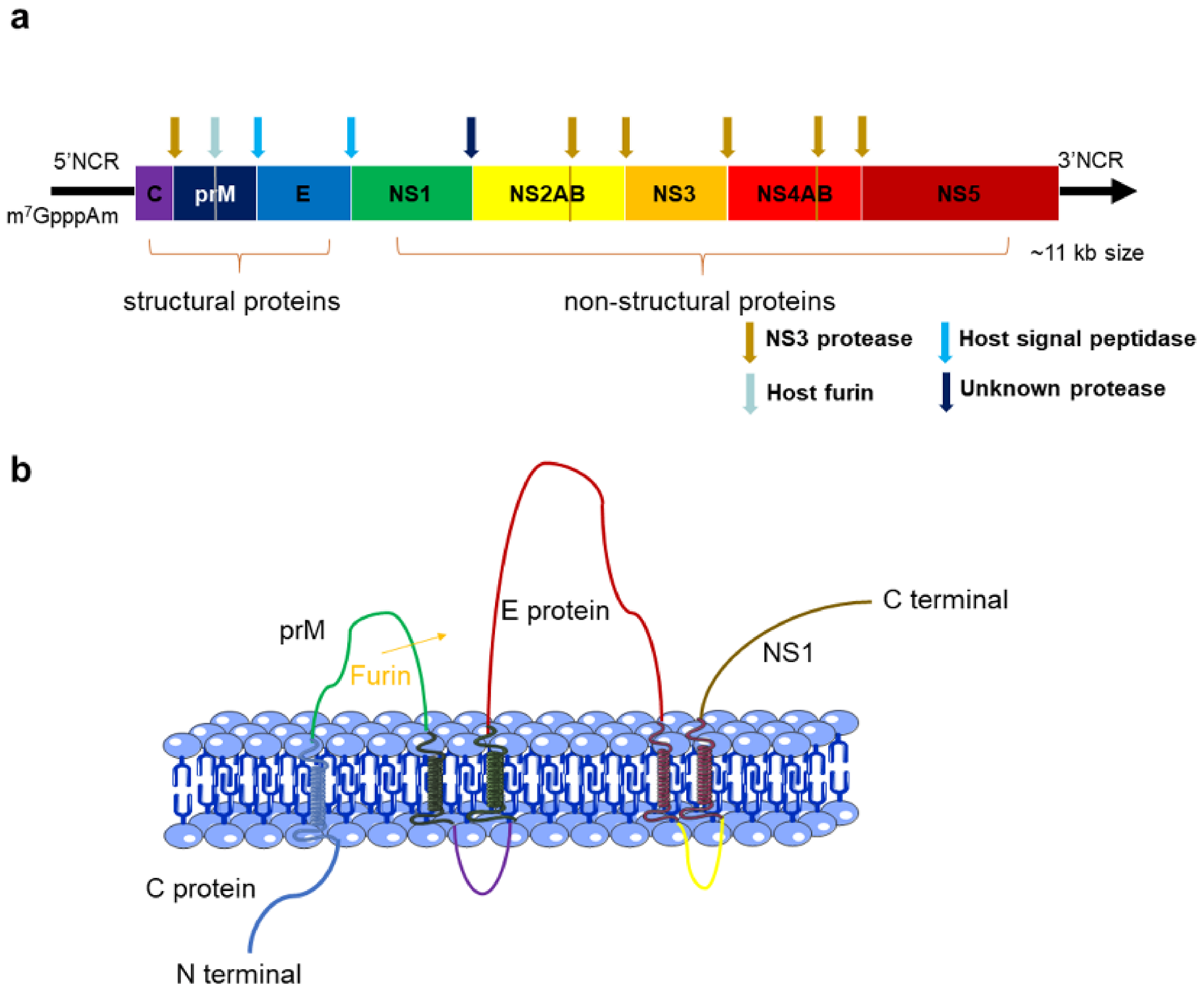
2. The Structures and Functions of Flavivirus Proteins
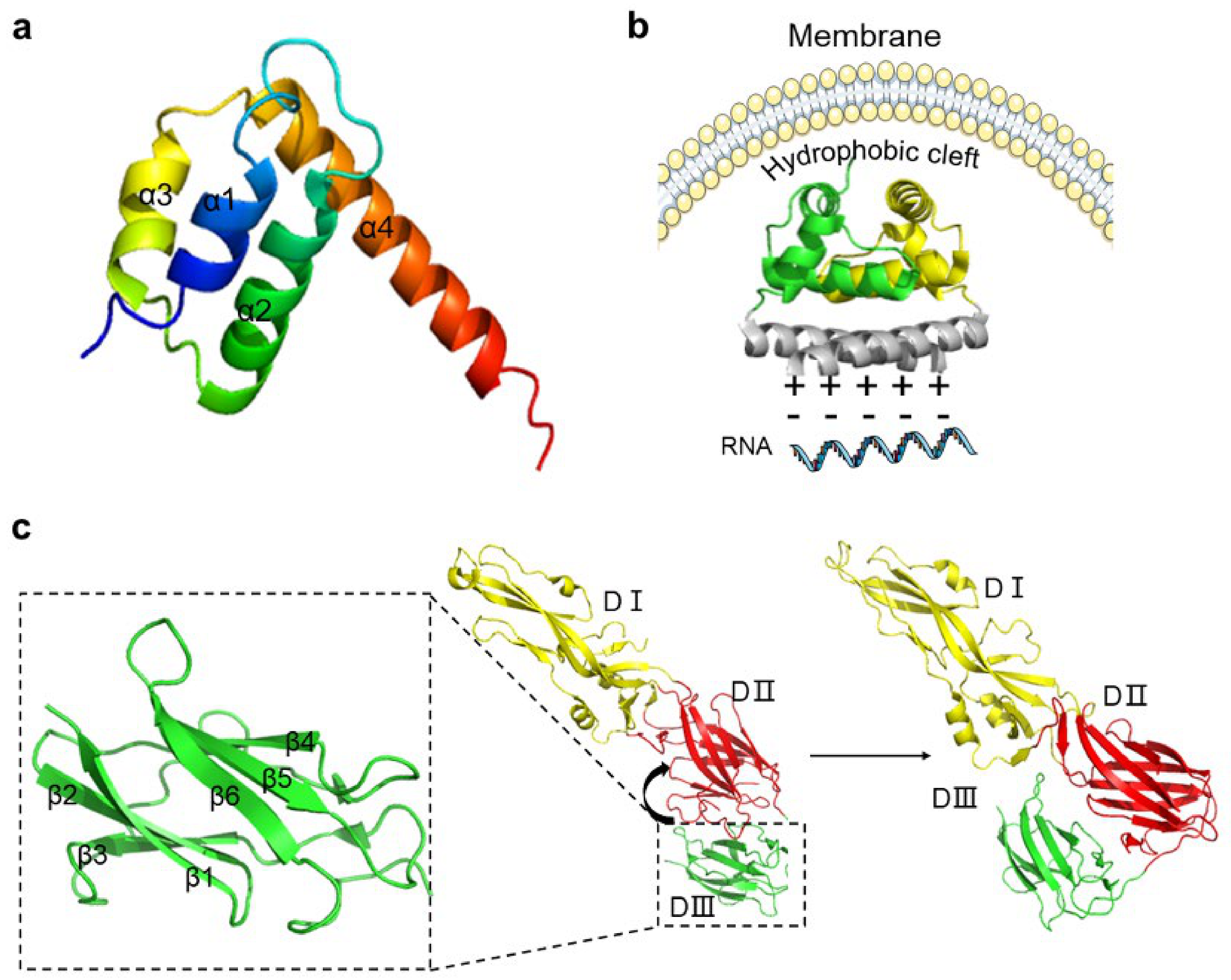
2.1. C Protein
2.2. PrM and E Proteins
2.3. NS1 Protein
2.4. NS3 Proteins
2.5. NS5 Protein
3. Vaccines of Flavivirus
3.1. Live Attenuated Vaccines
3.1.1. 17D Vaccine
3.1.2. SA14-14-2 Vaccine
3.2. Inactivated Vaccines
3.3. Molecularly Engineered Vaccines
3.3.1. Recombinant Vaccine
3.3.2. Molecularly Cloned Vaccine
3.3.3. DNA Vaccine
3.3.4. mRNA Vaccine
4. Structure-Based Anti-Flavivirus Drug Targets
4.1. E Protein as an Antiviral Drug Target
4.2. Non-Structural Proteins as Antiviral Drug Targets
4.2.1. NS1 Protein
4.2.2. NS3 Protein
4.2.3. NS5 Protein
4.3. Other Inhibitors of Non-Structural Proteins of Flaviviruses
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Dethoff, E.A.; Boerneke, M.A.; Gokhale, N.S.; Muhire, B.M.; Martin, D.P.; Sacco, M.T.; McFadden, M.J.; Weinstein, J.B.; Messer, W.B.; Horner, S.M.; et al. Pervasive tertiary structure in the dengue virus RNA genome. Proc. Natl. Acad. Sci. USA 2018, 115, 11513–11518. [Google Scholar] [CrossRef] [PubMed]
- Kummerer, B.M. Establishment and application of flavivirus replicons. Adv. Exp. Med. Biol. 2018, 1062, 165–173. [Google Scholar] [PubMed]
- Barnett, R. Dengue. Lancet 2017, 390, 1941. [Google Scholar] [CrossRef]
- Martin, E.; Borucki, M.K.; Thissen, J.; Garcia-Luna, S.; Hwang, M.; de Valdez, M.W.; Jaing, C.J.; Hamer, G.L.; Frank, M. Mosquito-Borne viruses and insect-specific viruses revealed in field-collected mosquitoes by a monitoring tool adapted from a microbial detection array. Appl. Environ. Microb. 2019, 85, e01202-19. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.S.; Amraoui, F.; Vega Rua, A.; Failloux, A.B. Aedes aegypti mosquitoes from Guadeloupe (French West Indies) are able to transmit yellow fever virus. PLoS ONE 2018, 13, e0204710. [Google Scholar] [CrossRef] [PubMed]
- Souza-Neto, J.A.; Powell, J.R.; Bonizzoni, M. Aedes aegypti vector competence studies: A review. Infect. Genet. Evol. 2019, 67, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.R. Historical perspectives on flavivirus research. Viruses 2017, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, J.J.; Rojas, A.; Pinsky, B.A. Yellow fever virus: Diagnostics for a persistent arboviral threat. J. Clin. Microbiol. 2018, 56, e00827-18. [Google Scholar] [CrossRef] [PubMed]
- Charlier, N.; Leyssen, P.; Pleij, C.W.A.; Lemey, P.; Billoir, F.; Van Laethem, K.; Vandamme, A.M.; De Clercq, E.; de Lamballerie, X.; Neyts, J. Complete genome sequence of Montana Myotis leukoencephalitis virus, phylogenetic analysis and comparative study of the 3′ untranslated region of flaviviruses with no known vector. J. Gen. Virol. 2002, 83, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Vasconcelos, P.F. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Ferreira-de-Lima, V.H.; Lima-Camara, T.N. Natural vertical transmission of dengue virus in Aedes aegypti and Aedes albopictus: A systematic review. Parasites Vectors 2018, 11, 77. [Google Scholar] [CrossRef]
- Danis-Lozano, R.; Diaz-Gonzalez, E.E.; Malo-Garcia, I.R.; Rodriguez, M.H.; Ramos-Castaneda, J.; Juarez-Palma, L.; Ramos, C.; Lopez-Ordonez, T.; Mosso-Gonzalez, C.; Fernandez-Salas, I. Vertical transmission of dengue virus in Aedes aegypti and its role in the epidemiological persistence of dengue in Central and Southern Mexico. Trop. Med. Int. Health 2019, 24, 1311–1319. [Google Scholar] [CrossRef]
- Holmes, E.C.; Twiddy, S.S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol 2003, 3, 19–28. [Google Scholar] [CrossRef]
- Carod-Artal, F.J.; Wichmann, O.; Farrar, J.; Gascon, J. Neurological complications of dengue virus infection. Lancet Neurol 2013, 12, 906–919. [Google Scholar] [CrossRef]
- Bodinayake, C.K.; Tillekeratne, L.G.; Nagahawatte, A.; Devasiri, V.; Kodikara Arachchi, W.; Strouse, J.J.; Sessions, O.M.; Kurukulasooriya, R.; Uehara, A.; Howe, S.; et al. Evaluation of the WHO 2009 classification for diagnosis of acute dengue in a large cohort of adults and children in Sri Lanka during a dengue-1 epidemic. PLoS Negl. Trop. Dis. 2018, 12, e0006258. [Google Scholar] [CrossRef]
- Morrone, S.R.; Lok, S.M. Structural perspectives of antibody-dependent enhancement of infection of dengue virus. Curr. Opin. Virol. 2019, 36, 1–8. [Google Scholar] [CrossRef]
- Narayan, R.; Tripathi, S. Intrinsic ADE: The dark side of antibody dependent enhancement during dengue infection. Front. Cell Infect. Microbiol. 2020, 10, 580096. [Google Scholar] [CrossRef]
- Bharaj, P.; Chahar, H.S.; Pandey, A.; Diddi, K.; Dar, L.; Guleria, R.; Kabra, S.K.; Broor, S. Concurrent infections by all four dengue virus serotypes during an outbreak of dengue in 2006 in Delhi, India. Virol. J. 2008, 5, 1. [Google Scholar] [CrossRef]
- Vinodkumar, C.S.; Kalapannavar, N.K.; Basavarajappa, K.G.; Sanjay, D.; Gowli, C.; Nadig, N.G.; Prasad, B.S. Episode of coexisting infections with multiple dengue virus serotypes in central Karnataka, India. J. Infect. Public Health 2013, 6, 302–306. [Google Scholar] [CrossRef]
- Vaddadi, K.; Gandikota, C.; Jain, P.K.; Prasad, V.S.V.; Venkataramana, M. Co-Circulation and co-infections of all dengue virus serotypes in Hyderabad, India 2014. Epidemiol. Infect. 2017, 145, 2563–2574. [Google Scholar] [CrossRef]
- Dhanoa, A.; Hassan, S.S.; Ngim, C.F.; Lau, C.F.; Chan, T.S.; Adnan, N.A.A.; Eng, W.W.H.; Gan, H.M.; Rajasekaram, G. Impact of dengue virus (DENV) co-infection on clinical manifestations, disease severity and laboratory parameters. BMC Infect. Dis. 2016, 16, 406. [Google Scholar] [CrossRef]
- Lardo, S.; Utami, Y.; Yohan, B.; Tarigan, S.M.M.U.; Santoso, W.D.; Nainggolan, L.; Sasmono, R.T. Concurrent infections of dengue viruses serotype 2 and 3 in patient with severe dengue from Jakarta, Indonesia. Asian Pac. J. Trop. Med. 2016, 9, 130–135. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengue antibody-dependent enhancement: Knowns and unknowns. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Rosen, L. The natural history of Janpanese encephalitis virus. Ann. Rev. Microbiol. 1986, 40, 395–414. [Google Scholar] [CrossRef]
- Chiu, H.P.; Chiu, H.; Yang, C.F.; Lee, Y.L.; Chiu, F.L.; Kuo, H.C.; Lin, R.J.; Lin, Y.L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef]
- Yun, S.I.; Lee, Y.M. Early events in Japanese encephalitis virus infection: Viral entry. Pathogens 2018, 7, 68. [Google Scholar] [CrossRef]
- Griffiths, M.J.; Turtle, L.; Solomon, T. Japanese encephalitis virus infection. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 123, pp. 561–576. [Google Scholar]
- Lindsey, N.P.; Lehman, J.A.; Staples, J.E.; Fischer, M. West Nile virus and other arboviral diseases—United States, 2013. MMWR Morb. Mortal. Wkly. Rep. 2014, 63, 521–526. [Google Scholar]
- Bolling, B.G.; Eisen, L.; Moore, C.G.; Blair, C.D. Insect-Specific flaviviruses from culex mosquitoes in Colorado, with evidence of vertical transmission. Am. J. Trop. Med. Hyg. 2011, 85, 169–177. [Google Scholar] [CrossRef]
- David, S.; Abraham, A.M. Epidemiological and clinical aspects on West Nile virus, a globally emerging pathogen. Infect. Dis. 2016, 48, 571–586. [Google Scholar] [CrossRef]
- Colmant, A.M.G.; Hobson-Peters, J.; Bielefeldt-Ohmann, H.; van den Hurk, A.F.; Hall-Mendelin, S.; Chow, W.K.; Johansen, C.A.; Fros, J.; Simmonds, P.; Watterson, D.; et al. A new clade of insect-specific flaviviruses from Australian anopheles mosquitoes displays species-specific host restriction. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile virus: Review of the literature. JAMA J. Am. Med. Assoc. 2013, 310, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Ross, T.M.; Evans, J.D. West Nile virus. Clin. Lab. Med. 2010, 30, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Dokland, T.; Walsh, M.; Mackenzie, J.M.; Khromykh, A.A.; Ee, K.H.; Wang, S. West Nile virus core protein; tetramer structure and ribbon formation. Structure 2004, 12, 1157–1163. [Google Scholar] [CrossRef]
- Gatherer, D.; Kohl, A. Zika virus: A previously slow pandemic spreads rapidly through the Americas. J. Gen. Virol. 2016, 97, 269–273. [Google Scholar] [CrossRef]
- MacNamara, F. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Reid, S.; Rimmer, K.; Thakur, K. Zika virus and neurologic disease. Neurol. Clin. 2018, 36, 767–787. [Google Scholar] [CrossRef]
- Main, B.J.; Nicholson, J.; Winokur, O.C.; Steiner, C.; Riemersma, K.K.; Stuart, J.; Takeshita, R.; Krasnec, M.; Barker, C.M.; Coffey, L.L. Vector competence of Aedes aegypti, Culex tarsalis, and Culex quinquefasciatus from California for Zika virus. PLoS Negl. Trop. Dis. 2018, 12, e0006524. [Google Scholar] [CrossRef]
- Kasprzykowski, J.I.; Fukutani, K.F.; Fabio, H.; Fukutani, E.R.; Costa, L.C.; Andrade, B.B.; Queiroz, A.T.L. A recursive sub-typing screening surveillance system detects the arising of the ZIKV African lineage in Brazil: Is there risk of a new epidemic? Int. J. Infect. Dis. 2020, 96, 579–581. [Google Scholar] [CrossRef]
- Plourde, A.R.; Bloch, E.M. A literature review of Zika virus. Emerg. Infect. Dis. 2016, 22, 1185–1192. [Google Scholar] [CrossRef]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Musso, D.; Gubler, D.J. Zika virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Liao, K.C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.C.; Chott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and molecular biology of flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef]
- Cruz-Oliveira, C.; Freire, J.M.; Conceicao, T.M.; Higa, L.M.; Castanho, M.A.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef]
- Choi, K.H.; Rossmann, M.G. RNA-Dependent RNA polymerases from flaviviridae. Curr. Opin. Struct. Biol. 2009, 19, 746–751. [Google Scholar] [CrossRef]
- Faustino, A.F.; Martins, A.S.; Karguth, N.; Artilheiro, V.; Enguita, F.J.; Ricardo, J.C.; Santos, N.C.; Martins, I.C. Structural and functional properties of the capsid protein of dengue and related flavivirus. Int. J. Mol. Sci. 2019, 20, 3870. [Google Scholar] [CrossRef]
- Lescar, J.; Soh, S.; Lee, L.T.; Vasudevan, S.G.; Kang, C.; Lim, S.P. The dengue virus replication complex: From RNA replication to protein-protein interactions to evasion of innate immunity. Adv. Exp. Med. Biol. 2018, 1062, 115–129. [Google Scholar]
- Li, L.; Lok, S.M.; Yu, I.M.; Zhang, Y.; Kuhn, R.J.; Chen, J.; Rossmann, M.G. The flavivirus precursor membrane-envelope protein complex: Structure and maturation. Science 2008, 319, 1830–1834. [Google Scholar] [CrossRef]
- Byk, L.A.; Gamarnik, A.V. Properties and functions of the dengue virus capsid protein. Annu. Rev. Virol. 2016, 3, 263–281. [Google Scholar] [CrossRef]
- Li, T.; Zhao, Q.; Yang, X.; Chen, C.; Yang, K.; Wu, C.; Zhang, T.; Duan, Y.; Xue, X.; Mi, K.; et al. Structural insight into the Zika virus capsid encapsulating the viral genome. Cell Res. 2018, 28, 497–499. [Google Scholar] [CrossRef]
- Tan, T.Y.; Fibriansah, G.; Lok, S.M. Capsid protein is central to the birth of flavivirus particles. PLoS Pathog. 2020, 16, e1008542. [Google Scholar] [CrossRef]
- Sirohi, D.; Kuhn, R.J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef]
- Ma, L.; Jones, C.T.; Groesch, T.D.; Richard, J.K.; Post, C.B. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. USA 2004, 101, 3414–3419. [Google Scholar] [CrossRef]
- Goertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef]
- Schrauf, S.; Mandl, C.W.; Bell-Sakyi, L.; Skern, T. Extension of flavivirus protein C differentially affects early RNA synthesis and growth in mammalian and arthropod host cells. J. Virol. 2009, 83, 11201–11210. [Google Scholar] [CrossRef]
- Oliveira, E.R.A.; Mohana-Borges, R.; de Alencastro, R.B.; Horta, B.A.C. The flavivirus capsid protein: Structure, function and perspectives towards drug design. Virus Res. 2017, 227, 115–123. [Google Scholar] [CrossRef]
- Zhang, X.; Jia, R.; Shen, H.; Wang, M.; Yin, Z.; Cheng, A. Structures and functions of the envelope glycoprotein in flavivirus infections. Viruses 2017, 9, 338. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Ogata, S.; Clements, D.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Conformational changes of the flavivirus E glycoprotein. Structure 2004, 12, 1607–1618. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Zhang, Q.; Tan, J.L.; Ng, T.S.; Lok, S.M. Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J. Virol. 2013, 87, 7700–7707. [Google Scholar] [CrossRef]
- Morrone, S.R.; Chew, V.S.Y.; Lim, X.N.; Ng, T.S.; Kostyuchenko, V.A.; Zhang, S.; Wirawan, M.; Chew, P.L.; Lee, J.; Tan, J.L.; et al. High flavivirus structural plasticity demonstrated by a non-spherical morphological variant. Nat. Commun. 2020, 11, 3112. [Google Scholar] [CrossRef] [PubMed]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef]
- Kang, C.; Keller, T.H.; Luo, D. Zika virus protease: An antiviral drug target. Trends Microbiol. 2017, 25, 797–808. [Google Scholar] [CrossRef]
- Lorenz, I.C.; Kartenbeck, J.; Mezzacasa, A.; Allison, S.L.; Heinz, F.X.; Helenius, A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J. Virol. 2003, 77, 4370–4382. [Google Scholar] [CrossRef]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Dutta, K.; White, M.A.; Cowburn, D.; Fox, R.O. NMR solution structure and backbone dynamics of domain III of the E protein of tick-borne Langat flavivirus suggests a potential site for molecular recognition. Protein Sci. 2006, 15, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Nie, K.; Du, S.; Qiu, J.; Pang, X.; Wang, P.; Cheng, G. Flavivirus NS1 protein in infected host sera enhances viral acquisition by mosquitoes. Nat. Microbiol. 2016, 1, 16087. [Google Scholar] [CrossRef]
- Kostyuchenko, V.A.; Chew, P.L.; Ng, T.S.; Lok, S.M. Near-Atomic resolution cryo-electron microscopic structure of dengue serotype 4 virus. J. Virol. 2014, 88, 477–482. [Google Scholar] [CrossRef]
- Edeling, M.A.; Diamond, M.S.; Fremont, D.H. Structural basis of Flavivirus NS1 assembly and antibody recognition. Proc. Natl. Acad. Sci. USA 2014, 111, 4285–4290. [Google Scholar] [CrossRef]
- Rastogi, M.; Sharma, N.; Singh, S.K. Flavivirus NS1: A multifaceted enigmatic viral protein. Virol. J. 2016, 13, 131. [Google Scholar] [CrossRef]
- Somnuke, P.; Hauhart, R.E.; Atkinson, J.P.; Diamond, M.S.; Avirutnan, P. N-Linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 2011, 413, 253–264. [Google Scholar] [CrossRef]
- Watterson, D.; Modhiran, N.; Young, P.R. The many faces of the flavivirus NS1 protein offer a multitude of options for inhibitor design. Antivir. Res. 2016, 130, 7–18. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Konwerski, J.R.; Ogata, C.M.; Smith, J.L. Use of massively multiple merged data for low-resolution S-SAD phasing and refinement of flavivirus NS1. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2719–2729. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; DelProposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, J.; Kim, M.; Do, J.; Lee, D.; Han, H. Low-Dielectric-Constant polyimide aerogel composite films with low water uptake. Polym. J. 2016, 48, 829–834. [Google Scholar] [CrossRef]
- Brand, C.; Bisaillon, M.; Geiss, B.J. Organization of the Flavivirus RNA replicase complex. Wiley Interdiscip. Rev. RNA 2017, 8, e1437. [Google Scholar] [CrossRef]
- Jayathilaka, D.; Gomes, L.; Jeewandara, C.; Jayarathna, G.S.B.; Herath, D.; Perera, P.A.; Fernando, S.; Wijewickrama, A.; Hardman, C.S.; Ogg, G.S.; et al. Role of NS1 antibodies in the pathogenesis of acute secondary dengue infection. Nat. Commun. 2018, 9, 5242. [Google Scholar] [CrossRef]
- Li, K.; Phoo, W.W.; Luo, D. Functional interplay among the flavivirus NS3 protease, helicase, and cofactors. Virol. Sin. 2014, 29, 74–85. [Google Scholar] [CrossRef][Green Version]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef]
- Nitsche, C. Proteases from dengue, West Nile and Zika viruses as drug targets. Biophys. Rev. 2019, 11, 157–165. [Google Scholar] [CrossRef]
- Noske, G.D.; Gawriljuk, V.O.; Fernandes, R.S.; Furtado, N.D.; Bonaldo, M.C.; Oliva, G.; Godoy, A.S. Structural characterization and polymorphism analysis of the NS2B-NS3 protease from the 2017 Brazilian circulating strain of Yellow Fever virus. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129521. [Google Scholar] [CrossRef]
- Wu, J.; Bera, A.K.; Kuhn, R.J.; Smith, J.L. Structure of the Flavivirus helicase: Implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 2005, 79, 10268–10277. [Google Scholar] [CrossRef]
- Kok, W.M. New developments in flavivirus drug discovery. Expert Opin. Drug Discov. 2016, 11, 433–445. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Milani, M.; Bollati, M.; Selisko, B.; Peyrane, F.; Pandini, V.; Sorrentino, G.; Canard, B.; Konarev, P.V.; Svergun, D.I.; et al. Crystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J. Mol. Biol. 2007, 372, 444–455. [Google Scholar] [CrossRef]
- Bollati, M.; Milani, M.; Mastrangelo, E.; de Lamballerie, X.; Canard, B.; Bolognesi, M. Crystal structure of a methyltransferase from a no-known-vector Flavivirus. Biochem. Biophys. Res. Commun. 2009, 382, 200–204. [Google Scholar] [CrossRef]
- Jansson, A.M.; Jakobsson, E.; Johansson, P.; Lantez, V.; Coutard, B.; de Lamballerie, X.; Unge, T.; Jones, T.A. Structure of the methyltransferase domain from the Modoc virus, a flavivirus with no known vector. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Chatrin, C.; Talapatra, S.K.; Canard, B.; Kozielski, F. The structure of the binary methyltransferase-SAH complex from Zika virus reveals a novel conformation for the mechanism of mRNA capping. Oncotarget 2018, 9, 3160–3171. [Google Scholar] [CrossRef] [PubMed]
- Hercik, K.; Brynda, J.; Nencka, R.; Boura, E. Structural basis of Zika virus methyltransferase inhibition by sinefungin. Arch. Virol. 2017, 162, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Geiss, B.J.; Thompson, A.A.; Andrews, A.J.; Sons, R.L.; Gari, H.H.; Keenan, S.M.; Peersen, O.B. Analysis of flavivirus NS5 methyltransferase cap binding. J. Mol. Biol. 2009, 385, 1643–1654. [Google Scholar] [CrossRef]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef]
- Assenberg, R.; Ren, J.; Verma, A.; Walter, T.S.; Alderton, D.; Hurrelbrink, R.J.; Fuller, S.D.; Bressanelli, S.; Owens, R.J.; Stuart, D.I.; et al. Crystal structure of the Murray Valley encephalitis virus NS5 methyltransferase domain in complex with cap analogues. J. Gen. Virol. 2007, 88, 2228–2236. [Google Scholar] [CrossRef]
- Muruato, A.E.; Shan, C.; Fontes-Garfias, C.R.; Liu, Y.; Cao, Z.G.; Gao, Q.; Weaver, S.C.; Shi, P.Y. Genetic stability of live-attenuated Zika vaccine candidates. Antivir. Res. 2019, 171, 104596. [Google Scholar] [CrossRef]
- Halstead, S.B. Safety issues from a phase 3 clinical trial of a live-attenuated chimeric yellow fever tetravalent dengue vaccine. Hum. Vaccines Immunother. 2018, 14, 2158–2162. [Google Scholar] [CrossRef]
- Tu, H.A.; Nivarthi, U.K.; Graham, N.R.; Eisenhauer, P.; Delacruz, M.J.; Pierce, K.K.; Whitehead, S.S.; Boyson, J.E.; Botten, J.W.; Kirkpatrick, B.D.; et al. Stimulation of B cell immunity in Flavivirus-Naive individuals by the tetravalent live attenuated dengue vaccine TV003. Cell Rep. Med. 2020, 1, 100155. [Google Scholar] [CrossRef]
- Whitehead, S.S. Development of TV003/TV005, a single dose, highly immunogenic live attenuated dengue vaccine; what makes this vaccine different from the Sanofi-Pasteur CYD (TM) vaccine? Expert Rev. Vaccines 2016, 15, 509–517. [Google Scholar] [CrossRef]
- Osorio, J.E.; Velez, I.D.; Thomson, C.; Lopez, L.; Jimenez, A.; Haller, A.A.; Silengo, S.; Scott, J.; Boroughs, K.L.; Stovall, J.L.; et al. Safety and immunogenicity of a recombinant live attenuated tetravalent dengue vaccine (DENVax) in flavivirus-naive healthy adults in Colombia: A randomised, placebo-controlled, phase 1 study. Lancet Infect. Dis. 2014, 14, 830–838. [Google Scholar] [CrossRef]
- Aguiar, M.; Stollenwerk, N. The impact of serotype cross-protection on vaccine trials: DENVax as a case study. Vaccines 2020, 8, 674. [Google Scholar] [CrossRef]
- Scherwitzl, I.; Mongkolsapaja, J.; Screaton, G. Recent advances in human flavivirus vaccines. Curr. Opin. Virol. 2017, 23, 95–101. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17D virus as a platform for new live attenuated vaccines. Hum. Vaccines Immunother. 2014, 10, 1256–1265. [Google Scholar] [CrossRef]
- Davis, E.H.; Beck, A.S.; Strother, A.E.; Thompson, J.K.; Widen, S.G.; Higgs, S.; Wood, T.G.; Barrett, A.D.T. Attenuation of live-attenuated yellow fever 17D vaccine virus is localized to a high-fidelity replication complex. mBio 2019, 10, e02294-19. [Google Scholar] [CrossRef]
- Barrett, A.D.T. Yellow fever live attenuated vaccine: A very successful live attenuated vaccine but still we have problems controlling the disease. Vaccine 2017, 35, 5951–5955. [Google Scholar] [CrossRef]
- Domingo, C.; Niedrig, M. Safety of 17D derived yellow fever vaccines. Expert Opin. Drug Saf. 2009, 8, 211–221. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- Yang, D.; Li, X.F.; Ye, Q.; Wang, H.J.; Deng, Y.Q.; Zhu, S.Y.; Zhang, Y.; Li, S.H.; Qin, C.F. Characterization of live-attenuated Japanese encephalitis vaccine virus SA14-14-2. Vaccine 2014, 32, 2675–2681. [Google Scholar] [CrossRef]
- Khou, C.; Pardigon, N. Identifying attenuating mutations: Tools for a new vaccine design against flaviviruses. Intervirology 2017, 60, 8–18. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, D.; Jia, L.; Xu, H.; Na, R.; Ge, Y.; Liu, S.; Yu, Y.; Li, Y. Genetic and neuroattenuation phenotypic characteristics and their stabilities of SA14-14-2 vaccine seed virus. Vaccine 2018, 36, 4650–4656. [Google Scholar] [CrossRef]
- Turtle, L.; Tatullo, F.; Bali, T.; Ravi, V.; Soni, M.; Chan, S.; Chib, S.; Venkataswamy, M.M.; Fadnis, P.; Yaich, M.; et al. Cellular immune responses to live attenuated Japanese encephalitis (JE) vaccine SA14-14-2 in adults in a JE/Dengue co-endemic area. PLoS Negl. Trop. Dis. 2017, 11, e0005263. [Google Scholar] [CrossRef] [PubMed]
- Beck, Y.; Fritz, R.; Orlinger, K.; Kiermayr, S.; Ilk, R.; Portsmouth, D.; Pollabauer, E.M.; Low-Baselli, A.; Hessel, A.; Kolch, D.; et al. Molecular basis of the divergent immunogenicity of two pediatric tick-borne Encephalitis virus vaccines. J. Virol. 2016, 90, 1964–1972. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Salat, J.; Mikulasek, K.; Larralde, O.; Pokorna Formanova, P.; Chrdle, A.; Haviernik, J.; Elsterova, J.; Teislerova, D.; Palus, M.; Eyer, L.; et al. Tick-Borne Encephalitis virus vaccines contain non-structural protein 1 antigen and may elicit NS1-specific antibody responses in vaccinated individuals. Vaccines 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Vorovitch, M.F.; Grishina, K.G.; Volok, V.P.; Chernokhaeva, L.L.; Grishin, K.V.; Karganova, G.G.; Ishmukhametov, A.A. Evervac: Phase I/II study of immunogenicity and safety of a new adjuvant-free TBE vaccine cultivated in Vero cell culture. Hum. Vaccines Immunother. 2020, 16, 2123–2130. [Google Scholar] [CrossRef]
- Guy, B.; Noriega, F.; Ochiai, R.L.; L’Azou, M.; Delore, V.; Skipetrova, A.; Verdier, F.; Coudeville, L.; Savarino, S.; Jackson, N. A recombinant live attenuated tetravalent vaccine for the prevention of dengue. Expert Rev. Vaccines 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Lin, H.H.; Yip, B.S.; Huang, L.M.; Wu, S.C. Zika virus structural biology and progress in vaccine development. Biotechnol. Adv. 2018, 36, 47–53. [Google Scholar] [CrossRef]
- Fibriansah, G.; Tan, J.L.; Smith, S.A.; de Alwis, R.; Ng, T.S.; Kostyuchenko, V.A.; Jadi, R.S.; Kukkaro, P.; de Silva, A.M.; Crowe, J.E.; et al. A highly potent human antibody neutralizes dengue virus serotype 3 by binding across three surface proteins. Nat. Commun. 2015, 6, 6341. [Google Scholar] [CrossRef]
- Whiteman, M.C.; Li, L.; Wicker, J.A.; Kinney, R.M.; Huang, C.; Beasley, D.W.; Chung, K.M.; Diamond, M.S.; Solomon, T.; Barrett, A.D. Development and characterization of non-glycosylated E and NS1 mutant viruses as a potential candidate vaccine for West Nile virus. Vaccine 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Larocca, R.A.; Abbink, P.; Peron, J.P.; Zanotto, P.M.; Iampietro, M.J.; Badamchi-Zadeh, A.; Boyd, M.; Ng’ang’a, D.; Kirilova, M.; Nityanandam, R.; et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536, 474–478. [Google Scholar] [CrossRef]
- Jimenez de Oya, N.; Escribano-Romero, E.; Blazquez, A.B.; Martin-Acebes, M.A.; Saiz, J.C. Current progress of avian vaccines against West Nile virus. Vaccines 2019, 7, 126. [Google Scholar] [CrossRef]
- Collins, M.H.; Metz, S.W. Progress and works in progress: Update on flavivirus vaccine development. Clin. Ther. 2017, 39, 1519–1536. [Google Scholar] [CrossRef]
- Urakami, A.; Ngwe Tun, M.M.; Moi, M.L.; Sakurai, A.; Ishikawa, M.; Kuno, S.; Ueno, R.; Morita, K.; Akahata, W. An envelope-modified tetravalent dengue virus-like-particle vaccine has implications for flavivirus vaccine design. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Boigard, H.; Alimova, A.; Martin, G.R.; Katz, A.; Gottlieb, P.; Galarza, J.M. Zika virus-like particle (VLP) based vaccine. PLoS Negl. Trop. Dis. 2017, 11, e0005608. [Google Scholar] [CrossRef]
- Khetarpal, N.; Khanna, I. Dengue fever: Causes, complications, and vaccine strategies. J. Immunol. Res. 2016, 2016, 6803098. [Google Scholar] [CrossRef]
- Reyes-Sandoval, A.; Ludert, J.E. The dual role of the antibody response against the Flavivirus non-structural protein 1 (NS1) in protection and immuno-pathogenesis. Front. Immunol. 2019, 10, 1651. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef]
- Ray, D.; Shi, P.Y. Recent advances in Flavivirus antiviral drug discovery and vaccine development. Anti Infect. Drug Discov. 2006, 1, 45–55. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Wong, G.; Gao, G.F. An mRNA-based vaccine strategy against Zika. Cell Res. 2017, 27, 1077–1078. [Google Scholar] [CrossRef][Green Version]
- Perera, R.; Khaliq, M.; Kuhn, R.J. Closing the door on flaviviruses: Entry as a target for antiviral drug design. Antivir. Res. 2008, 80, 11–22. [Google Scholar] [CrossRef]
- Li, P.C.; Jang, J.; Hsia, C.Y.; Groomes, P.V.; Lian, W.; de Wispelaere, M.; Pitts, J.D.; Wang, J.; Kwiatkowski, N.; Gray, N.S.; et al. Small molecules targeting the Flavivirus E protein with broad-spectrum activity and antiviral efficacy in vivo. ACS Infect. Dis. 2019, 5, 460–472. [Google Scholar] [CrossRef]
- Ivanova, T.; Hardes, K.; Kallis, S.; Dahms, S.O.; Than, M.E.; Kunzel, S.; Bottcher-Friebertshauser, E.; Lindberg, I.; Jiao, G.S.; Bartenschlager, R.; et al. Optimization of substrate-analogue furin inhibitors. ChemMedChem 2017, 12, 1953–1968. [Google Scholar] [CrossRef]
- Skrzypek, R.; Callaghan, R. The “pushmi-pullyu” of resistance to chloroquine in malaria. Essays Biochem. 2017, 61, 167–175. [Google Scholar]
- Dighe, S.N.; Ekwudu, O.; Dua, K.; Chellappan, D.K.; Katavic, P.L.; Collet, T.A. Recent update on anti-dengue drug discovery. Eur. J. Med. Chem. 2019, 176, 431–455. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Pezzullo, M.; De Burghgraeve, T.; Kaptein, S.; Pastorino, B.; Dallmeier, K.; de Lamballerie, X.; Neyts, J.; Hanson, A.M.; Frick, D.N.; et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: New prospects for an old drug. J. Antimicrob. Chemother. 2012, 67, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antivir. Res. 2010, 87, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.Y.; Li, H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef]
- Afaq, S.; Atiya, A.; Malik, A.; Alwabli, A.S.; Alzahrani, D.A.; Al-Solami, H.M.; Alzahrani, O.; Alam, Q.; Kamal, M.A.; Abulfaraj, A.A.; et al. Analysis of methyltransferase (MTase) domain from Zika virus (ZIKV). Bioinformation 2020, 16, 229–235. [Google Scholar] [CrossRef]
- Brecher, M.; Chen, H.; Li, Z.; Banavali, N.K.; Jones, S.A.; Zhang, J.; Kramer, L.D.; Li, H.M. Identification and characterization of novel broad-spectrum inhibitors of the flavivirus methyltransferase. ACS Infect. Dis. 2015, 1, 340–349. [Google Scholar] [CrossRef]
- Yadav, M.K.; Park, S.W.; Chae, S.W.; Song, J.J. Sinefungin, a natural nucleoside analogue of S-adenosylmethionine, inhibits Streptococcus pneumoniae biofilm growth. Biomed. Res. Int. 2014, 2014, 156987. [Google Scholar] [CrossRef]
- Noble, C.G.; Li, S.H.; Dong, H.P.; Chew, S.H.; Shi, P.Y. Crystal structure of dengue virus methyltransferase without S-adenosyl-L-methionine. Antivir. Res. 2014, 111, 78–81. [Google Scholar] [CrossRef]
- Wang, B.; Thurmond, S.; Hai, R.; Song, J. Structure and function of Zika virus NS5 protein: Perspectives for drug design. Cell. Mol. Life Sci. 2018, 75, 1723–1736. [Google Scholar] [CrossRef]
- Jain, R.; Butler, K.V.; Coloma, J.; Jin, J.; Aggarwal, A.K. Development of a S-adenosylmethionine analog that intrudes the RNA-cap binding site of Zika methyltransferase. Sci. Rep. 2017, 7, 1632. [Google Scholar] [CrossRef]
- Lim, S.V.; Rahman, M.B.; Tejo, B.A. Structure-based and ligand-based virtual screening of novel methyltransferase inhibitors of the dengue virus. BMC Bioinformatics 2011, 12 (Suppl. 13), S24. [Google Scholar] [CrossRef]
- Brecher, M.; Chen, H.; Liu, B.B.; Banavali, N.K.; Jones, S.A.; Zhang, J.; Li, Z.; Kramer, L.D.; Li, H.M. Novel broad spectrum inhibitors targeting the flavivirus methyltransferase. PLoS ONE 2015, 10, e0130062. [Google Scholar] [CrossRef]
- Geiss, B.J.; Stahla-Beek, H.J.; Hannah, A.M.; Gari, H.H.; Henderson, B.R.; Saeedi, B.J.; Keenan, S.M. A high-throughput screening assay for the identification of flavivirus NS5 capping enzyme GTP-binding inhibitors: Implications for antiviral drug development. J. Biomol. Screen. 2011, 16, 852–861. [Google Scholar] [CrossRef]
- Noreen; Ali, R.; Badshah, S.L.; Faheem, M.; Abbasi, S.W.; Ullah, R.; Bari, A.; Jamal, S.B.; Mahmood, H.M.; Haider, A.; et al. Identification of potential inhibitors of Zika virus NS5 RNA-dependent RNA polymerase through virtual screening and molecular dynamic simulations. Saudi Pharm. J. 2020, 28, 1580–1591. [Google Scholar]
- Lim, S.P.; Noble, C.G.; Seh, C.C.; Soh, T.S.; El Sahili, A.; Chan, G.K.; Lescar, J.; Arora, R.; Benson, T.; Nilar, S.; et al. Potent allosteric Dengue virus NS5 polymerase inhibitors: Mechanism of action and resistance profiling. PLoS Pathog. 2016, 12, e1005737. [Google Scholar] [CrossRef]
- Siqueira-Batista, R.; Bayao, T.D.; Cupertino, M.D.; Mayers, N.A.J.; Gomes, A.P. Sofosbuvir use for yellow fever: A new perspective treatment. Pathog. Glob. Health 2019, 113, 207–208. [Google Scholar] [CrossRef]
- Bullard-Feibelman, K.M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M.S.; Geiss, B.J. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antivir. Res. 2017, 137, 134–140. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Zaverucha-do-Valle, C.; Reis, P.A.; Barbosa-Lima, G.; Vieira, Y.R.; Mattos, M.; Silva, P.P.; Sacramento, C.; de Castro Faria Neto, H.C.; Campanati, L.; et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci. Rep. 2017, 7, 9409. [Google Scholar] [CrossRef]
- Jacobs, S.; Delang, L.; Verbeken, E.; Neyts, J.; Kaptein, S.J.F. A Viral polymerase inhibitor reduces Zika virus replication in the reproductive organs of male mice. Int. J. Mol. Sci. 2019, 20, 2122. [Google Scholar] [CrossRef]
- Sampath, A.; Padmanabhan, R. Molecular targets for flavivirus drug discovery. Antivir. Res. 2009, 81, 6–15. [Google Scholar] [CrossRef]
- Zakaria, M.K.; Carletti, T.; Marcello, A. Cellular targets for the treatment of flavivirus infections. Front. Cell Infect. Microbiol. 2018, 8, 398. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Song, H.; Liu, L.; Bletchly, C.; Brillault, L.; Amarilla, A.A.; Xu, X.; Qi, J.; Chai, Y.; Cheung, S.T.M.; et al. A broadly protective antibody that targets the flavivirus NS1 protein. Science 2021, 371, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Akey, D.L.; Wong, M.P.; Brown, W.C.; Lo, N.T.N.; Puerta-Guardo, H.; Tramontini Gomes de Sousa, F.; Wang, C.; Konwerski, J.R.; Espinosa, D.A.; et al. Structural basis for antibody inhibition of flavivirus NS1-triggered endothelial dysfunction. Science 2021, 371, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Houghton, M. Discovery of the hepatitis C virus. Liver Int. 2009, 29 (Suppl. 1), 82–88. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. Curr. Top. Microbiol. Immunol. 2013, 369, 113–142. [Google Scholar] [PubMed]
- Das, D.; Pandya, M. Recent advancement of Direct-acting Antiviral Agents (DAAs) in hepatitis C therapy. Mini Rev. Med. Chem. 2018, 18, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.; Abbasi, S.W.; Ahmad, S.; Zia, M.; Raza, A. Phylogenetic analysis, structure modeling and docking study of HCV NS3 protease for the identification of potent inhibitors. Infect. Genet. Evol. 2018, 59, 51–62. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine | Vaccine Type | Antigen | An-Virus | Stage |
|---|---|---|---|---|
| 17D | Live attenuated vaccine | E protein | YFV | licensed |
| SA14-14-2 | Live attenuated vaccine | E protein | JEV | licensed |
| TV003 | Live attenuated vaccine | prM-E | DENV1–4 | Phase III |
| DENVax | Live attenuated vaccine | prM-E | DENV1–4 | Phase III |
| FSME-IMMUN | Inactivated vaccine | E protein | TBEV | licensed |
| Encepur | Inactivated vaccine | E protein | TBEV | licensed |
| Evervac | Inactivated vaccine | E protein | TBEV | Phase II |
| TDENV-PIV | Inactivated vaccine | C-prM-E-NS1/3/5 | DENV1–4 | Phase I |
| ZPIV | Inactivated vaccine | E protein | ZIKV | Phase I |
| CYD-TDV(Dengvaxia) | Recombinant vaccine | prM-E | DENV1–4 | licensed |
| ChimeriVax-WN02 | Recombinant vaccine e | prM-E | WNV | Phase II |
| V180 | Recombinant vaccine | E protein | DENV1–4 | Phase I |
| ZIKV-VLP | VLPs(Virus-like particles) | C-prM-E-NS2B/NS3 | ZIKV | Animal |
| DENV-VLP | VLPs(Virus-like particles) | prM-E | DENV1–4 | preclinical |
| GLS-5700 | DNA vaccine | prM-E/NS1 | ZIKV | Phase I |
| IgEsig-prM-E-LNP | mRNA vaccine | prM-E | ZIKV | Animal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Wang, M.; Cao, J.; Shen, J.; Zhou, X.; Wang, D.; Cao, J. Flavivirus: From Structure to Therapeutics Development. Life 2021, 11, 615. https://doi.org/10.3390/life11070615
Zhao R, Wang M, Cao J, Shen J, Zhou X, Wang D, Cao J. Flavivirus: From Structure to Therapeutics Development. Life. 2021; 11(7):615. https://doi.org/10.3390/life11070615
Chicago/Turabian StyleZhao, Rong, Meiyue Wang, Jing Cao, Jing Shen, Xin Zhou, Deping Wang, and Jimin Cao. 2021. "Flavivirus: From Structure to Therapeutics Development" Life 11, no. 7: 615. https://doi.org/10.3390/life11070615
APA StyleZhao, R., Wang, M., Cao, J., Shen, J., Zhou, X., Wang, D., & Cao, J. (2021). Flavivirus: From Structure to Therapeutics Development. Life, 11(7), 615. https://doi.org/10.3390/life11070615