
Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly


Abstract
1. Introduction
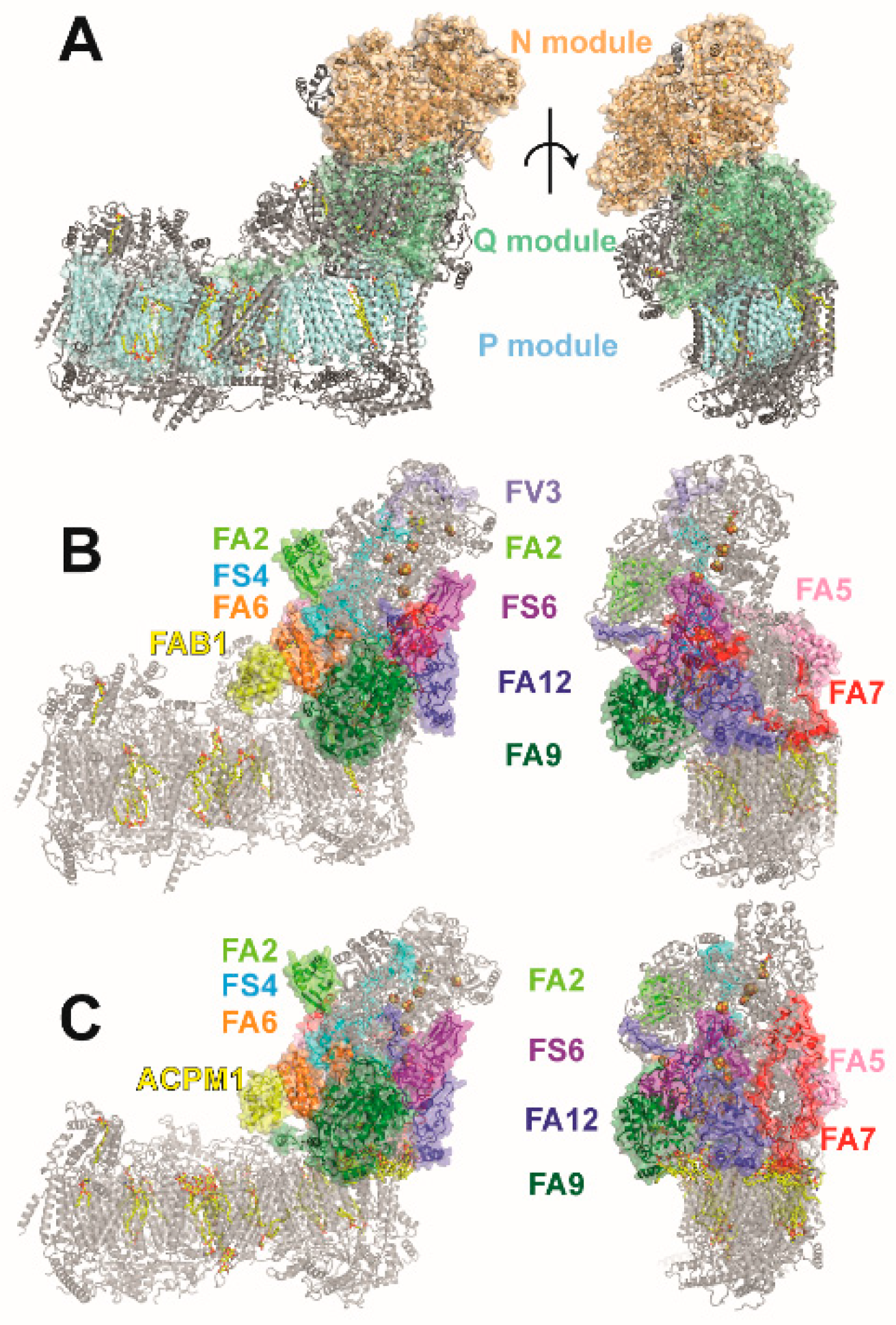
2. Accessory Subunits of the Matrix Arm in Yeast and Mammalian Complex I
3. Leigh Syndrome and the Ndufs4 KO Mouse Model
4. NDUFS4-Linked Complex I Dysfunction at the Molecular Level
5. The Role of NDUFS4, NDUFS6, and NDUFA12 in Complex I Assembly
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory Complex I—Structure, Mechanism and Evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yoga, E.G.; Angerer, H.; Parey, K.; Zickermann, V. Respiratory Complex I—Mechanistic Insights and Advances in Structure Determination. Biochim. et Biophys. Acta (BBA) Bioenerg. 2020, 1861, 148153. [Google Scholar] [CrossRef]
- Hirst, J. Mitochondrial Complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef]
- Sazanov, L.A. A Giant Molecular Proton Pump: Structure and Mechanism of Respiratory Complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, A.B.; Vinogradov, A.D. Slow Active/Inactive Transition of the Mitochondrial NADH-Ubiquinone Reductase. Biochim. et Biophys. Acta (BBA) Bioenerg. 1990, 1019, 151–158. [Google Scholar] [CrossRef]
- Maklashina, E.; Kotlyar, A.B.; Cecchini, G. Active/De-Active Transition of Respiratory Complex I in Bacteria, Fungi, and Animals. Biochim. et Biophys. Acta (BBA) Bioenerg. 2003, 1606, 95–103. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-Nitrosation of a Cysteine Switch on Mitochondrial Complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Stepanova, A.; Galkin, A. Ischemic A/D Transition of Mitochondrial Complex I and its Role in ROS Generation. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1857, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal Structure of the Entire Respiratory Complex I. Nat. Cell Biol. 2013, 494, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Mechanistic Insight from the Crystal Structure of Mitochondrial Complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef]
- Agip, A.-N.A.; Blaza, J.N.; Fedor, J.G.; Hirst, J. Mammalian Respiratory Complex I through the Lens of Cryo-EM. Annu. Rev. Biophys. 2019, 48, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Kampjut, D.; Sazanov, L.A. The Coupling Mechanism of Mammalian Respiratory Complex I. Science 2020, 370, eabc4209. [Google Scholar] [CrossRef]
- Grba, D.N.; Hirst, J. Mitochondrial Complex I Structure Reveals Ordered Water Molecules for Catalysis and Proton Translocation. Nat. Struct. Mol. Biol. 2020, 27, 1–9. [Google Scholar] [CrossRef]
- Klusch, N.; Senkler, J.; Yildiz, Ö.; Kühlbrandt, W.; Braun, H.-P. A Ferredoxin Bridge Connects the Two Arms of Plant Mitochondrial Complex I. Plant Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Soufari, H.; Parrot, C.; Kuhn, L.; Waltz, F.; Hashem, Y. Specific Features and Assembly of the Plant Mitochondrial Complex I Revealed by Cryo-EM. Nat. Commun. 2020, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Fearnley, I.M.; Skehel, J.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Bovine Complex I Is a Complex of 45 Different Subunits. J. Biol. Chem. 2006, 281, 32724–32727. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, S.; Drose, S.; Zwicker, K.; Zickermann, V.; Brandt, U. Yarrowia Lipolytica, a Yeast Genetic System to Study Mito-chondrial Complex I. Biochim. Biophys. Acta 2002, 1555, 83–91. [Google Scholar] [CrossRef]
- Parey, K.; Haapanen, O.; Sharma, V.; Köfeler, H.; Züllig, T.; Prinz, S.; Siegmund, K.; Wittig, I.; Mills, D.J.; Vonck, J.; et al. High-Resolution cryo-EM Structures of Respiratory Complex I: Mechanism, Assembly, and Disease. Sci. Adv. 2019, 5, eaax9484. [Google Scholar] [CrossRef]
- Kmita, K.; Zickermann, V. Accessory Subunits of Mitochondrial Complex I. Biochem. Soc. Trans. 2013, 41, 1272–1279. [Google Scholar] [CrossRef]
- Brandt, U. Energy Converting NADH: Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional Modules and Structural Basis of Conformational Coupling in Mitochondrial Complex I. Science 2010, 329, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T. Iron–Sulfur Clusters/Semiquinones in Complex I. Biochim. et Biophys. Acta (BBA) Bioenerg. 1998, 1364, 186–206. [Google Scholar] [CrossRef]
- Roessler, M.M.; King, M.S.; Robinson, A.J.; Armstrong, F.A.; Harmer, J.; Hirst, J. Direct Assignment of EPR Spectra to Structurally Defined Iron-Sulfur Clusters in Complex I by Double Electron–Electron Resonance. Proc. Natl. Acad. Sci. USA 2010, 107, 1930–1935. [Google Scholar] [CrossRef]
- Schulte, M.; Frick, K.; Gnandt, E.; Jurkovic, S.; Burschel, S.; Labatzke, R.; Aierstock, K.; Fiegen, D.; Wohlwend, D.; Gerhardt, S.; et al. A Mechanism to Prevent Production of Reactive Oxygen Species by Escherichia Coli Respiratory Complex I. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Sazanov, L.A. Structure of the Hydrophilic Domain of Respiratory Complex I from Thermus Thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [CrossRef]
- Zickermann, V.; Bostina, M.; Hunte, C.; Ruiz, T.; Radermacher, M.; Brandt, U. Functional Implications from an Unexpected Position of the 49-kDa Subunit of NADH:Ubiquinone Oxidoreductase. J. Biol. Chem. 2003, 278, 29072–29078. [Google Scholar] [CrossRef] [PubMed]
- Warnau, J.; Sharma, V.; Gamiz-Hernandez, A.P.; Di Luca, A.; Haapanen, O.; Vattulainen, I.; Wikström, M.; Hummer, G.; Kaila, V.R.I. Redox-Coupled Quinone Dynamics in the Respiratory Complex I. Proc. Natl. Acad. Sci. USA 2018, 115, E8413–E8420. [Google Scholar] [CrossRef]
- Fedor, J.G.; Jones, A.J.Y.; Di Luca, A.; Kaila, V.R.I.; Hirst, J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. USA 2017, 114, 12737–12742. [Google Scholar] [CrossRef]
- Yip, C.-Y.; Harbour, M.E.; Jayawardena, K.; Fearnley, I.M.; Sazanov, L.A. Evolution of Respiratory Complex I. J. Biol. Chem. 2011, 286, 5023–5033. [Google Scholar] [CrossRef]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J.Z.J. Structure of Mammalian Respiratory Complex I. Nat. Cell Biol. 2016, 536, 354–358. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, G.D.M.; Sazanov, L.A. Atomic Structure of the Entire Mammalian Mitochondrial Complex I. Nat. Cell Biol. 2016, 538, 406–410. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory Subunits are Integral for Assembly and Function of Human Mitochondrial Complex I. Nat. Cell Biol. 2016, 538, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.-C.L.; Phan, D.H.; Johnson, A.N.; Pasapuleti, M.; AlKhaldi, H.A.; Zhang, F.; Vik, S.B. Analysis of Human Mutations in the Supernumerary Subunits of Complex I. Life 2020, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Rodenburg, R.J. Mitochondrial Complex I-Linked Disease. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human Diseases Associated with Defects in Assembly of OXPHOS Complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef]
- Bridges, H.R.; Mohammed, K.; Harbour, M.E.; Hirst, J. Subunit NDUFV3 is Present in Two Distinct Isoforms in Mammalian Complex I. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1858, 197–207. [Google Scholar] [CrossRef]
- Dibley, M.G.; Ryan, M.T.; Stroud, D.A. A Novel Isoform of the Human Mitochondrial Complex I Subunit NDUFV3. FEBS Lett. 2016, 591, 109–117. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Cabrera-Orefice, A.; Huynen, M.A.; Arnold, S. Identification and Evolutionary Analysis of Tissue-Specific Isoforms of Mitochondrial Complex I Subunit NDUFV3. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1858, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Abdrakhmanova, A.; Dobrynin, K.; Zwicker, K.; Kerscher, S.; Brandt, U. Functional Sulfurtransferase is Associated with Mitochondrial Complex I fromYarrowia Lipolytica, but is Not Required for Assembly of its Iron-Sulfur Clusters. FEBS Lett. 2005, 579, 6781–6785. [Google Scholar] [CrossRef]
- Parey, K.; Brandt, U.; Xie, H.; Mills, D.J.; Siegmund, K.; Vonck, J.; Kühlbrandt, W.; Zickermann, V. Cryo-EM Structure of Respiratory Complex I at Work. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Jörnvall, H.; Persson, B.; Krook, M.; Atrian, S.; Gonzalez-Duarte, R.; Jeffery, J.; Ghosh, D. Short-Chain Dehydrogenases/Reductases (SDR). Biochemistry 1995, 34, 6003–6013. [Google Scholar] [CrossRef]
- Abdrakhmanova, A.; Zwicker, K.; Kerscher, S.; Zickermann, V.; Brandt, U. Tight Binding of NADPH to the 39-kDa Subunit of Complex I is Not Required for Catalytic Activity but Stabilizes the Multiprotein Complex. Biochim. et Biophys. Acta (BBA) Bioenerg. 2006, 1757, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Babot, M.; Labarbuta, P.; Birch, A.; Kee, S.; Fuszard, M.; Botting, C.H.; Wittig, I.; Heide, H.; Galkin, A. ND3, ND1 and 39kDa Subunits are more Exposed in the De-Active form of Bovine Mitochondrial Complex I. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1837, 929–939. [Google Scholar] [CrossRef]
- Agip, A.-N.A.; Blaza, J.N.; Bridges, H.R.; Viscomi, C.; Rawson, S.; Muench, S.P.; Hirst, J. Cryo-EM Structures of Complex I from Mouse Heart Mitochondria in Two Biochemically Defined States. Nat. Struct. Mol. Biol. 2018, 25, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, K.; Abdrakhmanova, A.; Richers, S.; Hunte, C.; Kerscher, S.; Brandt, U. Characterization of Two Different Acyl Carrier Proteins in Complex I from Yarrowia Lipolytica. Biochim. et Biophys. Acta (BBA) Bioenerg. 2010, 1797, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Angerer, H.; Schönborn, S.; Gorka, J.; Bahr, U.; Karas, M.; Wittig, I.; Heidler, J.; Hoffmann, J.; Morgner, N.; Zickermann, V. Acyl Modification and Binding of Mitochondrial ACP to Multiprotein Complexes. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1913–1920. [Google Scholar] [CrossRef]
- Carroll, J.; Fearnley, I.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Analysis of the Subunit Composition of Complex I from Bovine Heart Mitochondria*S. Mol. Cell. Proteom. 2003, 2, 117–126. [Google Scholar] [CrossRef]
- Angerer, H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology 2015, 4, 133–150. [Google Scholar] [CrossRef]
- Angerer, H.; Radermacher, M.; Mańkowska, M.; Steger, M.; Zwicker, K.; Heide, H.; Wittig, I.; Brandt, U.; Zickermann, V. The LYR Protein Subunit NB4M/NDUFA6 of Mitochondrial Complex I Anchors an Acyl Carrier Protein and is Essential for Catalytic Activity. Proc. Natl. Acad. Sci. USA 2014, 111, 5207–5212. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.C.; Bornhövd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 Interacting Protein Isd11 has an Essential Role in Fe/S Cluster Biogenesis in Mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef]
- Zhu, J.; King, M.S.; Yu, M.; Klipcan, L.; Leslie, A.G.W.; Hirst, J. Structure of Subcomplex Iβ of Mammalian Respiratory Complex I Leads to New Supernumerary Subunit Assignments. Proc. Natl. Acad. Sci. USA 2015, 112, 12087–12092. [Google Scholar] [CrossRef] [PubMed]
- Kastaniotis, A.J.; Autio, K.J.; Kerätär, J.M.; Monteuuis, G.; Mäkelä, A.M.; Nair, R.R.; Pietikäinen, L.P.; Shvetsova, A.; Chen, Z.; Hiltunen, J.K. Mitochondrial Fatty Acid Synthesis, Fatty Acids and Mitochondrial Physiology. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 39–48. [Google Scholar] [CrossRef]
- Nowinski, S.M.; Van Vranken, J.G.; Dove, K.K.; Rutter, J. Impact of Mitochondrial Fatty Acid Synthesis on Mitochondrial Biogenesis. Curr. Biol. 2018, 28, R1212–R1219. [Google Scholar] [CrossRef] [PubMed]
- Yoga, E.G.; Parey, K.; Djurabekova, A.; Haapanen, O.; Siegmund, K.; Zwicker, K.; Sharma, V.; Zickermann, V.; Angerer, H. Essential Role of Accessory Subunit LYRM6 in the Mechanism of Mitochondrial Complex I. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Ingraham, C.A.; Burwell, L.S.; Skalska, J.; Brookes, P.S.; Howell, R.L.; Sheu, S.-S.; Pinkert, C.A. NDUFS4: Creation of a Mouse Model Mimicking a Complex I Disorder. Mitochondrion 2009, 9, 204–210. [Google Scholar] [CrossRef]
- Breuer, M.E.; Willems, P.H.; Smeitink, J.A.; Koopman, W.J.; Nooteboom, M. Cellular and Animal Models for Mitochondrial Complex I Deficiency: A Focus on the NDUFS4 Subunit. IUBMB Life 2013, 65, 202–208. [Google Scholar] [CrossRef]
- Papa, S.; Sardanelli, A.M.; Cocco, T.; Speranza, F.; Scacco, S.C.; Technikova-Dobrova, Z. The Nuclear-Encoded 18 kDa (IP) AQDQ Subunit of Bovine Heart Complex I is Phosphorylated by the Mitochondrial cAMP-Dependent Protein Kinase. FEBS Lett. 1996, 379, 299–301. [Google Scholar] [CrossRef]
- De Rasmo, D.; Palmisano, G.; Scacco, S.; Technikova-Dobrova, Z.; Panelli, D.; Cocco, T.M.; Sardanelli, A.M.; Gnoni, A.; Micelli, L.; Trani, A.; et al. Phosphorylation Pattern of the NDUFS4 Subunit of Complex I of the Mammalian Respiratory Chain. Mitochondrion 2010, 10, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fearnley, I.M.; Peak-Chew, S.Y.; Walker, J.E. The Phosphorylation of Subunits of Complex I from Bovine Heart Mitochondria. J. Biol. Chem. 2004, 279, 26036–26045. [Google Scholar] [CrossRef]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-Dependent Protein Kinase Regulates the Mitochondrial Import of the Nuclear Encoded NDUFS4 Subunit of Complex I. Cell. Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef]
- Papa, S.; De Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian Complex I: A Regulable and Vulnerable Pacemaker in Mitochondrial Respiratory Function. Biochim. et Biophys. Acta (BBA) Bioenerg. 2008, 1777, 719–728. [Google Scholar] [CrossRef]
- Kmita, K.; Wirth, C.; Warnau, J.; Guerrero-Castillo, S.; Hunte, C.; Hummer, G.; Kaila, V.R.I.; Zwicker, K.; Brandt, U.; Zickermann, V. Accessory NUMM (NDUFS6) Subunit Harbors a Zn-Binding Site and is Essential for Biogenesis of Mitochondrial Complex I. Proc. Natl. Acad. Sci. USA 2015, 112, 5685–5690. [Google Scholar] [CrossRef] [PubMed]
- Kensche, P.R.; Duarte, I.; Huynen, A.M. A Three-Dimensional Topology of Complex I Inferred from Evolutionary Correlations. BMC Struct. Biol. 2012, 12, 19. [Google Scholar] [CrossRef]
- Petruzzella, V.; Vergari, R.; Puzziferri, I.; Boffoli, D.; Lamantea, E.; Zeviani, M.; Papa, S. A Nonsense Mutation in the NDUFS4 gene Encoding the 18 kDa (AQDQ) Subunit of Complex I Abolishes Assembly and Activity of the Complex in a Patient with Leigh-Like Syndrome. Hum. Mol. Genet. 2001, 10, 529–535. [Google Scholar] [CrossRef]
- Petruzzella, V.; Papa, S. Mutations in Human Nuclear Genes Encoding for Subunits of Mitochondrial Respiratory Complex I: The NDUFS4 Gene. Gene 2002, 286, 149–154. [Google Scholar] [CrossRef]
- Petruzzella, V.; Panelli, D.; Torraco, A.; Stella, A.; Papa, S. Mutations in the NDUFS4 Gene of Mitochondrial Complex I Alter Stability of the Splice Variants. FEBS Lett. 2005, 579, 3770–3776. [Google Scholar] [CrossRef][Green Version]
- Scacco, S.; Petruzzella, V.; Budde, S.; Vergari, R.; Tamborra, R.; Panelli, D.; Heuvel, L.P.V.D.; Smeitink, J.A.; Papa, S. Pathological Mutations of the Human NDUFS4 Gene of the 18-kDa (AQDQ) Subunit of Complex I Affect the Expression of the Protein and the Assembly and Function of the Complex. J. Biol. Chem. 2003, 278, 44161–44167. [Google Scholar] [CrossRef]
- Anderson, S.L.; Chung, W.K.; Frezzo, J.; Papp, J.C.; Ekstein, J.; DiMauro, S.; Rubin, B.Y. A Novel Mutation in NDUFS4 Causes Leigh Syndrome in an Ashkenazi Jewish Family. J. Inherit. Metab. Dis. 2008, 31, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.E.; Beaulieu, C.L.; Bernier, F.P.; Sparkes, R.; Innes, A.M.; Jackel-Cram, C.; Ober, C.; Parboosingh, J.S.; Lemire, E.G. A Novel NDUFS4 Frameshift Mutation Causes Leigh Disease in the Hutterite Population. Am. J. Med. Genet. Part A 2016, 173, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Budde, S.M.S.; Heuvel, L.P.W.J.V.D.; Smeets, R.J.P.; Skladal, D.; Mayr, J.A.; Boelen, C.; Petruzzella, V.; Papa, S.; Smeitink, J.A.M. Clinical Heterogeneity in Patients with Mutations in the NDUFS4 Gene of Mitochondrial Complex I. J. Inherit. Metab. Dis. 2003, 26, 813–815. [Google Scholar] [CrossRef]
- Leshinsky-Silver, E.; Lebre, A.-S.; Minai, L.; Saada, A.; Steffann, J.; Cohen, S.; Rötig, A.; Munnich, A.; Lev, D.; Lerman-Sagie, T. NDUFS4 Mutations cause Leigh Syndrome with Predominant Brainstem Involvement. Mol. Genet. Metab. 2009, 97, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Escobar, J.D.; Oyarzabal, A.; Montero, R.; Artuch, R.; Jou, C.; Jiménez, C.; Gort, L.; Briones, P.; Muchart, J.; López-Gallardo, E.; et al. Ndufs4 Related Leigh Syndrome: A Case Report and Review of the Literature. Mitochondrion 2016, 28, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S. NDUFS4-Related Leigh Syndrome in Hutterites. Am. J. Med. Genet. Part A 2017, 173, 1450–1451. [Google Scholar] [CrossRef]
- Heuvel, L.V.D.; Ruitenbeek, W.; Smeets, R.; Gelman-Kohan, Z.; Elpeleg, O.; Loeffen, J.; Trijbels, F.; Mariman, E.; de Bruijn, D.; Smeitink, J. Demonstration of a New Pathogenic Mutation in Human Complex I Deficiency: A 5-bp Duplication in the Nuclear Gene Encoding the 18-kD (AQDQ) Subunit. Am. J. Hum. Genet. 1998, 62, 262–268. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.-H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical Features and Biochemical and DNA Abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D. Subacute Necrotizing Encephalomyelopathy in an Infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; Sallevelt, S.C.; Smeets, H.J. Leigh Syndrome: Resolving the Clinical and Genetic Heterogeneity Paves the Way for Treatment Options. Mol. Genet. Metab. 2016, 117, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A Meta-Analysis and Systematic Review of Leigh Syndrome: Clinical Manifestations, Respiratory Chain Enzyme Complex Deficiency, and Gene Mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef]
- Bénit, P.; Steffann, J.; Lebon, S.; Chretien, D.; Kadhom, N.; De Lonlay, P.; Goldenberg, A.; Dumez, Y.; Dommergues, M.; Rustin, P.; et al. Genotyping Microsatellite DNA Markers at Putative Disease Loci in Inbred/Multiplex Families with Respiratory Chain Complex I Deficiency Allows Rapid Identification of a Novel Nonsense Mutation (IVS1nt −1) in the NDUFS4 Gene in Leigh Syndrome. Qual. Life Res. 2003, 112, 563–566. [Google Scholar] [CrossRef]
- Lombardo, B.; Ceglia, C.; Tarsitano, M.; Pierucci, I.; Salvatore, F.; Pastore, L. Identification of a Deletion in the NDUFS4 Gene Using Array-Comparative Genomic Hybridization in a Patient with Suspected Mitochondrial Respiratory Disease. Gene 2014, 535, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Assouline, Z.; Jambou, M.; Rio, M.; Bole-Feysot, C.; De Lonlay, P.; Barnerias, C.; Desguerre, I.; Bonnemains, C.; Guillermet, C.; Steffann, J.; et al. A Constant and Similar Assembly Defect of Mitochondrial Respiratory Chain Complex I Allows Rapid Identification of NDUFS4 Mutations in Patients with Leigh Syndrome. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1062–1069. [Google Scholar] [CrossRef]
- Catania, A.; Iuso, A.; Bouchereau, J.; Kremer, L.S.; Paviolo, M.; Terrile, C.; Bénit, P.; Rasmusson, A.G.; Schwarzmayr, T.; Tiranti, V.; et al. Arabidopsis Thaliana Alternative Dehydrogenases: A Potential Therapy for Mitochondrial Complex I Deficiency? Perspectives and Pitfalls. Orphanet J. Rare Dis. 2019, 14, 236. [Google Scholar] [CrossRef] [PubMed]
- De Haas, R.; Das, D.; Garanto, A.; Renkema, H.G.; Greupink, R.; Broek, P.V.D.; Pertijs, J.; Collin, R.W.J.; Willems, P.; Beyrath, J.; et al. Therapeutic Effects of the Mitochondrial ROS-Redox Modulator KH176 in a Mammalian Model of Leigh Disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial Complex I Deficiency Increases Protein Acetylation and Accelerates Heart Failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Gowda, G.A.N.; Tian, R. Targeting NAD+ Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Kayser, E.-B.; Bornstein, R.; Stokes, J.; Bitto, A.; Park, K.Y.; Pan, A.; Sun, G.; Raftery, D.; Kaeberlein, M.; et al. Regional Metabolic Signatures in the Ndufs4(KO) Mouse Brain Implicate Defective Glutamate/α-Ketoglutarate Metabolism in Mitochondrial Disease. Mol. Genet. Metab. 2020, 130, 118–132. [Google Scholar] [CrossRef]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G. Region-Specific Defects of Respiratory Capacities in the Ndufs4(KO) Mouse Brain. PLoS ONE 2016, 11, e0148219. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Martin-Perez, M.; Grillo, A.S.; Ito, T.K.; Valente, A.S.; Han, J.; Entwisle, S.W.; Huang, H.Z.; Kim, D.; Yajima, M.; Kaeberlein, M.; et al. PKC Downregulation upon Rapamycin Treatment Attenuates Mitochondrial Disease. Nat. Metab. 2020, 2, 1472–1481. [Google Scholar] [CrossRef]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.-H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia Treatment Reverses Neurodegenerative Disease in a Mouse Model of Leigh Syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a Therapy for Mitochondrial Disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Grange, R.M.; Sharma, R.; Shah, H.; Reinstadler, B.; Goldberger, O.; Cooper, M.K.; Nakagawa, A.; Miyazaki, Y.; Hindle, A.G.; Batten, A.J.; et al. Hypoxia Ameliorates Brain Hyperoxia and NAD+ Deficiency in a Murine Model of Leigh Syndrome. Mol. Genet. Metab. 2021, 133, 83–93. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. 2019, 30, 824–832.e3. [Google Scholar] [CrossRef] [PubMed]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Pentimalli, T.M.; Jüttner, R.; Glažar, P.; Uppal, K.; Bottani, E.; Brunetti, D.; Secker, C.; et al. Defective Metabolic Programming Impairs Early Neuronal Morphogenesis in Neural Cultures and an Organoid Model of Leigh Syndrome. Nat. Commun. 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Calvo, E.S.; Tucker, E.J.; Compton, A.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, A.O.; et al. High-Throughput, Pooled Sequencing Identifies Mutations in NUBPL and FOXRED1 in Human Complex I Deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Budde, S.; Heuvel, L.V.D.; Janssen, A.; Smeets, R.; Buskens, C.; DeMeirleir, L.; Van Coster, R.; Baethmann, M.; Voit, T.; Trijbels, J.; et al. Combined Enzymatic Complex I and III Deficiency Associated with Mutations in the Nuclear Encoded NDUFS4 Gene. Biochem. Biophys. Res. Commun. 2000, 275, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the Mitochondrial ND1 Gene as a Cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New Perspective in Diagnostics of Mitochondrial Disorders: Two Years’ Experience with Whole-Exome Sequencing at a National Paediatric Centre. J. Transl. Med. 2016, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical Validity of Biochemical and Molecular Analysis in Diagnosing Leigh Syndrome: A Study of 106 Japanese Patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Shaag, A.; Mandel, H.; Reich, D.S.; Penyakov, M.; Hujeirat, Y.; Saada, A.; Elpeleg, O.; Shalev, A.S. Mutated NDUFS6 is the Cause of Fatal Neonatal Lactic Acidemia in Caucasus Jews. Eur. J. Hum. Genet. 2009, 17, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Torraco, A.; Nasca, A.; Verrigni, D.; Pennisi, A.; Zaki, M.S.; Olivieri, G.; Assouline, Z.; Martinelli, D.; Maroofian, R.; Rizza, T.; et al. Novel NDUFA12 Variants are Associated with Isolated Complex I Defect and Variable Clinical Manifestation. Hum. Mutat. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Rodenburg, R.J.; Brand, M.V.D.; Thomsen, L.L.; Duno, M.; Batbayli, M.; Wibrand, F.; Nijtmans, L. Respiratory Chain Complex I Deficiency due to NDUFA12 Mutations as a New Cause of Leigh Syndrome. J. Med. Genet. 2011, 48, 737–740. [Google Scholar] [CrossRef]
- Speer, R.R.; Ezeanya, U.C.; Beaudoin, S.J.; Glass, K.M.; Oji-Mmuo, C.N. Term Neonate Presenting with the Combined Occurrence of Mucolipidosis Type II and Leigh Syndrome. J. Pediatr. Genet. 2019, 9, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Barghuti, F.; Elian, K.; Gomori, J.M.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. The Unique Neuroradiology of Complex I Deficiency Due to NDUFA12L Defect. Mol. Genet. Metab. 2008, 94, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Herzer, M.; Koch, J.; Prokisch, H.; Rodenburg, R.; Rauscher, C.; Radauer, W.; Forstner, R.; Pilz, P.; Rolinski, B.; Freisinger, P.; et al. Leigh Disease with Brainstem Involvement in Complex I Deficiency due to Assembly Factor NDUFAF2 Defect. Neuropediatrics 2010, 41, 30–34. [Google Scholar] [CrossRef]
- Hoefs, S.J.; Dieteren, C.E.; Rodenburg, R.J.; Naess, K.; Bruhn, H.; Wibom, R.; Wagena, E.; Willems, P.H.; Smeitink, J.A.; Nijtmans, L.G.; et al. Baculovirus Complementation Restores a Novel NDUFAF2 Mutation Causing Complex I Deficiency. Hum. Mutat. 2009, 30, E728–E736. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, I.; Kennaway, N.G.; Shoubridge, E.A. A Molecular Chaperone for Mitochondrial Complex I Assembly is Mutated in a Progressive Encephalopathy. J. Clin. Investig. 2005, 115, 2784–2792. [Google Scholar] [CrossRef]
- Reynaud-Dulaurier, R.; Benegiamo, G.; Marrocco, E.; Al-Tannir, R.; Surace, E.M.; Auwerx, J.; Decressac, M. Gene Replacement Therapy Provides Benefit in an Adult Mouse Model of Leigh Syndrome. Brain 2020, 143, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Cerutti, R.; Luna-Sanchez, M.; Zeviani, M.; Viscomi, C. A Single Intravenous Injection of AAV-PHP.B-hNDUFS4 Ameliorates the Phenotype of Ndufs4 Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 1071–1078. [Google Scholar] [CrossRef]
- Lazarou, M.; McKenzie, M.; Ohtake, A.; Thorburn, D.R.; Ryan, M.T. Analysis of the Assembly Profiles for Mitochondrial- and Nuclear-DNA-Encoded Subunits into Complex I. Mol. Cell. Biol. 2007, 27, 4228–4237. [Google Scholar] [CrossRef]
- Pereira, B.; Videira, A.; Duarte, M. Novel Insights into the Role of Neurospora crassa NDUFAF2, an Evolutionarily Conserved Mitochondrial Complex I Assembly Factor. Mol. Cell. Biol. 2013, 33, 2623–2634. [Google Scholar] [CrossRef]
- Adjobo-Hermans, M.J.; de Haas, R.; Willems, P.H.; Wojtala, A.; Vries, S.E.V.E.-D.; Wagenaars, J.A.; Brand, M.V.D.; Rodenburg, R.J.; Smeitink, J.A.; Nijtmans, L.G.; et al. NDUFS4 Deletion Triggers Loss of NDUFA12 in Ndufs4 Mice and Leigh Syndrome Patients: A Stabilizing Role for NDUFAF2. Biochim. et Biophys. Acta (BBA) Bioenerg. 2020, 1861, 148213. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.E.; Watt, W.C.; Marcinek, D.J.; Kapur, R.P.; Schenkman, K.A.; Palmiter, R.D. Mice with Mitochondrial Complex I Deficiency Develop a Fatal Encephalomyopathy. Cell Metab. 2008, 7, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, M.A.; Willems, P.; Brand, M.V.D.; Valsecchi, F.; Kruse, S.; Palmiter, R.; Smeitink, J.; Nijtmans, L. Mitochondrial Complex III Stabilizes Complex I in the Absence of NDUFS4 to Provide Partial Activity. Hum. Mol. Genet. 2011, 21, 115–120. [Google Scholar] [CrossRef]
- Kahlhöfer, F.; Kmita, K.; Wittig, I.; Zwicker, K.; Zickermann, V. Accessory Subunit NUYM (NDUFS4) is Required for Stability of the Electron Input Module and Activity of Mitochondrial Complex I. Biochim. et Biophys. Acta (BBA) Bioenerg. 2017, 1858, 175–181. [Google Scholar] [CrossRef]
- Galkin, A.; Brandt, U. Superoxide Radical Formation by Pure Complex I (NADH:Ubiquinone Oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef]
- Formosa, L.E.; Dibley, M.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of Mitochondrial Respiratory Chain Complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Muellner-Wong, L.; Reljic, B.; Sharpe, A.J.; Jackson, T.D.; Beilharz, T.H.; Stojanovski, D.; Lazarou, M.; Stroud, D.A.; Ryan, M.T. Dissecting the Roles of Mitochondrial Complex I Intermediate Assembly Complex Factors in the Biogenesis of Complex I. Cell Rep. 2020, 31, 107541. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef]
- Tsuneoka, M.; Teye, K.; Arima, N.; Soejima, M.; Otera, H.; Ohashi, K.; Koga, Y.; Fujita, H.; Shirouzu, K.; Kimura, H.; et al. A Novel Myc-Target Gene, mimitin, That Is Involved in Cell Proliferation of Esophageal Squamous Cell Carcinoma*. J. Biol. Chem. 2005, 280, 19977–19985. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Sosunov, S.; Niatsetskaya, Z.; Konrad, C.; Starkov, A.A.; Manfredi, G.; Wittig, I.; Ten, V.; Galkin, A.; Sosunov, S. Redox-Dependent Loss of Flavin by Mitochondrial Complex I in Brain Ischemia/Reperfusion Injury. Antioxid. Redox Signal. 2019, 31, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Kussmaul, L.; Hirst, J. The Mechanism of Superoxide Production by NADH:Ubiquinone Oxidoreductase (complex I) from Bovine Heart Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Homo sapiens | Yarrowia lipolytica | Bos taurus | Comment |
|---|---|---|---|
| central subunits peripheral arm | |||
| NDUFS1 | NUAM | 75-kDa | 2x Fe4S4; 1x Fe2S2 |
| NDUFV1 | NUBM | 51-kDa | FMN; NADH; 1x Fe4S4 |
| NDUFS2 | NUCM | 49-kDa | Q-binding |
| NDUFS3 | NUGM | 30-kDa | |
| NDUFV2 | NUHM | 24-kDa | 1x Fe2S2 |
| NDUFS8 | NUIM | TYKY | 2x Fe4S4 |
| NDUFS7 | NUKM | PSST | Q-binding; 1x Fe4S4 |
| accessory subunits peripheral arm | |||
| NDUFA2 | NI8M | B8 | |
| NDUFS4 | NUYM | 18-kDa/AQDQ | |
| NDUFS6 | NUMM | 13-kDa | Zn2+ |
| NDUFA12 | N7BM | B17.2 | paralog of assembly factor NDUFAF2 |
| NDUFA7 | NUZM | B14.5a | |
| NDUFA5 | NUFM | B13 | |
| NDUFA9 | NUEM | 39-kDa | NADPH |
| NDUFA6 | NB4M | B14 | LYRM6 |
| NDUFAB1 | ACPM1 | SDAP | ACPM |
| NDUFV3 | 9-kDa | ||
| ST1 | sulfur transferase | ||
| Subunit | Mutation DNA | Mutation Protein | Disease | Reference |
|---|---|---|---|---|
| NDUFS4 | c.44 G > A | p.Trp15* | Leigh like syndrome | [65] |
| c.44 G > A | no complex I assembly | Leigh like syndrome | [68] | |
| c.99-1 G > A c.462delA | p.Ser34Ilefs*4 p.Lys154Asnfs*34 | Leigh syndrome | [97] | |
| c.221delC | p.Thr74Ilefs*17 | Complex I deficiency | [97] | |
| c.289delG | p.Tyr97* | Leigh like syndrome | [68] | |
| c.291delG | p.Trp97* | Leigh syndrome | [73] | |
| c.316 C > T | p.Arg106* | Leigh like syndrome | [98] | |
| c.340 T > C | p.Trp114Arg | Leigh syndrome | [99] | |
| c.355 G > C c.462delA | p.Asp119His p.Lys154Asnfs*34 | Leigh syndrome | [72] | |
| c.393dupA | p.Glu132Argfs*15 | Leigh syndrome | [70] | |
| c.462delA | p.Lys154Asnfs*34 | Leigh syndrome | [69] | |
| c.466-470 AAGTC duplication | frameshift, elongation of the carboxyl terminus by 14 residues | Leigh like syndrome | [68,75] | |
| NDUFS6 | c.186+2 T > A | splicing abnormality, deletion | Complex I deficiency | [100] |
| c.313_315delAAAG | p.104Lys_106Thrfs | Complex I deficiency | [101] | |
| c.343 C > A c.309 + 5 G > A | p.Cys115Arg | Leigh syndrome | [102] | |
| c.344 G > A | p.Cys115Tyr | lactic acidemia | [103] | |
| NDUFA12 | c.86G > A | p.Arg29Lys | Leigh syndrome | [104] |
| c.178C > T | p.Arg60* | Leigh syndrome | [105] | |
| c.178C > T | p.Arg60* | Mucolipidosis Type II, Leigh syndrome | [106] | |
| c.224G > A | p.Trp75* | Leigh syndrome | [104] | |
| c.253G > T | p.Glu85* | Leigh syndrome | [104] | |
| c.395delA | p.Lys132Argfs*50 | Leigh syndrome | [104] | |
| NDUFA2 | c.1A > T | p.M1L | hypotonia, nystagmus, ataxia, acute episodes of encephalopathy | [107] |
| c.9G > A | p.Trp3* | Leigh syndrome | [108] | |
| c.103delA | p.Ile35Serfs* | Leigh syndrome | [97] | |
| c.114C > G | p.Y38* | Leigh syndrome | [109] | |
| c.182C > T | p.R45* | progressive encephalopathy | [110] | |
| c.221G > A | p.Trp74* | Leigh syndrome | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahlhöfer, F.; Gansen, M.; Zickermann, V. Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly. Life 2021, 11, 455. https://doi.org/10.3390/life11050455
Kahlhöfer F, Gansen M, Zickermann V. Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly. Life. 2021; 11(5):455. https://doi.org/10.3390/life11050455
Chicago/Turabian StyleKahlhöfer, Flora, Max Gansen, and Volker Zickermann. 2021. "Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly" Life 11, no. 5: 455. https://doi.org/10.3390/life11050455
APA StyleKahlhöfer, F., Gansen, M., & Zickermann, V. (2021). Accessory Subunits of the Matrix Arm of Mitochondrial Complex I with a Focus on Subunit NDUFS4 and Its Role in Complex I Function and Assembly. Life, 11(5), 455. https://doi.org/10.3390/life11050455