
Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Studied Cohort
2.2. Molecular Genetic Analysis of GAA Gene
2.3. In Silico Analysis
2.4. GAA Enzyme Activity Measurement
2.5. Determination of CRIM Status
2.6. Statistical Analysis
3. Results
3.1. Clinical Assessment
3.2. Genetic Testing and Distribution of GAA Genotype
3.3. Correlation between α-Glucosidase Enzyme Activity and GAA Genotypes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018, 15, 928–942. [Google Scholar] [CrossRef]
- Dasouki, M.; Jawdat, O.; Almadhoun, O.; Pasnoor, M.; McVey, A.L.; Abuzinadah, A.; Herbelin, L.; Barohn, R.J.; Dimachkie, M.M. Pompe disease: Literature review and case series. Neurol. Clin. 2014, 32, 751–776. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet Med. 2006, 8, 267–288. [Google Scholar] [CrossRef]
- Bay, L.B.; Denzler, I.; Durand, C.; Eiroa, H.; Frabasil, J.; Fainboim, A.; Maxit, C.; Schenone, A.; Spécola, N. Infantile-onset Pompe disease: Diagnosis and management. Arch. Argent Pediatr. 2019, 117, 271–278. [Google Scholar]
- Toscano, A.; Rodolico, C.; Musumeci, O. Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann. Transl. Med. 2019, 7, 284. [Google Scholar] [CrossRef]
- Musumeci, O.; Toscano, A. Diagnostic tools in late onset Pompe disease (LOPD). Ann. Transl. Med. 2019, 7, 286. [Google Scholar] [CrossRef]
- Van der Ploeg, A.T.; Reuser, A.J. Pompe’s disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Labrousse, P.; Chien, Y.H.; Pomponio, R.J.; Keutzer, J.; Lee, N.C.; Akmaev, V.R.; Scholl, T.; Hwu, W.L. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol. Genet Metab. 2010, 99, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, C.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J. Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2015, 38, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Nicolino, M.; Voit, T.; Rogers, R.C.; Tsai, A.C.; Waterson, J.; Herman, G.E.; Amalfitano, A.; Thurberg, B.L.; Richards, S.; et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J. Pediatr. 2006, 149, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Molnár, M.J.; Borsos, B.; Várdi, K.V.; Grosz, Z.; Sebők, Á.; Dézsi, L.; Almássy, Z.; Kerényi, L.; Jobbágy, Z.; Jávor, L.; et al. The long-term follow-up of enzyme replacement treatment in late onset Pompe disease. Ideggyogy Sz. 2020, 73, 151–159. [Google Scholar] [CrossRef]
- Colella, P.; Mingozzi, F. Gene Therapy for Pompe Disease: The Time is now. Hum. Gene Ther. 2019, 30, 1245–1262. [Google Scholar] [CrossRef] [PubMed]
- Van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual Pascual, S.I.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur. J. Neurol. 2017, 24, 768-e31. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Maloney, C.L.; Vaccaro, C.; Afonso, K.; Tsai, A.C.; Bossen, E.; Kishnani, P.S.; O’Callaghan, M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab. Investig. 2006, 86, 1208–1220. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Kobayashi, H.; Kawagoe, S.; Aoki, K.; Kaneshiro, E.; Shimizu, H.; Eto, Y.; Ida, H.; Ohashi, T. Endoplasmic reticulum stress induces autophagy through activation of p38 MAPK in fibroblasts from Pompe disease patients carrying c.546G>T mutation. Mol. Genet. Metab. 2011, 104, 566–573. [Google Scholar] [CrossRef]
- Meikle, P.J.; Yan, M.; Ravenscroft, E.M.; Isaac, E.L.; Hopwood, J.J.; Brooks, D.A. Altered trafficking and turnover of LAMP-1 in Pompe disease-affected cells. Mol. Genet. Metab. 1999, 66, 179–188. [Google Scholar] [CrossRef]
- Fukuda, T.; Roberts, A.; Ahearn, M.; Zaal, K.; Ralston, E.; Plotz, P.H.; Raben, N. Autophagy and lysosomes in Pompe disease. Autophagy 2006, 2, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Roberts, A.; Plotz, P.H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 2007, 26, 45–48. [Google Scholar]
- Feeney, E.J.; Austin, S.; Chien, Y.H.; Mandel, H.; Schoser, B.; Prater, S.; Hwu, W.L.; Ralston, E.; Kishnani, P.S.; Raben, N. The value of muscle biopsies in Pompe disease: Identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol. Commun. 2014, 2, 2. [Google Scholar] [CrossRef]
- Gray, D.A.; Woulfe, J. Lipofuscin and aging: A matter of toxic waste. Sci. Aging Knowl. Environ. 2005, 2005, re1. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Niño, M.Y.; Int Groen, S.L.M.; Bergsma, A.J.; van der Beek, N.A.M.E.; Kroos, M.; Hoogeveen-Westerveld, M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.K.; Elbin, C.S.; Chuang, W.L.; Cooper, S.K.; Marashio, C.A.; Beauregard, C.; Keutzer, J.M. Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clin. Chem. 2008, 54, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, V.R.; Zhang, X.K.; Keutzer, J.; Bodamer, O.A.; Mühl, A.; Orsini, J.J.; Caggana, M.; Vogt, R.F.; Hannon, W.H. Development and evaluation of quality control dried blood spot materials in newborn screening for lysosomal storage disorders. Clin. Chem. 2009, 55, 158–164. [Google Scholar] [CrossRef]
- Elbin, C.S.; Olivova, P.; Marashio, C.A.; Cooper, S.K.; Cullen, E.; Keutzer, J.M.; Zhang, X.K. The effect of preparation, storage and shipping of dried blood spots on the activity of five lysosomal enzymes. Clin. Chim. Acta 2011, 11, 1207–1212. [Google Scholar] [CrossRef]
- Schedule of Accreditation. Available online: https://www.ukas.com/wp-content/uploads/schedule_uploads/00007/8661-Medical-Single.pdf (accessed on 25 May 2021).
- Ficicioglu, C.; Ahrens-Nicklas, R.C.; Barch, J.; Cuddapah, S.R.; DiBoscio, B.S.; DiPerna, J.C.; Gordon, P.L.; Henderson, N.; Menello, C.; Luongo, N.; et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int. J. Neonatal. Screen 2020, 6, 89. [Google Scholar] [CrossRef]
- Herzog, A.; Hartung, R.; Reuser, A.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: Molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Pittis, M.G.; Donnarumma, M.; Montalvo, A.L.; Dominissini, S.; Kroos, M.; Rosano, C.; Stroppiano, M.; Bianco, M.G.; Donati, M.A.; Parenti, G.; et al. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum. Mutat. 2008, 29, E27–E36. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.M.; van Leenen, D.; Kroos, M.A.; Beesley, C.E.; Van Der Ploeg, A.T.; Sakuraba, H.; Wevers, R.; Kleijer, W.; Michelakakis, H.; Kirk, E.P.; et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum. Mutat. 2004, 23, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.A.; van Leenen, D.; Verbiest, J.; Reuser, A.J.; Hermans, M.M. Glycogen storage disease type II: Identification of a dinucleotide deletion and a common missense mutation in the lysosomal alpha-glucosidase gene. Clin. Genet. 1998, 53, 379–382. [Google Scholar] [CrossRef]
- Gort, L.; Coll, M.J.; Chabás, A. Glycogen storage disease type II in Spanish patients: High frequency of c.1076-1G>C mutation. Mol. Genet. Metab. 2007, 92, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.; Manta, P.; Mavridou, I.; Muntoni, F.; Halley, D.; Van der Helm, R.; Zaifeiriou, D.; Van der Ploeg, A.; Reuser, A.; Michelakakis, H. Seven cases of Pompe disease from Greece. J. Inherit. Metab. Dis. 2006, 29, 556–563. [Google Scholar] [CrossRef]
- Kroos, M.A.; Van der Kraan, M.; Van Diggelen, O.P.; Kleijer, W.J.; Reuser, A.J.; Van den Boogaard, M.J.; Ausems, M.G.; Ploos van Amstel, H.K.; Poenaru, L.; Nicolino, M.; et al. Glycogen storage disease type II: Frequency of three common mutant alleles and their associated clinical phenotypes studied in 121 patients. J. Med. Genet. 1995, 32, 836–837. [Google Scholar] [CrossRef]
- Parini, R.; De Lorenzo, P.; Dardis, A.; Burlina, A.; Cassio, A.; Cavarzere, P.; Concolino, D.; Della Casa, R.; Deodato, F.; Donati, M.A.; et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J. Rare Dis. 2018, 13, 32. [Google Scholar] [CrossRef]
- Hermans, M.M.; de Graaff, E.; Kroos, M.A.; Wisselaar, H.A.; Oostra, B.A.; Reuser, A.J. Identification of a point mutation in the human lysosomal alpha-glucosidase gene causing infantile glycogenosis type II. Biochem. Biophys. Res. Commun. 1991, 179, 919–926. [Google Scholar] [CrossRef]
- Hermans, M.M.; Kroos, M.A.; de Graaff, E.; Oostra, B.A.; Reuser, A.J. Two mutations affecting the transport and maturation of lysosomal alpha-glucosidase in an adult case of glycogen storage disease type II. Hum. Mutat. 1993, 2, 268–273. [Google Scholar] [CrossRef]
- Müller-Felber, W.; Horvath, R.; Gempel, K.; Podskarbi, T.; Shin, Y.; Pongratz, D.; Walter, M.C.; Baethmann, M.; Schlotter-Weigel, B.; Lochmüller, H.; et al. Late onset Pompe disease: Clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul. Disord. 2007, 17, 698–706. [Google Scholar] [CrossRef]
- Markic, J.; Polic, B.; Stricevic, L.; Metlicic, V.; Kuzmanic-Samija, R.; Kovacevic, T.; Ivkosic, I.E.; Mestrovic, J. Effects of immune modulation therapy in the first Croatian infant diagnosed with Pompe disease: A 3-year follow-up study. Wien. Klin Wochenschr. 2014, 126, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, Y.; Fuji, N.; Yamazaki, N.; Hirakiyama, A.; Kamioka, T.; Seo, J.H.; Mashima, R.; Kosuga, M.; Okuyama, T. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol. Genet. Metab. Rep. 2017, 14, 3–9. [Google Scholar] [CrossRef]
- Reuser, A.J.; Kroos, M.A.; Hermans, M.M.; Bijvoet, A.G.; Verbeet, M.P.; Van Diggelen, O.P.; Kleijer, W.J.; Van der Ploeg, A.T. Glycogenosis type II (acid maltase deficiency). Muscle Nerve Suppl. 1995, 3, S61–S69. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular genetics of Pompe disease: A comprehensive overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, O.; Thieme, A.; Claeys, K.G.; Wenninger, S.; Kley, R.A.; Kuhn, M.; Lukacs, Z.; Deschauer, M.; Gaeta, M.; Toscano, A.; et al. Homozygosity for the common GAA gene splice site mutation c.-32-13T > G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul. Disord. 2015, 25, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Huie, M.L.; Chen, A.S.; Tsujino, S.; Shanske, S.; DiMauro, S.; Engel, A.G.; Hirschhorn, R. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): Molecular identification of an IVS1 (-13T-->G) mutation in a majority of patients and a novel IVS10 (+1GT-->CT) mutation. Hum. Mol. Genet. 1994, 3, 2231–2236. [Google Scholar] [PubMed]
- Boerkoel, C.F.; Exelbert, R.; Nicastri, C.; Nichols, R.C.; Miller, F.W.; Plotz, P.H.; Raben, N. Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am. J. Hum. Genet. 1995, 56, 887–897. [Google Scholar]
- Dardis, A.; Zanin, I.; Zampieri, S.; Stuani, C.; Pianta, A.; Romanello, M.; Baralle, F.E.; Bembi, B.; Buratti, E. Functional characterization of the common c.-32-13T > G mutation of GAA gene: Identification of potential therapeutic agents. Nucleic Acids Res. 2014, 42, 1291–1302. [Google Scholar] [CrossRef]
- Raben, N.; Nichols, R.C.; Martiniuk, F.; Plotz, P.H. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum. Mol. Genet. 1996, 5, 995–1000. [Google Scholar] [CrossRef]
- Markic, J.; Polic, B.; Kuzmanic-Samija, R.; Marusic, E.; Stricevic, L.; Metlicic, V.; Mestrovic, J. Immune Modulation Therapy in a CRIM-Positive and IgG Antibody-Positive Infant with Pompe Disease Treated with Alglucosidase Alfa: A Case Report. JIMD Rep. 2012, 2, 11–15. [Google Scholar]
- van Capelle, C.I.; van der Meijden, J.C.; van den Hout, J.M.; Jaeken, J.; Baethmann, M.; Voit, T.; Kroos, M.A.; Derks, T.G.; Rubio-Gozalbo, M.E.; Willemsen, M.A.; et al. Childhood Pompe disease: Clinical spectrum and genotype in 31 patients. Orphanet J. Rare Dis. 2016, 11, 65. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Rossi, B.; Tang, K.; Wu, X.; Mascioli, K.; Donaudy, F.; Tuzzi, M.R.; Fontana, F.; Cubellis, M.V.; Porto, C.; et al. The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum. Mutat. 2009, 30, 1683–1692. [Google Scholar] [CrossRef]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.L.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef]
- Deming, D.; Lee, K.; McSherry, T.; Wei, R.R.; Edmunds, T.; Garman, S.C. The molecular basis for Pompe disease revealed by the structure of human acid α-glucosidase. BioRxiv 2017. [Google Scholar] [CrossRef]
- Tarnopolsky, M.; Katzberg, H.; Petrof, B.J.; Sirrs, S.; Sarnat, H.B.; Myers, K.; Dupré, N.; Dodig, D.; Genge, A.; Venance, S.L.; et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. Can. J. Neurol. Sci. 2016, 43, 472–485. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
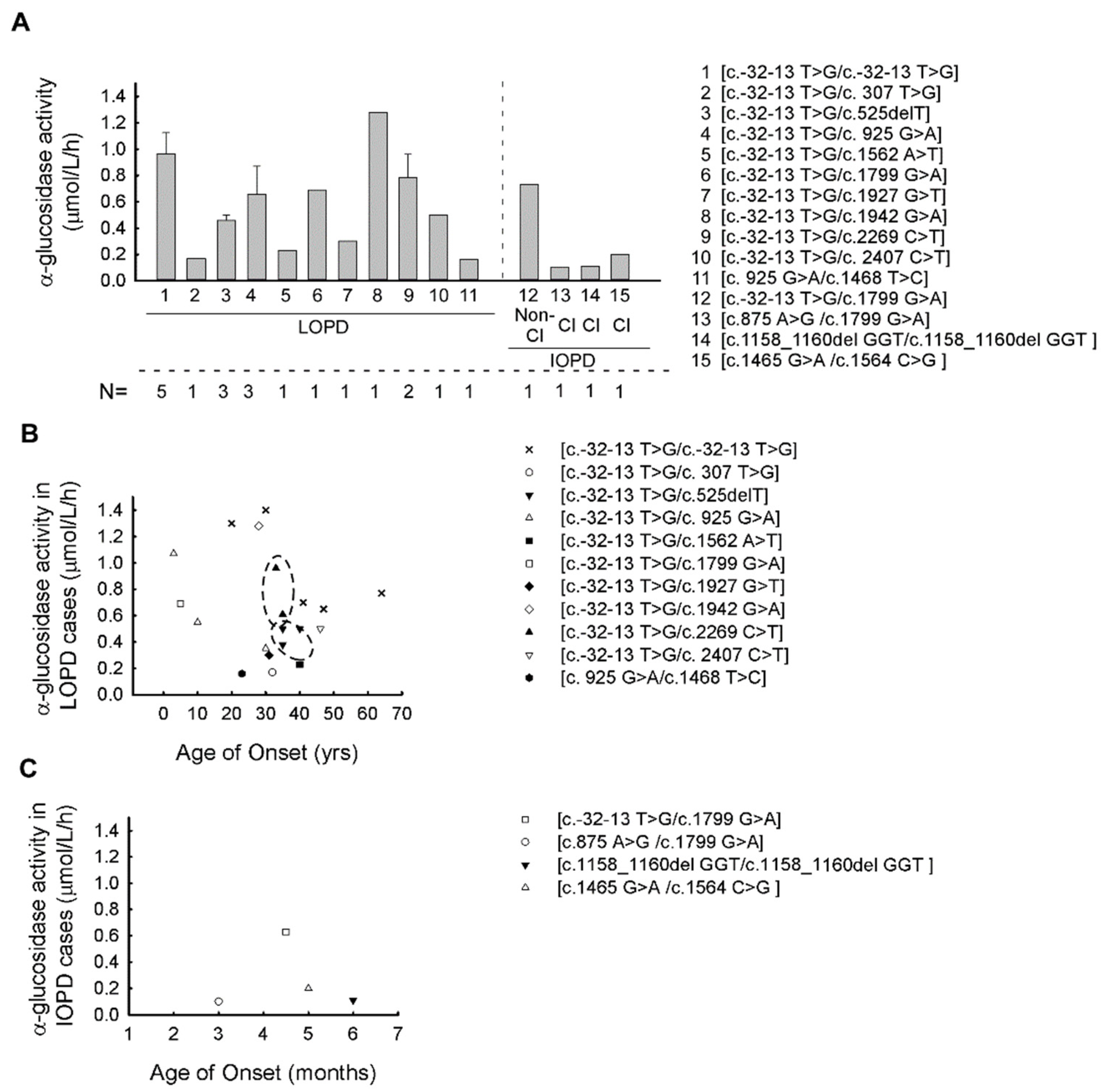
| Patient Number | Family Number | Genotype | GAA Activity (μmol/L/h) (Average ± SD) | Gender | AOO (ys) | Type of PD |
|---|---|---|---|---|---|---|
| P1 | F1 | c.-32-13 T > G/c.-32-13 T > G | 1.40 ± 0.1 | f | 30 | LOPD |
| P2 | F1 | c.-32-13 T > G/c.-32-13 T > G | 1.30 ± 0.15 | m | 20 | LOPD |
| P3 | F2 | c.-32-13 T > G/c.-32-13 T > G | 0.65 ± 0.23 | f | 47 | LOPD |
| P4 | F2 | c.-32-13 T > G/c.-32-13 T > G | 0.77 ± 0.15 | f | 64 | LOPD |
| P5 | F3 | c.-32-13 T > G/c.-32-13 T > G | 0.70 ± 0.1 | m | 41 | LOPD |
| P6 | F4 | c.-32-13 T > G/c.307 T > G | 0.17 ± 0.2 | f | 32 | LOPD |
| P7 | F5 | c.-32-13 T > G/c.525delT | 0.50 ± 0.1 | f | 35 | LOPD |
| P8 | F5 | c.-32-13 T > G/c.525delT | 0.50 ± 0.2 | f | 40 | LOPD |
| P9 | F6 | c.-32-13 T > G/c.525delT | 0.38 ± 0.41 | f | 35 | LOPD |
| P10 | F7 | c.-32-13 T > G/c.925 G > A | 1.07 ± 0.23 | m | 3 | LOPD |
| P11 | F7 | c.-32-13 T > G/c.925 G > A | 0.55 ± 0.05 | f | 10 | LOPD |
| P12 | F8 | c.-32-13 T > G/c.925 G > A | 0.35 ± 0.08 | f | 30 | LOPD |
| P13 | F9 | c.-32-13 T > G/c.1562 A > T | 0.23 ± 0.08 | f | 40 | LOPD |
| P14 | F10 | c.-32-13 T > G/c.1799 G > A | 0.69 ± 0.55 | m | 5 | LOPD |
| P15 | F11 | c.-32-13 T > G/c.1799 G > A | 0.73 ± 0.15 | m | 0.37 | IOPD |
| P16 | F12 | c.-32-13 T > G/c.1927G > T | 0.30 ± 0 | m | 31 | LOPD |
| P17 | F13 | c.-32-13 T > G/c.1942 G > A | 1.28 ± 0.12 | m | 28 | LOPD |
| P18 | F14 | c.-32-13 T > G/c.2269 C > T | 0.96 ± 0.23 | f | 33 | LOPD |
| P19 | F14 | c.-32-13 T > G/c.2269 C > T | 0.61 ± 0.1 | f | 35 | LOPD |
| P20 | F15 | c.-32-13 T > G/c.2407 C > T | 0.50 ± 0.07 | m | 46 | LOPD |
| P21 | F16 | c.875 A > G/c.1799 G > A | 0.10 ± 0.05 | f | 0.25 | IOPD |
| P22 | F17 | c.925 G > A/c.1468 T > C | 0.16 ± 0.13 | f | 23 | LOPD |
| P23 | F18 | c.1158_1160del GGT/c.1158_1160del GGT | 0.11 ± 0.02 | f | 0.5 | IOPD |
| P24 | F19 | c.1465 G > A/c.1564 C > G | 0.20 ± 0.1 | f | 0.4 | IOPD |
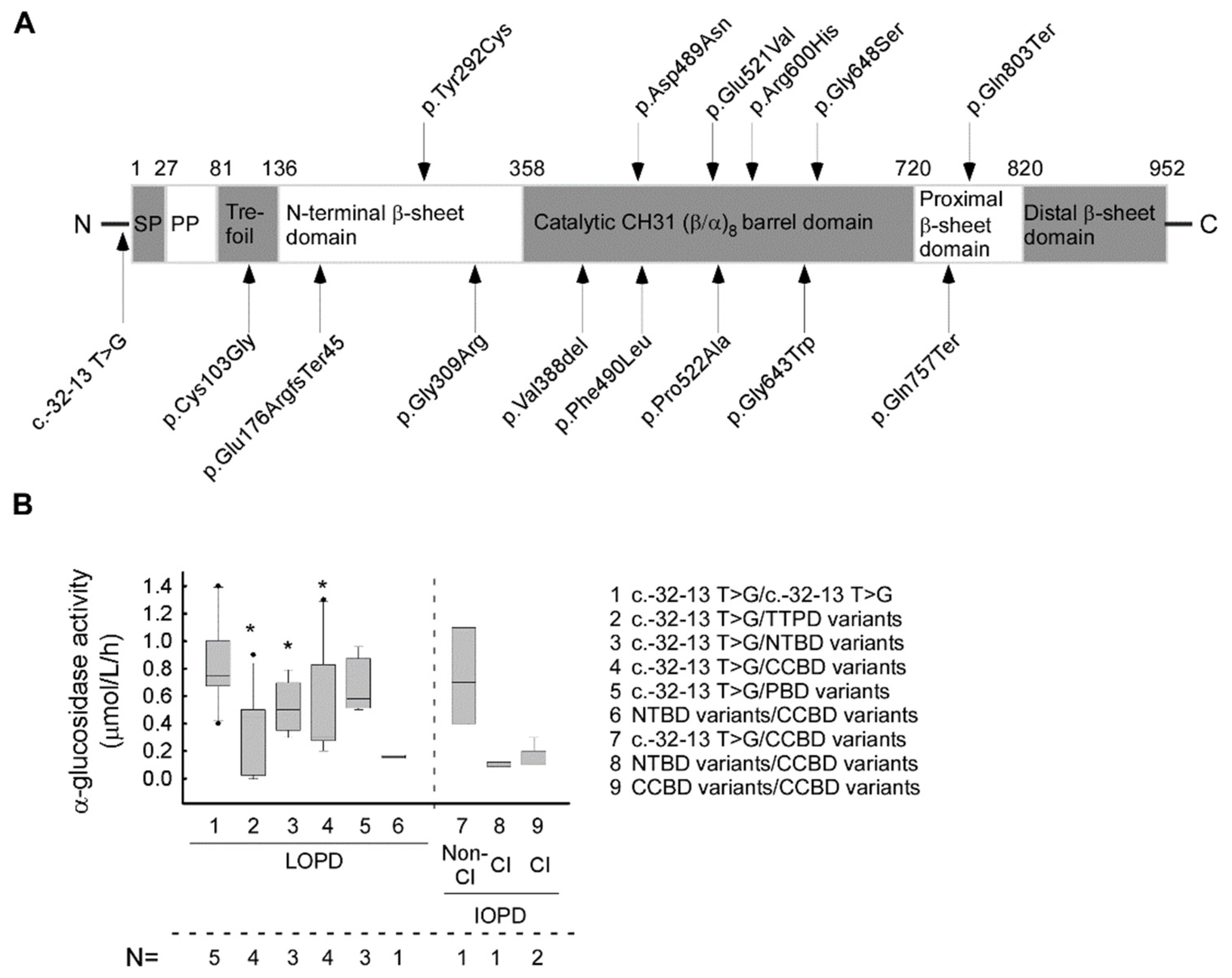
| No | Nt. Change | AA Change | Affec-ted Allele Number | Domain | Rsid | ACMG Score | Gnomad AF | Mutation Type | Pathogenety Scores by Vasrsome Database | Predic-ted Severity | Phenoty-pe with Null Allele | CRIM Status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.-32-13 T > G | - | 25 | - | rs386834236 | P | <0.001 | splice site | 1P | PM | C/A | pos | [30,31] |
| 2 | c. 307 T > G | p.Cys103Gly | 1 | TTPD | rs398123174 | P | <0.001 | missense | 19P/3B | PLS | CI | pos | [32] |
| 3 | c.525delT | p.Glu176Arg fsTer45 | 3 | TTPD | rs386834235 | P | <0.001 | INDEL | na | VS | CI | neg | [33] |
| 4 | c.875A > G | p.Tyr292Cys | 1 | NTBD | rs1057516600 | P/LP | <0.001 | missense | 20P/1B | PM | CI | pos | [32,34] |
| 5 | c.925 G > A | p.Gly309Arg | 4 | NTBD | rs543300039 | P | <0.001 | missense | 20P/0B | PLS | CI | pos | [35,36] |
| 6 | c.1158_1160del GGT | p.Val388del | 2 | CCBD | na | LP | na | INDEL | na | na | na | pos | ps |
| 7 | c.1465G > A | p.Asp489Asn | 1 | CCBD | rs398123169 | P/LP | <0.001 | missense | 20P/1B | PLS | CI | pos | [37] |
| 8 | c.1468 T > C | p.Phe490Leu | 1 | CCBD | na | LP | na | missense | 21P | LS | unkn | pos | [11] |
| 9 | c.1562 A > T | p.Glu521Val | 1 | CCBD | rs1455277014 | LP | <0.001 | missense | 20P/1B | unkn | CI | unk | [38] |
| 10 | c.1564C > G | p.Pro522Ala | 1 | CCBD | rs892129065 | P/LP | <0.001 | missense | 20P/1B | PLS | CI | pos | [39] |
| 11 | c.1799 G > A | p.Arg600His | 3 | CCBD | rs377544304 | LP | <0.001 | missense | 19P/1B | PLS | CI | pos | [39] |
| 12 | c.1927G > T | p.Gly643Trp | 1 | CCBD | rs28937909 | P | <0.001 | missense | 20P/1B | na | na | na | [32] |
| 13 | c.1942 G > A | p.Gly648Ser | 1 | CCBD | rs536906561 | P | <0.001 | missense | 19P/1B | PLS | CI | unk | [40] |
| 14 | c.2269 C > T | p.Gln757Ter | 2 | PBD | rs200483245 | P | <0.001 | nonsense | 4P/4B | VS | CI | neg | [41] |
| 15 | c. 2407 C > T | p.Gln803Ter | 1 | PBD | rs1344266804 | LP | <0.001 | nonsense | 7P/1B | VS | unkn | unk | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gal, A.; Grosz, Z.; Borsos, B.; Szatmari, I.; Sebők, A.; Jávor, L.; Harmath, V.; Szakszon, K.; Dezsi, L.; Balku, E.; et al. Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease. Life 2021, 11, 507. https://doi.org/10.3390/life11060507
Gal A, Grosz Z, Borsos B, Szatmari I, Sebők A, Jávor L, Harmath V, Szakszon K, Dezsi L, Balku E, et al. Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease. Life. 2021; 11(6):507. https://doi.org/10.3390/life11060507
Chicago/Turabian StyleGal, Aniko, Zoltán Grosz, Beata Borsos, Ildikó Szatmari, Agnes Sebők, Laszló Jávor, Veronika Harmath, Katalin Szakszon, Livia Dezsi, Eniko Balku, and et al. 2021. "Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease" Life 11, no. 6: 507. https://doi.org/10.3390/life11060507
APA StyleGal, A., Grosz, Z., Borsos, B., Szatmari, I., Sebők, A., Jávor, L., Harmath, V., Szakszon, K., Dezsi, L., Balku, E., Jobbagy, Z., Herczegfalvi, A., Almássy, Z., Kerényi, L., & Molnar, M. J. (2021). Correlation of GAA Genotype and Acid-α-Glucosidase Enzyme Activity in Hungarian Patients with Pompe Disease. Life, 11(6), 507. https://doi.org/10.3390/life11060507