
Immunoreactivity of Muscarinic Acetylcholine M2 and Serotonin 5-HT2B Receptors, Norepinephrine Transporter and Kir Channels in a Model of Epilepsy

 ,
, 
 ,
,  , ,
, ,  and
and 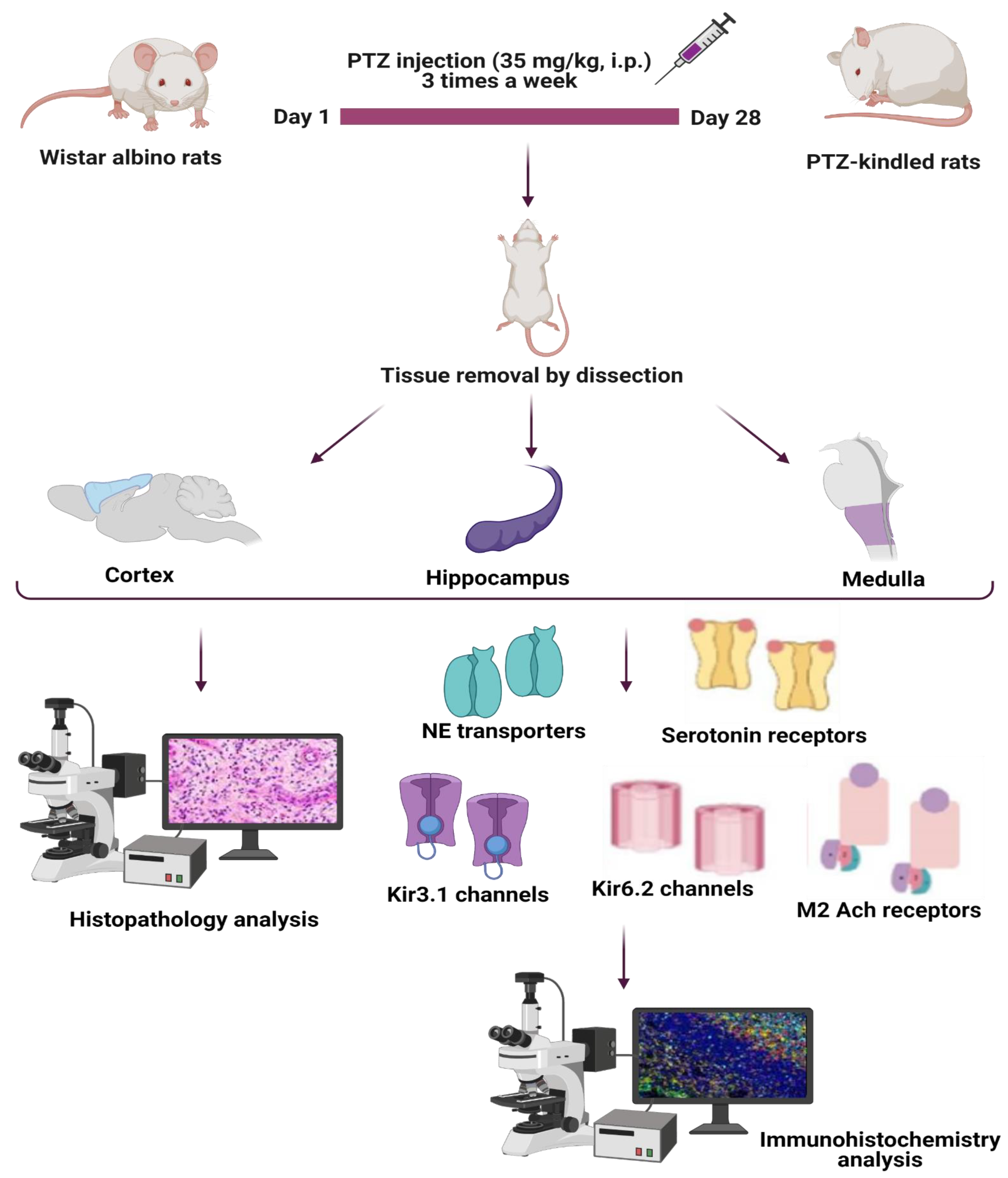{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. PTZ-Kindling Epilepsy Model
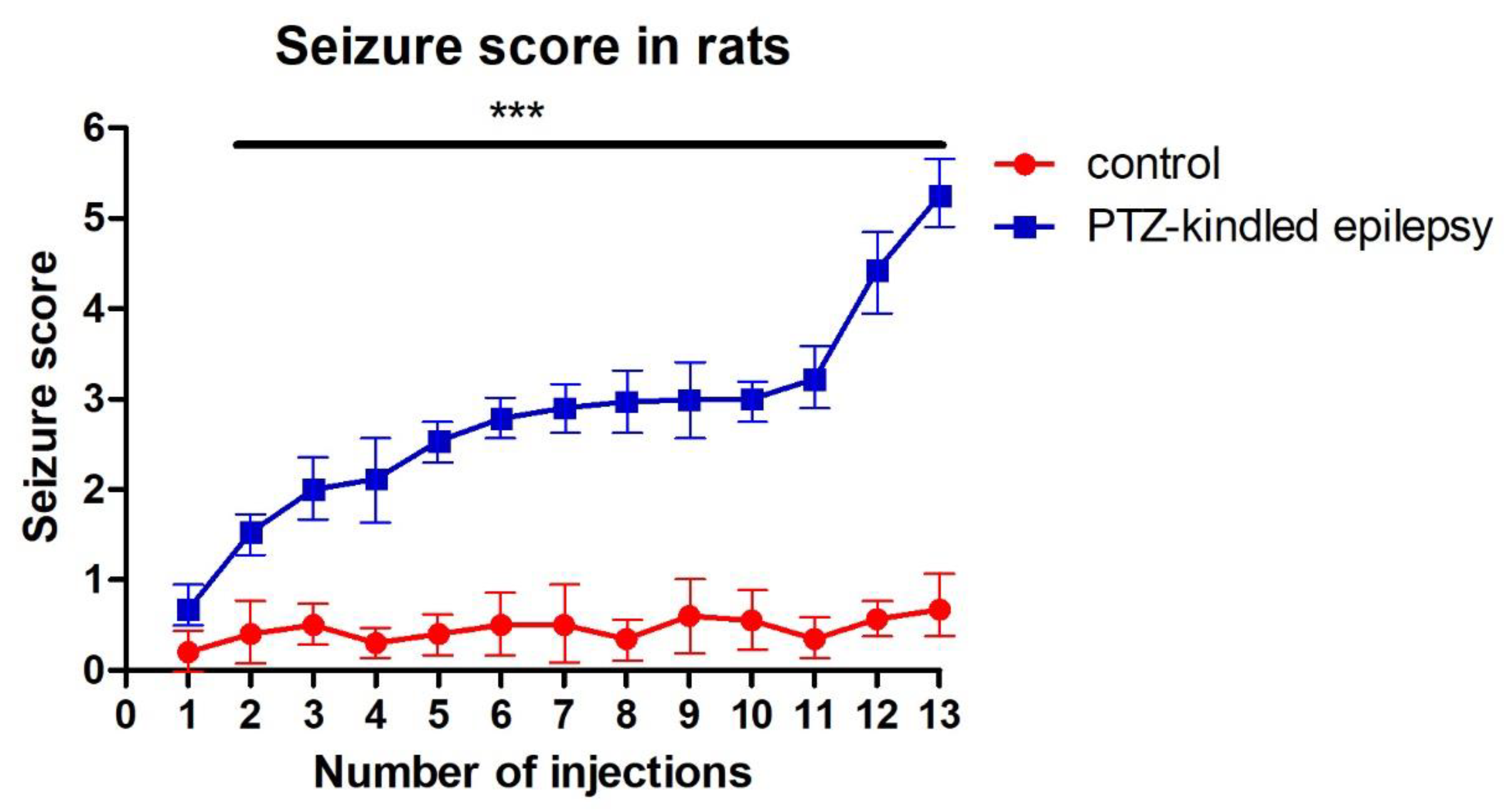
| Score | Criteria |
| Score 0 | No response |
| Score 1 | Facial movements, ear and whisker twitching |
| Score 2 | Myoclonic convulsions without rearing |
| Score 3 | Myoclonic jerks, upright position with clonic forelimb convulsions |
| Score 4 | Clonic-tonic convulsions |
| Score 5 | Generalized clonic-tonic seizures with loss of postural control |
| Score 6 | Death |
2.2. Histological and Pathological Staining
2.3. Statistical Analysis
3. Results
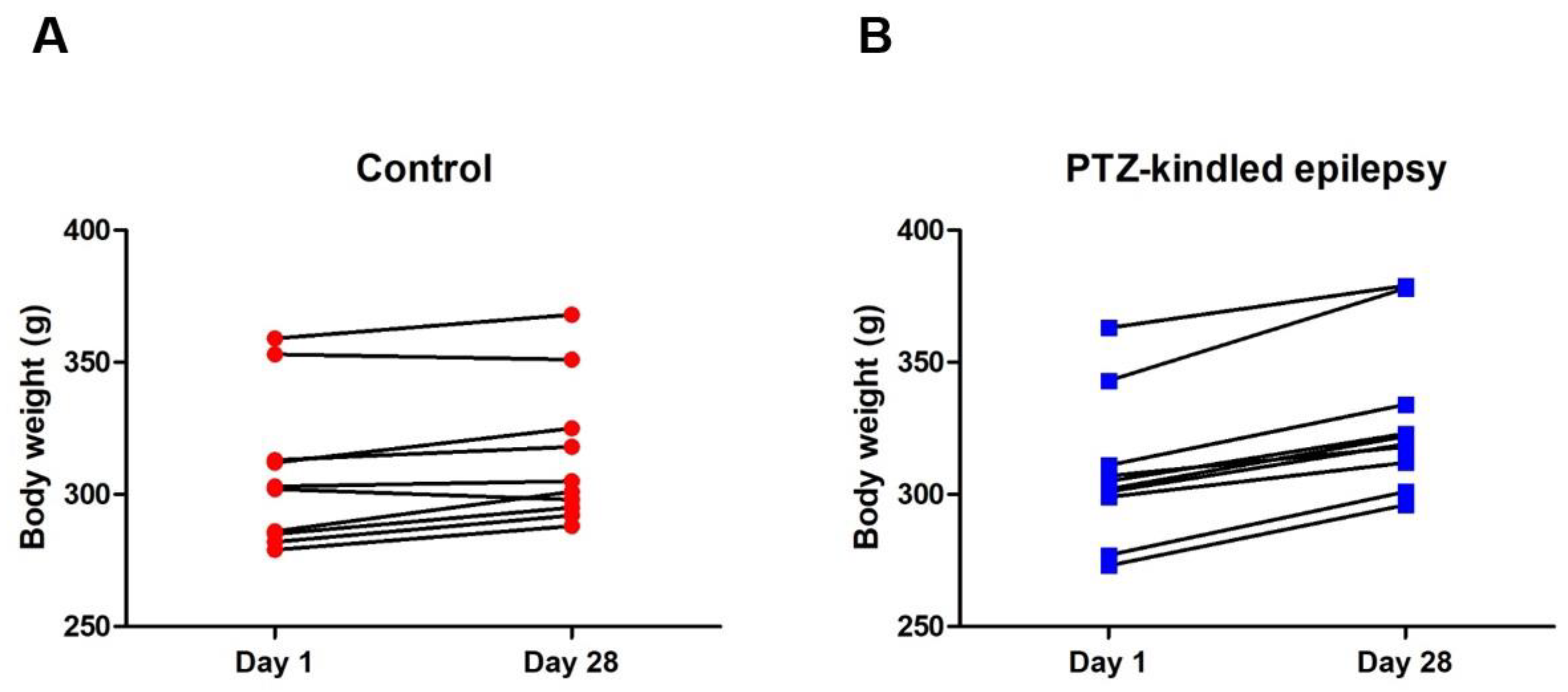
3.1. Racine-Scoring of PTZ-Kindling Model in Animals
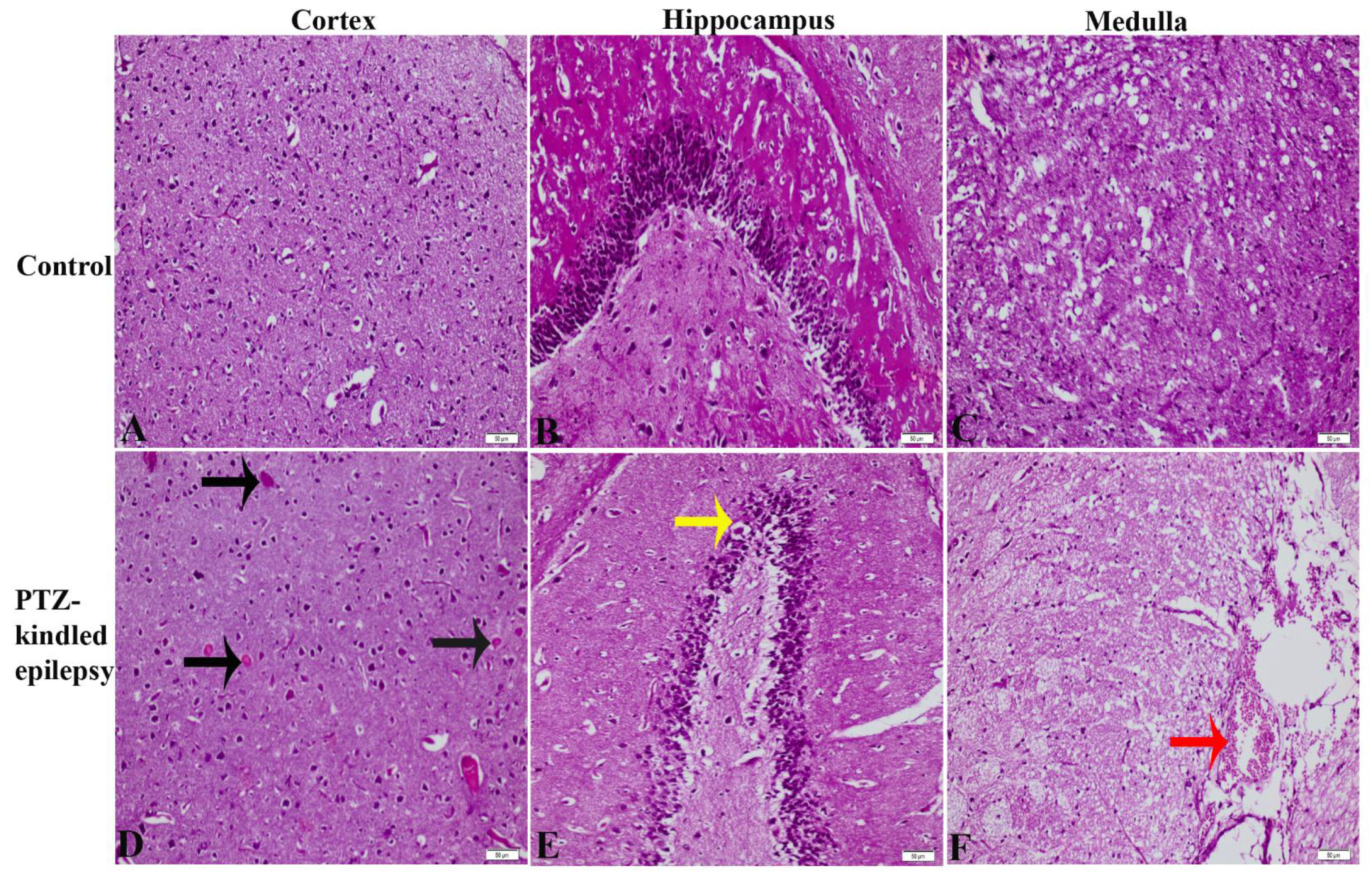
3.2. Gross Histopathology Findings
3.3. Immunohistochemistry Findings
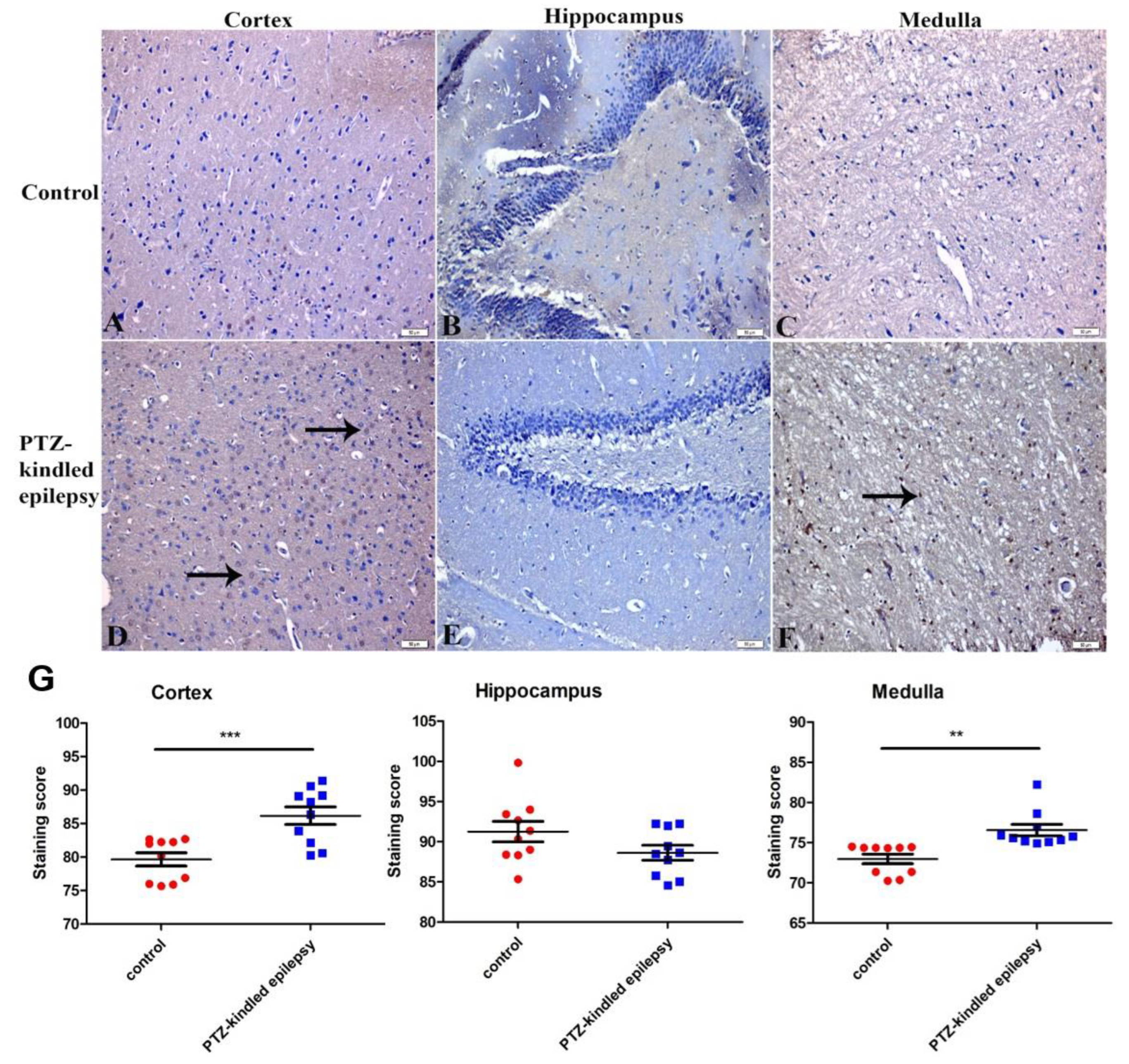
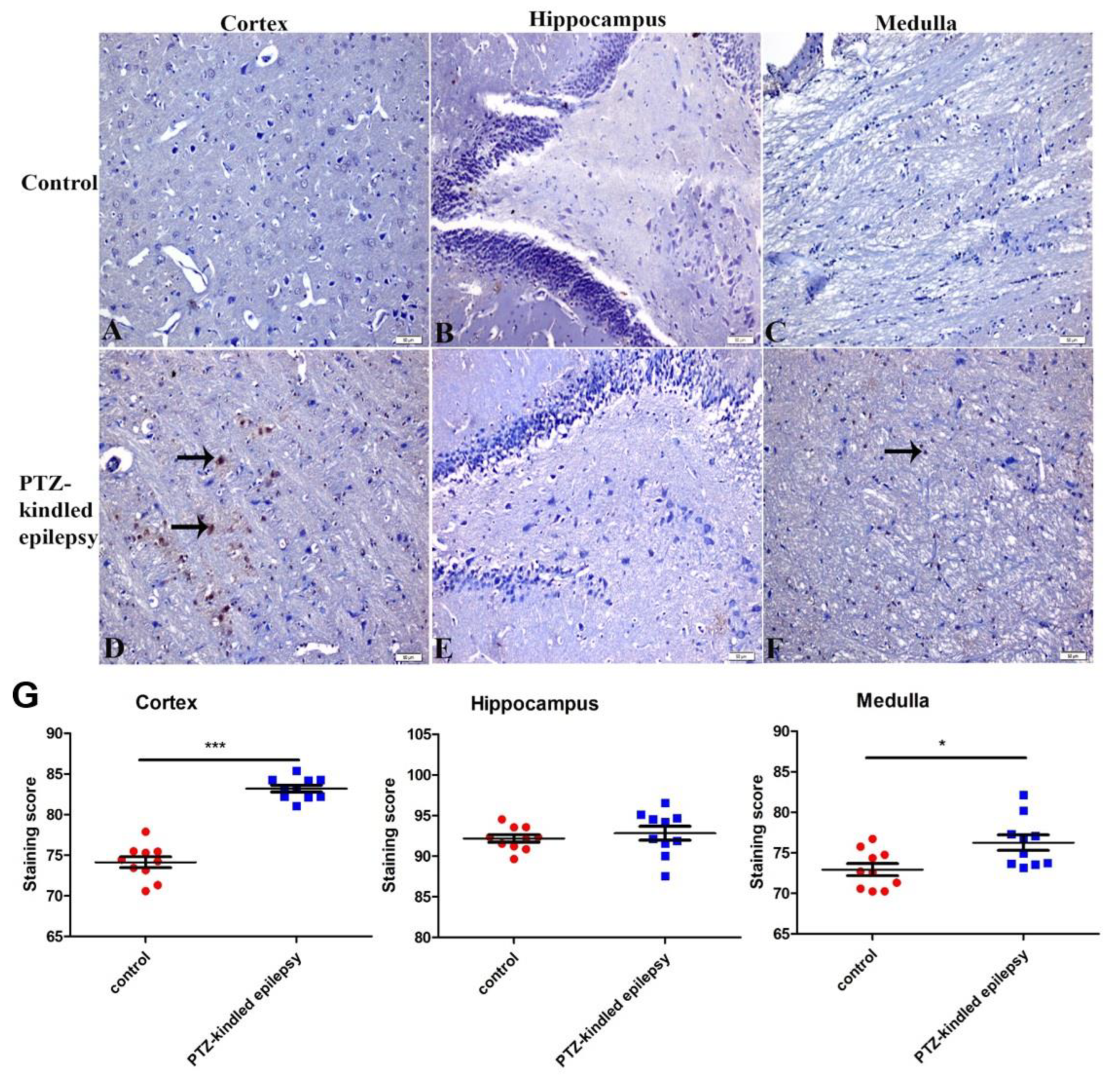
3.3.1. Upregulation of the Muscarinic ACh Receptor M2
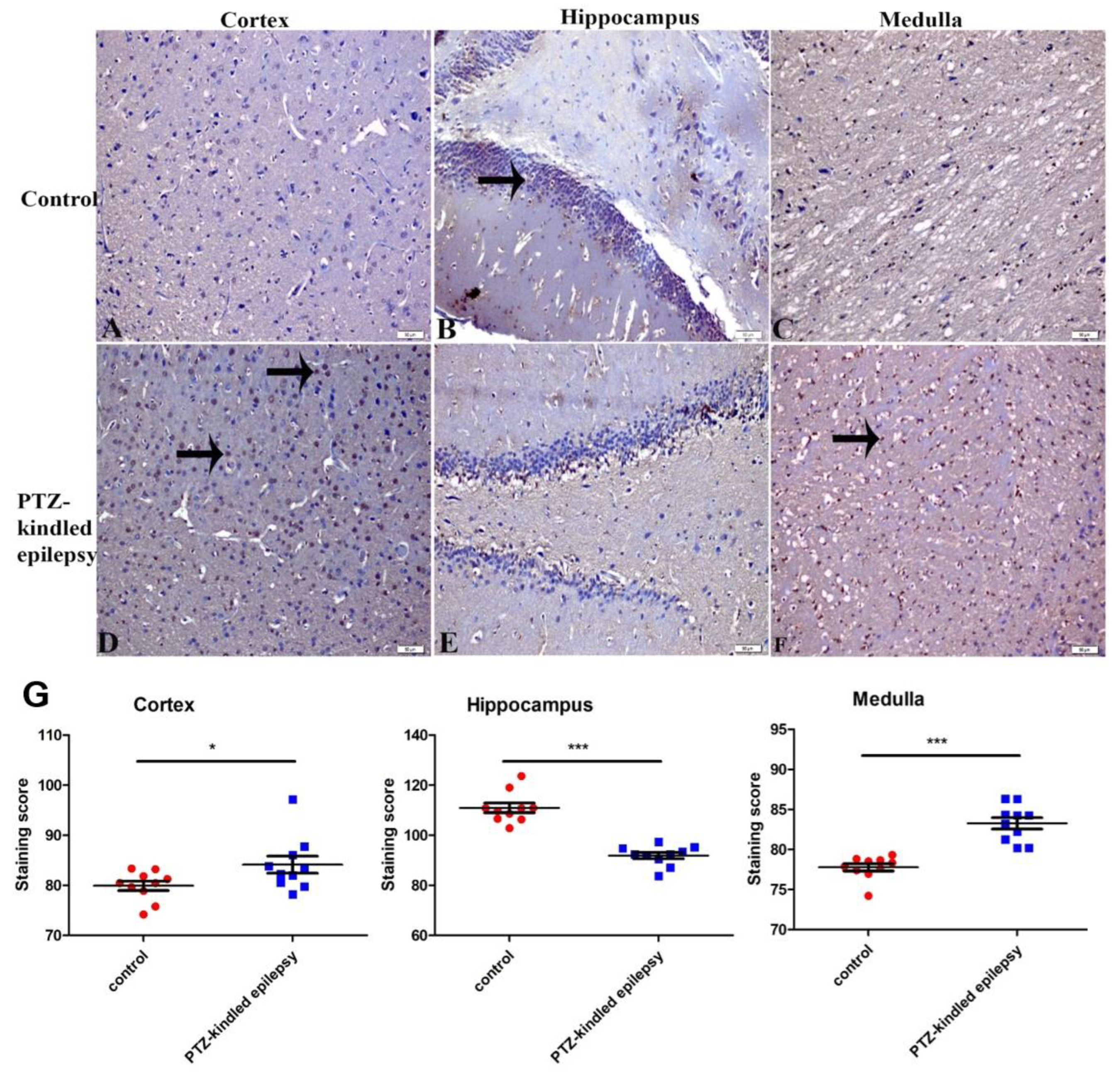
3.3.2. Upregulation of the Serotonin Receptor 2B (5-HT2B)
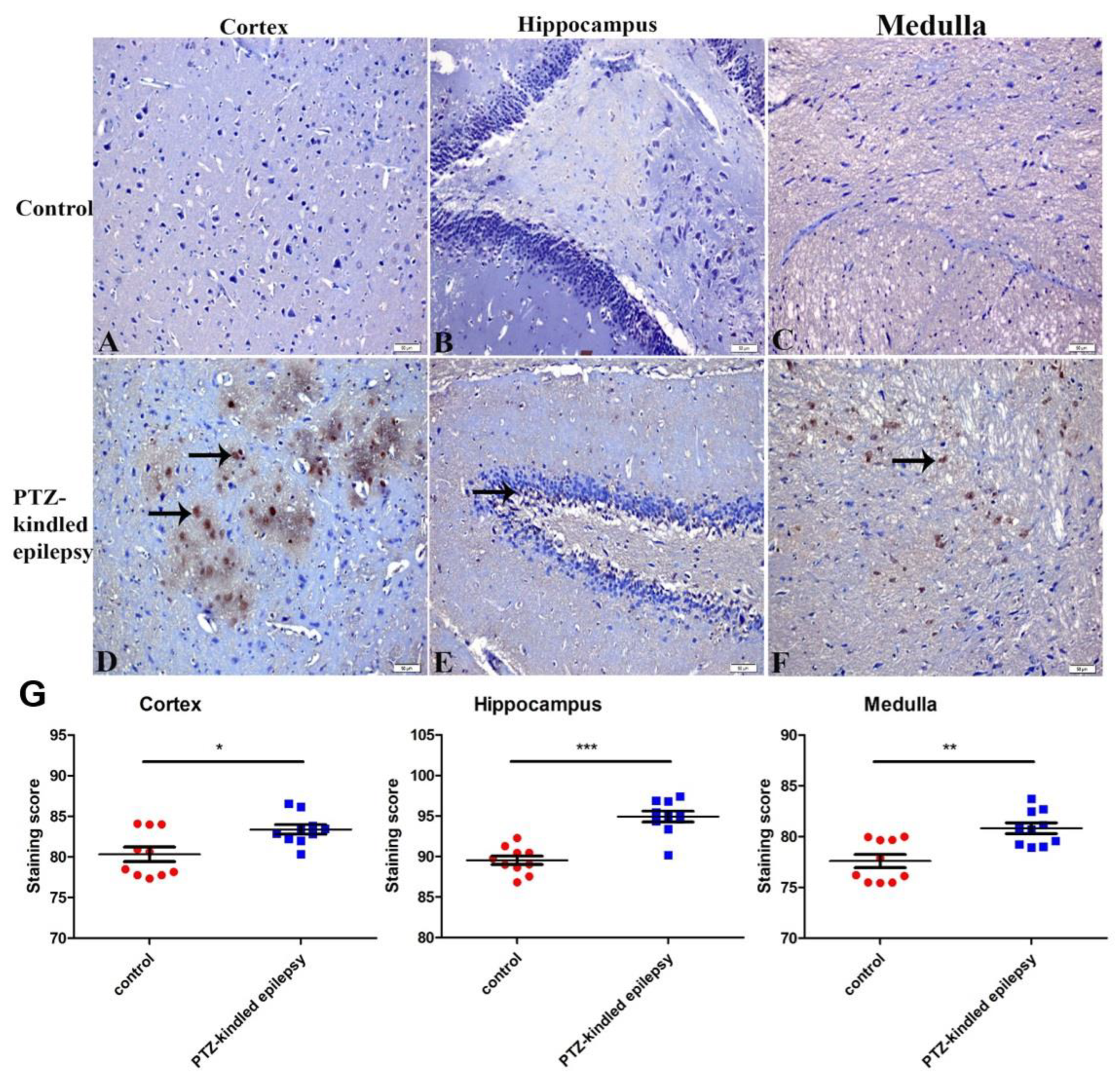
3.3.3. Increased Immunoreactivity of Norepinephrine Transporter
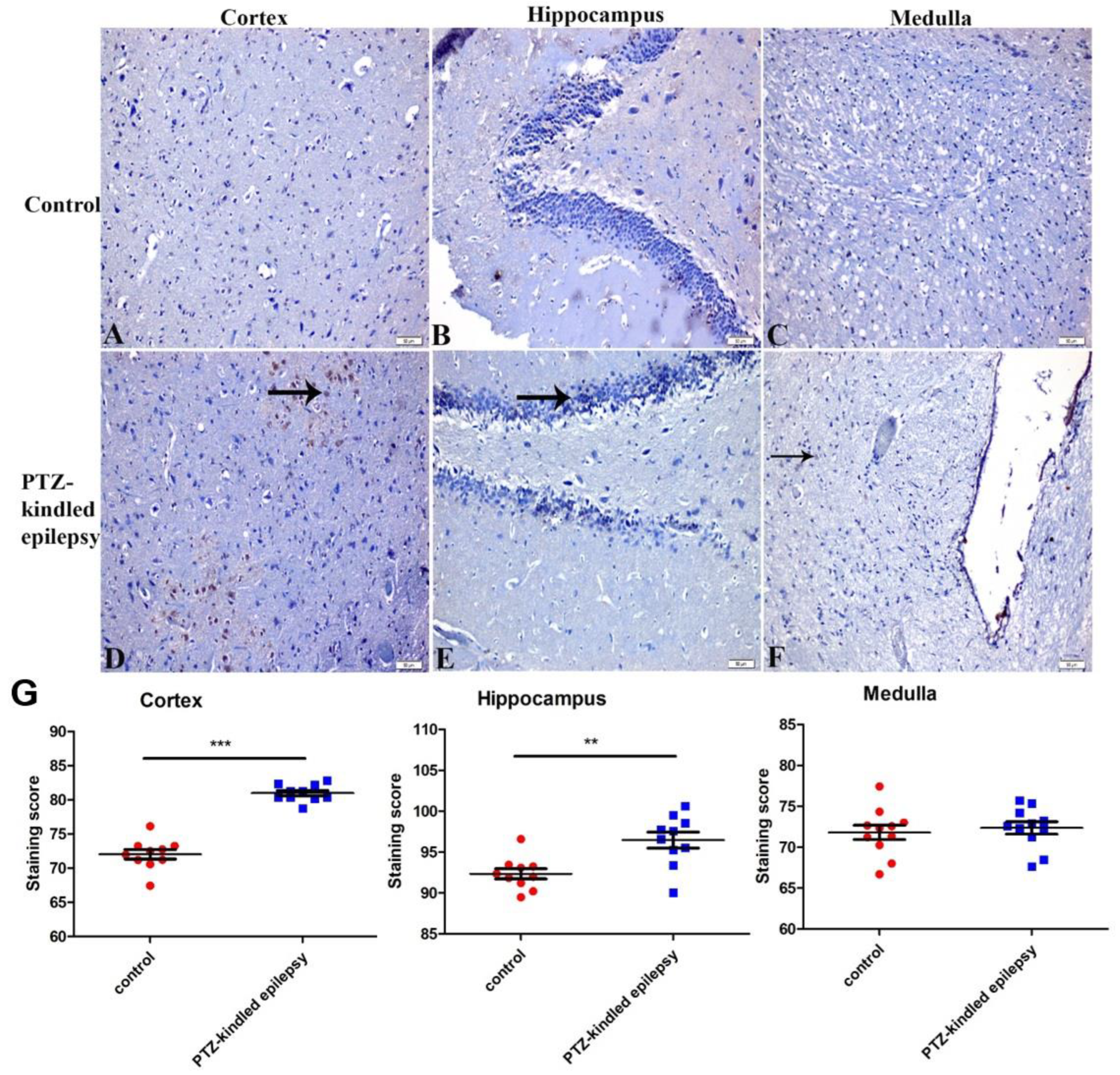
3.3.4. Ach-Activated Kir3.1 Channel and ATP-Dependent Kir6.2 Channel
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Vossler, D.G.; Weingarten, M.; Gidal, B.E.; American Epilepsy Society Treatments Committee. Summary of Antiepileptic Drugs Available in the United States of America: WORKING TOWARD A WORLD WITHOUT EPILEPSY. Epilepsy Curr. 2018, 18, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Dalic, L.; Cook, M.J. Managing drug-resistant epilepsy: Challenges and solutions. Neuropsychiatr. Dis. Treat. 2016, 12, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Fukata, M. Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 2017, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Svob Strac, D.; Pivac, N.; Smolders, I.J.; Fogel, W.A.; De Deurwaerdere, P.; Di Giovanni, G. Monoaminergic mechanisms in epilepsy may offer innovative therapeutic opportunity for monoaminergic multi-target drugs. Front. Neurosci. 2016, 10, 492. [Google Scholar] [CrossRef]
- Lascano, A.M.; Korff, C.M.; Picard, F. Seizures and epilepsies due to channelopathies and neurotransmitter receptor dysfunction: A parallel between genetic and immune aspects. Mol. Syndromol. 2016, 7, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Schousboe, A. Glutamate/Gaba-Glutamine Cycle; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Deransart, C.; Riban, V.; Le, B.; Marescaux, C.; Depaulis, A. Dopamine in the striatum modulates seizures in a genetic model of absence epilepsy in the rat. Neuroscience 2000, 100, 335–344. [Google Scholar] [CrossRef]
- Celada, P.; Puig, M.V.; Artigas, F. Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 2013, 7, 25. [Google Scholar] [CrossRef]
- Upton, N.; Stean, T.; Middlemiss, D.; Blackburn, T.; Kennett, G. Studies on the role of 5-HT2C and 5-HT2B receptors in regulating generalised seizure threshold in rodents. Eur. J. Pharmacol. 1998, 359, 33–40. [Google Scholar] [CrossRef]
- Tolete, P.; Knupp, K.; Karlovich, M.; DeCarlo, E.; Bluvstein, J.; Conway, E.; Friedman, D.; Dugan, P.; Devinsky, O. Lorcaserin therapy for severe epilepsy of childhood onset: A case series. Neurology 2018, 91, 837–839. [Google Scholar] [CrossRef]
- Kim, D. Modulation of acetylcholine-activated K+ channel function in rat atrial cells by phosphorylation. J. Physiol. 1991, 437, 133–155. [Google Scholar] [CrossRef]
- Ohno, Y.; Hibino, H.; Lossin, C.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007, 1178, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ten Eick, R.; Nawrath, H.; McDonald, T.F.; Trautwein, W. On the mechanism of the negative inotropic effect of acetylcholine. Pflugers Arch. 1976, 361, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wiser, O.; Qian, X.; Ehlers, M.; Ja, W.W.; Roberts, R.W.; Reuveny, E.; Jan, Y.N.; Jan, L.Y. Modulation of basal and receptor-induced GIRK potassium channel activity and neuronal excitability by the mammalian PINS homolog LGN. Neuron 2006, 50, 561–573. [Google Scholar] [CrossRef]
- Downes, G.B.; Granato, M. Acetylcholinesterase function is dispensable for sensory neurite growth but is critical for neuromuscular synapse stability. Dev. Biol. 2004, 270, 232–245. [Google Scholar] [CrossRef]
- Sun, H.S.; Feng, Z.P. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol. Sin. 2013, 34, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.R.; Chang, J.; Ding, J.H.; Fan, Y.; Sun, X.L.; Hu, G. Kir6.2 knockout alters neurotransmitter release in mouse striatum: An in vivo microdialysis study. Neurosci. Lett. 2008, 439, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Bateman, L.M.; Laxer, K.D. Evidence for brainstem network disruption in temporal lobe epilepsy and sudden unexplained death in epilepsy. Neuroimage Clin. 2014, 5, 208–216. [Google Scholar] [CrossRef]
- Hattiangady, B.; Shetty, A.K. Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia 2008, 49 (Suppl. 5), 26–41. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Akyuz, E.; Polat, K.; Ates, S.; Unalmis, D.; Tokpinar, A.; Yilmaz, S.; Kaymak, E.; Doganyigit, Z.; Villa, C. Investigating Cardiac Morphological Alterations in a Pentylenetetrazol-Kindling Model of Epilepsy. Diagnostics 2020, 10, 388. [Google Scholar] [CrossRef]
- Popova, I.; Malkov, A.; Ivanov, A.I.; Samokhina, E.; Buldakova, S.; Gubkina, O.; Osypov, A.; Muhammadiev, R.S.; Zilberter, T.; Molchanov, M.; et al. Metabolic correction by pyruvate halts acquired epilepsy in multiple rodent models. Neurobiol. Dis. 2017, 106, 244–254. [Google Scholar] [CrossRef]
- Akyuz, E.; Doganyigit, Z.; Paudel, Y.N.; Kaymak, E.; Yilmaz, S.; Uner, A.; Shaikh, M.F. Increased ACh-Associated Immunoreactivity in Autonomic Centers in PTZ Kindling Model of Epilepsy. Biomedicines 2020, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A.; Aracri, P.; Meneghini, S.; Brusco, S.; Amadeo, A. The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 2015, 6, 22. [Google Scholar] [CrossRef]
- Nirwan, N.; Vyas, P.; Vohora, D. Animal models of status epilepticus and temporal lobe epilepsy: A narrative review. Rev. Neurosci. 2018, 29, 757–770. [Google Scholar] [CrossRef]
- López-Meraz, M.-L.; González-Trujano, M.-E.; Neri-Bazán, L.; Hong, E.; Rocha, L.L. 5-HT1A receptor agonists modify epileptic seizures in three experimental models in rats. Neuropharmacology 2005, 49, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Taskiran, M.; Tasdemir, A.; Ayyildiz, N.; Ayyildiz, M.; Agar, E. The effect of serotonin on penicillin-induced epileptiform activity. Int. J. Neurosci. 2019, 129, 687–697. [Google Scholar] [CrossRef]
- Murugesan, A.; Rani, M.R.S.; Vilella, L.; Lacuey, N.; Hampson, J.P.; Faingold, C.L.; Friedman, D.; Devinsky, O.; Sainju, R.K.; Schuele, S.; et al. Postictal serotonin levels are associated with peri-ictal apnea. Neurology 2019, 93, e1485–e1494. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, N.C.; Joaquim, H.P.; Talib, L.L.; de Vincentiis, S.; Gattaz, W.F.; Valente, K.D. Hippocampal serotonin depletion is related to the presence of generalized tonic–clonic seizures, but not to psychiatric disorders in patients with temporal lobe epilepsy. Epilepsy Res. 2015, 111, 18–25. [Google Scholar] [CrossRef]
- Venzi, M.; David, F.; Bellet, J.; Cavaccini, A.; Bombardi, C.; Crunelli, V.; Di Giovanni, G. Role for serotonin2A (5-HT2A) and 2C (5-HT2C) receptors in experimental absence seizures. Neuropharmacology 2016, 108, 292–304. [Google Scholar] [CrossRef]
- Goral, I.; Latka, K.; Bajda, M. Structure Modeling of the Norepinephrine Transporter. Biomolecules 2020, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Pizzanelli, C.; Biagioni, F.; Murri, L.; Fornai, F. The role of norepinephrine in epilepsy: From the bench to the bedside. Neurosci. Biobehav. Rev. 2004, 28, 507–524. [Google Scholar] [CrossRef]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2019, 12, 334. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Sun, Z.; Zhang, J.; Gao, N.; Qi, F.; Che, F.; Ma, G. Diazoxide preconditioning antagonizes cytotoxicity induced by epileptic seizures. Neural Regen. Res. 2013, 8, 1000–1006. [Google Scholar]
- Yamada, K.; Ji, J.J.; Yuan, H.; Miki, T.; Sato, S.; Horimoto, N.; Shimizu, T.; Seino, S.; Inagaki, N. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science 2001, 292, 1543–1546. [Google Scholar] [CrossRef]
- D’Adamo, M.C.; Catacuzzeno, L.; Di Giovanni, G.; Franciolini, F.; Pessia, M. K(+) channelepsy: Progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 2013, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Barot, N.; Nei, M. Autonomic aspects of sudden unexpected death in epilepsy (SUDEP). Clin. Auton. Res. 2019, 29, 151–160. [Google Scholar] [CrossRef]
- Patodia, S.; Somani, A.; O’Hare, M.; Venkateswaran, R.; Liu, J.; Michalak, Z.; Ellis, M.; Scheffer, I.E.; Diehl, B.; Sisodiya, S.M.; et al. The ventrolateral medulla and medullary raphe in sudden unexpected death in epilepsy. Brain 2018, 141, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Richerson, G.B.; Buchanan, G.F. The serotonin axis: Shared mechanisms in seizures, depression, and SUDEP. Epilepsia 2011, 52 (Suppl. 1), 28–38. [Google Scholar] [CrossRef]
- Allen, L.A.; Harper, R.M.; Lhatoo, S.; Lemieux, L.; Diehl, B. Neuroimaging of Sudden Unexpected Death in Epilepsy (SUDEP): Insights From Structural and Resting-State Functional MRI Studies. Front. Neurol. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Patodia, S.; Paradiso, B.; Ellis, M.; Somani, A.; Sisodiya, S.M.; Devinsky, O.; Thom, M. Characterisation of medullary astrocytic populations in respiratory nuclei and alterations in sudden unexpected death in epilepsy. Epilepsy Res. 2019, 157, 106213. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.; Mironov, S.L.; Karschin, A.; Richter, D.W. Dynamic activation of K(ATP) channels in rhythmically active neurons. J. Physiol. 2001, 537, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Donaire, A.; Carreno, M.; Gomez, B.; Fossas, P.; Bargallo, N.; Agudo, R.; Falip, M.; Setoain, X.; Boget, T.; Raspall, T.; et al. Cortical laminar necrosis related to prolonged focal status epilepticus. J. Neurol. Neurosurg. Psychiatry 2006, 77, 104–106. [Google Scholar] [CrossRef]
- Samokhina, E.; Samokhin, A. Neuropathological profile of the pentylenetetrazol (PTZ) kindling model. Int. J. Neurosci. 2018, 128, 1086–1096. [Google Scholar] [CrossRef]
- Ali, S.O.; Shahin, N.N.; Safar, M.M.; Rizk, S.M. Therapeutic potential of endothelial progenitor cells in a rat model of epilepsy: Role of autophagy. J. Adv. Res. 2019, 18, 101–112. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akyuz, E.; Doganyigit, Z.; Paudel, Y.N.; Koklu, B.; Kaymak, E.; Villa, C.; Arulsamy, A.; Shaikh, M.F.; Devinsky, O. Immunoreactivity of Muscarinic Acetylcholine M2 and Serotonin 5-HT2B Receptors, Norepinephrine Transporter and Kir Channels in a Model of Epilepsy. Life 2021, 11, 276. https://doi.org/10.3390/life11040276
Akyuz E, Doganyigit Z, Paudel YN, Koklu B, Kaymak E, Villa C, Arulsamy A, Shaikh MF, Devinsky O. Immunoreactivity of Muscarinic Acetylcholine M2 and Serotonin 5-HT2B Receptors, Norepinephrine Transporter and Kir Channels in a Model of Epilepsy. Life. 2021; 11(4):276. https://doi.org/10.3390/life11040276
Chicago/Turabian StyleAkyuz, Enes, Zuleyha Doganyigit, Yam Nath Paudel, Betul Koklu, Emin Kaymak, Chiara Villa, Alina Arulsamy, Mohd. Farooq Shaikh, and Orrin Devinsky. 2021. "Immunoreactivity of Muscarinic Acetylcholine M2 and Serotonin 5-HT2B Receptors, Norepinephrine Transporter and Kir Channels in a Model of Epilepsy" Life 11, no. 4: 276. https://doi.org/10.3390/life11040276
APA StyleAkyuz, E., Doganyigit, Z., Paudel, Y. N., Koklu, B., Kaymak, E., Villa, C., Arulsamy, A., Shaikh, M. F., & Devinsky, O. (2021). Immunoreactivity of Muscarinic Acetylcholine M2 and Serotonin 5-HT2B Receptors, Norepinephrine Transporter and Kir Channels in a Model of Epilepsy. Life, 11(4), 276. https://doi.org/10.3390/life11040276