
Comparative Genomics Analysis of Repetitive Elements in Ten Gymnosperm Species: “Dark Repeatome” and Its Abundance in Conifer and Gnetum Species

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequencing Data
2.2. Bioinformatics Analysis
2.3. Read Cleaning and Quality Assurance
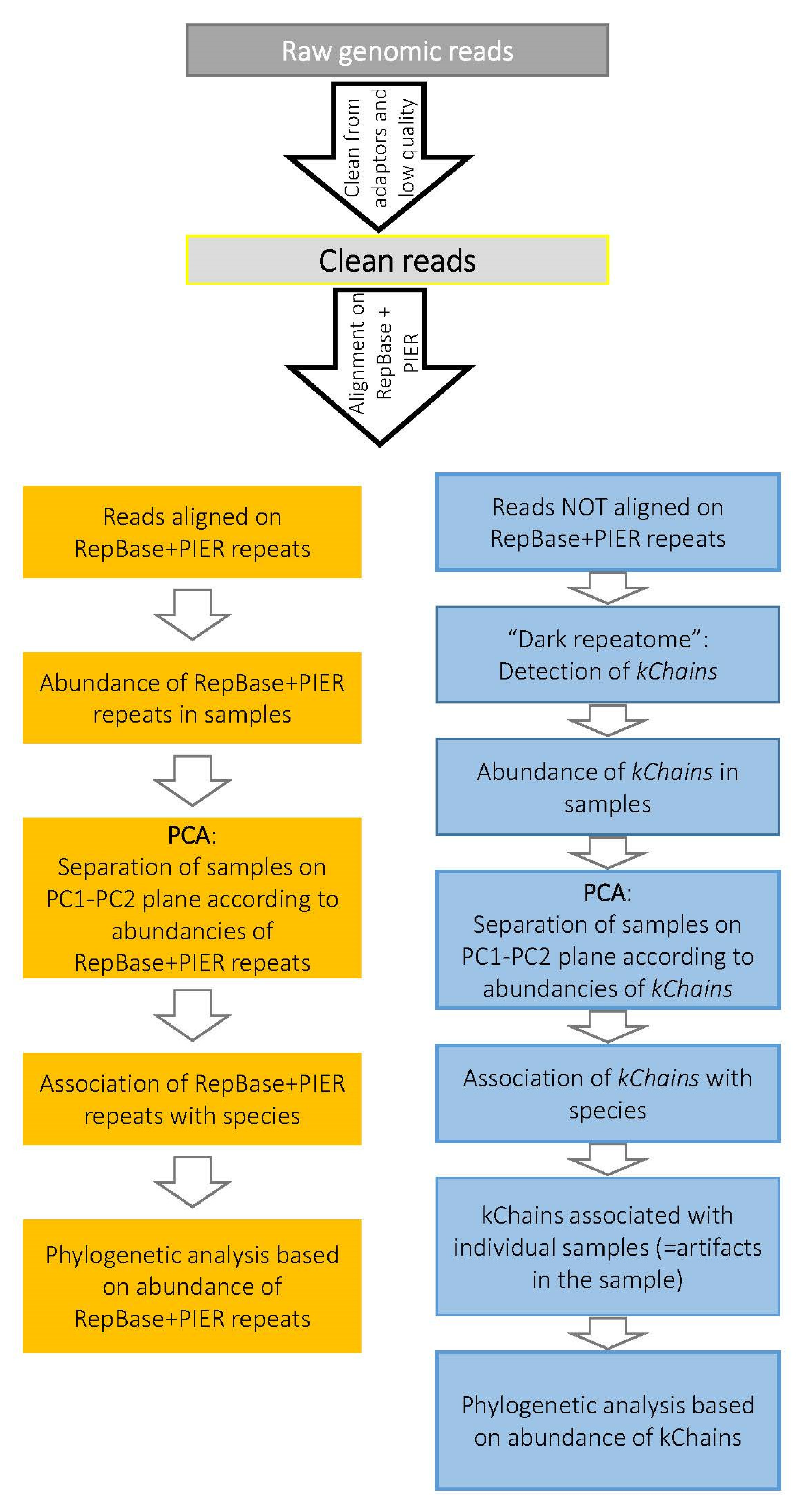
2.4. Alignment of Genomics Reads onto Repeat Sequences: RE Abundance Calculation
2.5. Putative Repetitive Elements—kChains
2.6. Principal Component Analysis (PCA)
2.7. Phylogenetic Analysis Based on Repeat Abundance
3. Results
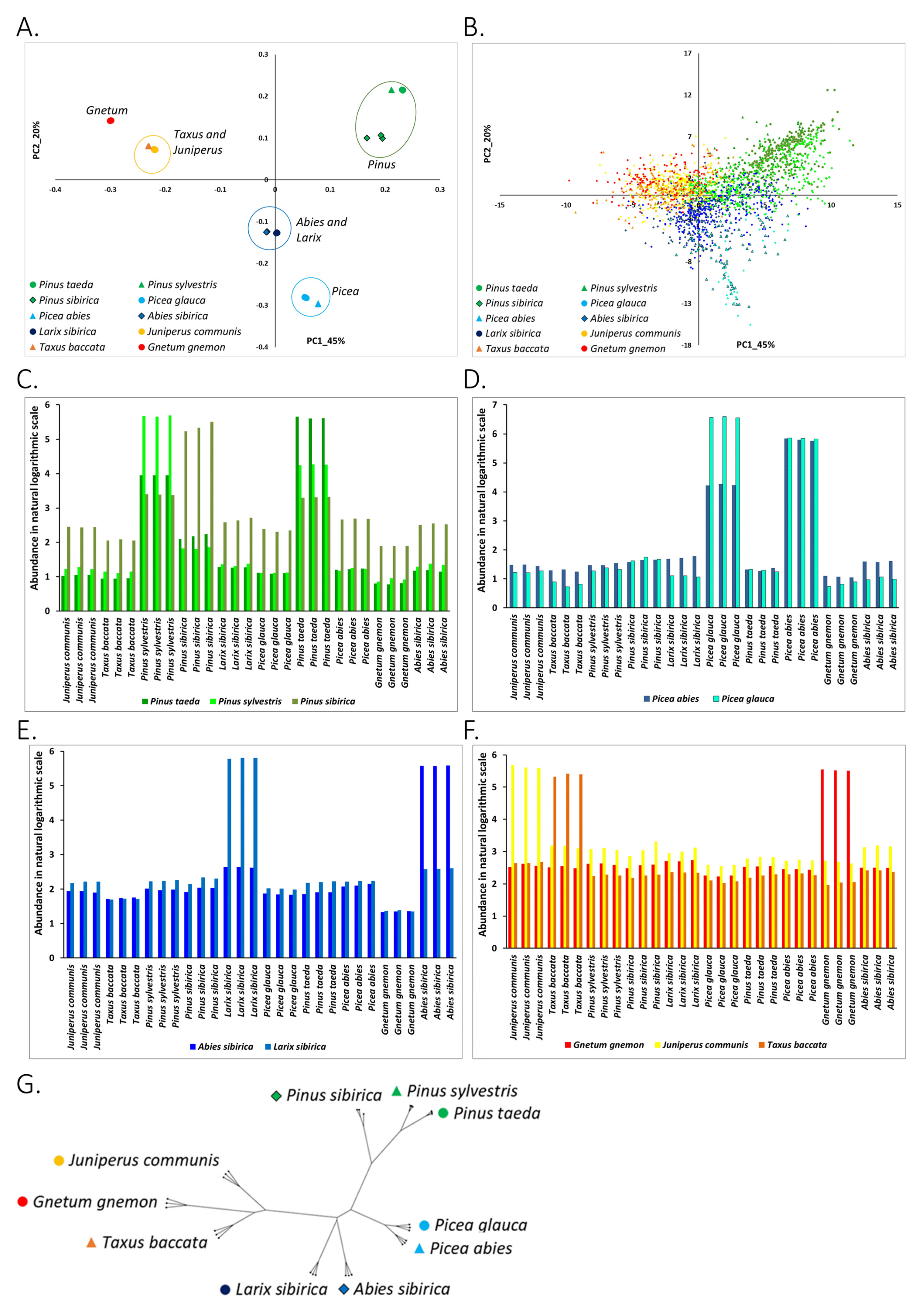
3.1. Abundance of Known RE in Conifers and Gnetum Species
3.1.1. Clustering of Species on the PC1-PC2 Plane Agrees with the Taxonomy
3.1.2. Association of Types of Known RE with the Studied Plants
3.1.3. Phylogenetic Analysis of Conifer and Gnetum Species Based on the Genomic Abundance of Known RE
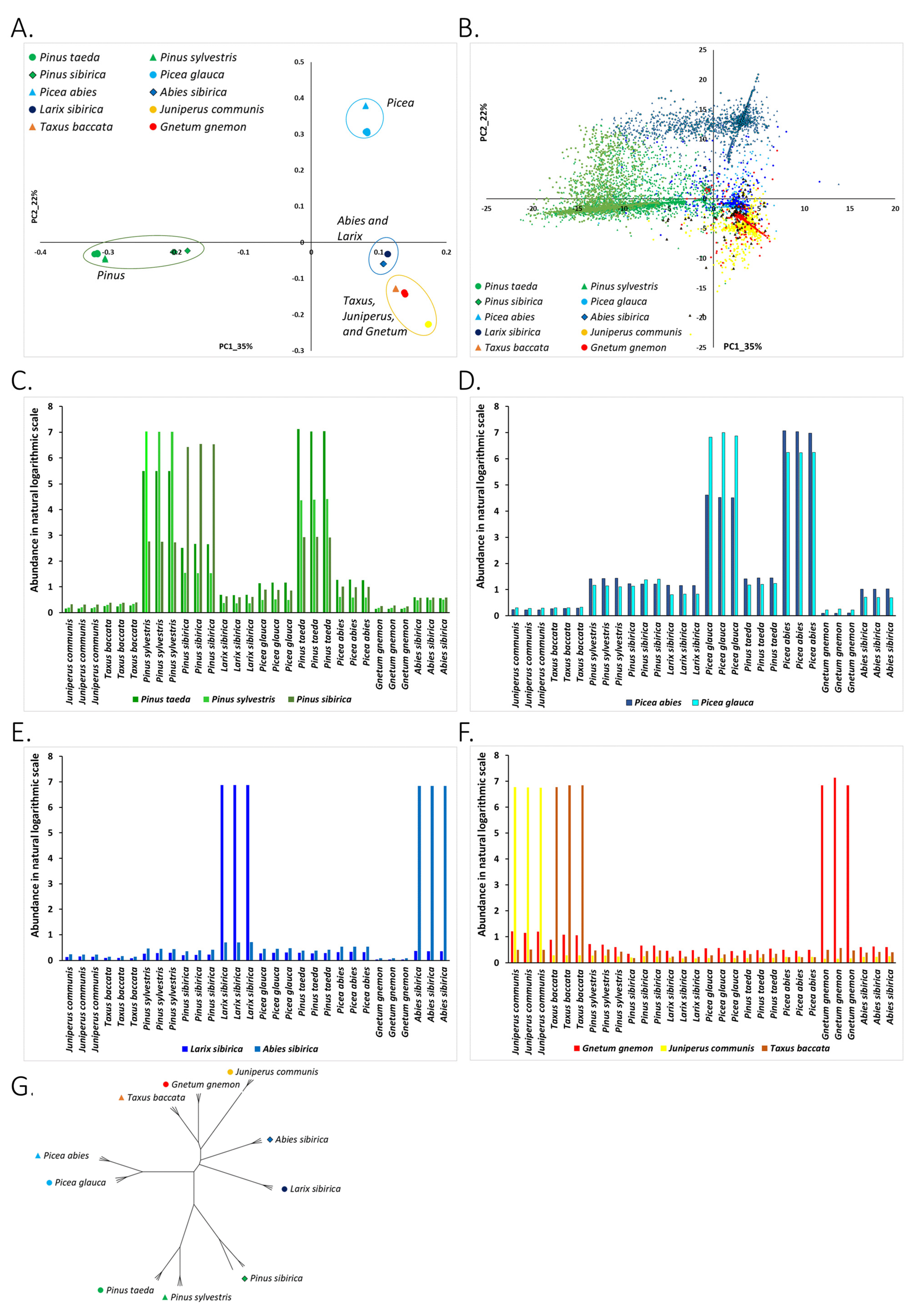
3.2. “Dark Repeatome”: Its Abundance in Genomes of Studied Plants
3.2.1. Clustering of Species on the PC1-PC2 Plane and Association of Species with Nuclear DNA kChains
3.2.2. Phylogenetic Analysis of Conifer and Gnetum Species Based on The Genomic Abundance of Putative Repetitive Elements (kChains)
3.2.3. Analysis of kChains Associated with Chloroplast Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birol, I.; Raymond, A.; Jackman, S.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.; Brand, D.; Vandervalk, B.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Leitch, I.J. The Plant DNA C-values database (release 7.1): An updated online repository of plant genome size data for comparative studies. New Phytol. 2020, 226, 301–305. [Google Scholar] [CrossRef]
- Kuzmin, D.; Feranchuk, S.I.; Sharov, V.; Cybin, A.N.; Makolov, S.V.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Stepwise large genome assembly approach: A case of Siberian larch (Larix sibirica Ledeb). BMC Bioinform. 2019, 20, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Mosca, E.; Cruz, F.; Gómez-Garrido, J.; Bianco, L.; Rellstab, C.; Brodbeck, S.; Csilléry, K.; Fady, B.; Fladung, M.; Fussi, B.; et al. A Reference Genome Sequence for the European Silver Fir (Abies alba Mill.): A Community-Generated Genomic Resource. G3: Genes Genomes Genet. 2019, 9, 2039–2049. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Lin, B.Y.; Zieve, J.J.; Dougherty, W.M.; Martínez-García, P.J.; Koriabine, M.; Holtz-Morris, A.; DeJong, P.; Crepeau, M.; Langley, C.H.; et al. Insights into the Loblolly Pine Genome: Characterization of BAC and Fosmid Sequences. PLoS ONE 2013, 8, e72439. [Google Scholar] [CrossRef]
- Zimin, A.; Stevens, K.A.; Crepeau, M.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.; de Jong, P.J.; et al. Sequencing and Assembly of the 22-Gb Loblolly Pine Genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef]
- Li, Z.; Baniaga, A.E.; Sessa, E.B.; Scascitelli, M.; Graham, S.W.; Rieseberg, L.H.; Barker, M.S. Early genome duplications in conifers and other seed plants. Sci. Adv. 2015, 1, e1501084. [Google Scholar] [CrossRef]
- Krutovsky, K.V.; Oreshkova, N.V.; Putintseva, Y.A.; Ibe, A.A.; Deich, K.O.; Shilkina, E.A. Preliminary results of de novo whole genome sequencing of Siberian larch (Larix sibirica Ledeb.) and Siberian stone pine (Pinus sibirica Du Tour.). Siberian J. For. Sci. 2014, 1, 79–83, (In Russian with English abstract). [Google Scholar]
- Warren, R.; Keeling, C.; Yuen, M.M.S.; Raymond, A.; Taylor, G.A.; Vandervalk, B.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Sundaramurthi, J.C.; Lee, J.; Kandimalla, M.A.; Chen, I.-M.; Kyrpides, N.C.; Reddy, T.B.K. Genomes OnLine Database (GOLD) v.8: Overview and updates. Nucleic Acids Res. 2020, 49, D723–D733. [Google Scholar] [CrossRef]
- Williams, C.G.; Savolainen, O. Inbreeding depression in conifers: Implications for breeding strategy. For. Sci. 1996, 42, 102–117. [Google Scholar] [CrossRef]
- González-Martínez, S.C.; Dillon, S.; Garnier-Géré, P.H.; Krutovsky, K.V.; Alía, R.; Burgarella, C.; Eckert, A.J.; García-Gil, M.R.; Grivet, D.; Heuetz, M.; et al. Patterns of nucleotide diversity and association mapping. In Genetics, Genomics and Breeding of Conifers; Plomion, C., Bousquet, J., Kole, C., Eds.; CRC Press, Science Publishers, Inc.: Enfield, NH, USA, 2011; pp. 239–275. [Google Scholar]
- Ritland, K.; Krutovsky, K.V.; Tsumura, Y.; Pelgas, B.; Isabel, N.; Bousquet, J. Genetic mapping in conifers. In Genetics, Genomics and Breeding of Conifers; Plomion, C., Bousquet, J., Kole, C., Eds.; CRC Press, Science Publishers, Inc.: Enfield, NH, USA, 2011; pp. 196–238. [Google Scholar]
- Krutovsky, K.; Neale, D.B. Nucleotide Diversity and Linkage Disequilibrium in Cold-Hardiness- and Wood Quality-Related Candidate Genes in Douglas Fir. Genetics 2005, 171, 2029–2041. [Google Scholar] [CrossRef]
- Kinlaw, C.S.; Neale, D.B. Complex gene families in pine genomes. Trends Plant Sci. 1997, 2, 356–359. [Google Scholar] [CrossRef]
- Rigault, P.; Boyle, B.; Lepage, P.; Cooke, J.; Bousquet, J.; MacKay, J. A White Spruce Gene Catalog for Conifer Genome Analyses. Plant Physiol. 2011, 157, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.-S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martínez-García, P.J.; et al. Unique Features of the Loblolly Pine (Pinus taeda L.) Megagenome Revealed Through Sequence Annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Belyayev, A.; Kalendar, R.; Brodsky, L.; Nevo, E.; Schulman, A.H.; Raskina, O. Transposable elements in a marginal plant population: Temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob. DNA 2010, 1, 6. [Google Scholar] [CrossRef]
- Grandbastien, M.-A.; Audeon, C.; Bonnivard, E.; Casacuberta, J.; Chalhoub, B.; Costa, A.-P.; Le, Q.; Melayah, D.; Petit, M.; Poncet, C.; et al. Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 2005, 110, 229–241. [Google Scholar] [CrossRef]
- Kidwell, M.G.; Lisch, D. Transposons unbound. Nature 1998, 393, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G.; Lisch, D.R. Transposable elements and host genome evolution. Trends Ecol. Evol. 2000, 15, 95–99. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant Retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Kunze, R.; Saedler, H.; Lonnig, W. Plant Transposable Elements. Adv. Bot. Res. 1997, 27, 331–470. [Google Scholar] [CrossRef]
- Schwilk, D.W.; Ackerly, D.D. Flammability and serotiny as strategies: Correlated evolution in pines. Oikos 2001, 94, 326–336. [Google Scholar] [CrossRef]
- Kellogg, E.A.; Bennetzen, J.L. The evolution of nuclear genome structure in seed plants. Am. J. Bot. 2004, 91, 1709–1725. [Google Scholar] [CrossRef]
- Ugarković, Ð.; Plohl, M. Variation in satellite DNA profiles—Causes and effects. EMBO J. 2002, 21, 5955–5959. [Google Scholar] [CrossRef]
- Ugarković, D. Satellite DNA Libraries and Centromere Evolution. Open Evol. J. 2008, 2, 1–6. [Google Scholar] [CrossRef][Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 1–6. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhang, S.; Song, X.; Weyrich, A.; Wang, Y.; Liu, X.; Wan, N.; Liu, J.; Lövy, M.; Cui, H.; et al. Genome evolution of blind subterranean mole rats: Adaptive peripatric versus sympatric speciation. Proc. Natl. Acad. Sci. USA 2020, 117, 32499–32508. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Lin, C.-P.; Huang, J.-P.; Wu, C.-S.; Hsu, C.-Y.; Chaw, S.-M. Comparative Chloroplast Genomics Reveals the Evolution of Pinaceae Genera and Subfamilies. Genome Biol. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef]
- Uddenberg, D.; Akhter, S.; Ramachandran, P.; Sundstrom, J.F.; Carlsbecker, A. Sequenced genomes and rapidly emerging technologies pave the way for conifer evolutionary developmental biology. Front. Plant Sci. 2015, 6, 1602. [Google Scholar] [CrossRef]
- Dodsworth, S.; Chase, M.W.; Kelly, L.J.; Leitch, I.J.; Macas, J.; Novak, P.; Piednoël, M.; Schneeweiss, H.; Leitch, A.R. Genomic Repeat Abundances Contain Phylogenetic Signal. Syst. Biol. 2015, 64, 112–126. [Google Scholar] [CrossRef]
- Won, H.; Renner, S.S. Dating Dispersal and Radiation in the Gymnosperm Gnetum (Gnetales)—Clock Calibration When Outgroup Relationships Are Uncertain. Syst. Biol. 2006, 55, 610–622. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Li, Z.; Van De Peer, Y.; Ingvarsson, P. Contrasting Rates of Molecular Evolution and Patterns of Selection among Gymnosperms and Flowering Plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef]
- BLAST® Command Line Applications User Manual; National Center for Biotechnology Information: Bethesda, MD, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK569850/ (accessed on 14 September 2021).
- Boratyn, G.M.; Camacho, C.; Cooper, P.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Shahzad, R.; Lubna; Kang, S.M.; Al-Harrasi, A.; Al-Rawahi, A.; Lee, I.-J. Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species. PLoS ONE 2018, 13, e0192966. [Google Scholar] [CrossRef] [PubMed]
- Pyke, K.A. Plastid Division and Development. Plant Cell 1999, 11, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Bioinformatics Bits and Bobs: Why You Should QC Your Reads and Your Assembly. Available online: http://grahametherington.blogspot.com/2014/09/why-you-should-qc-your-reads-and-your.html (accessed on 20 September 2021).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titievsky, A.; Putintseva, Y.A.; Taranenko, E.A.; Baskin, S.; Oreshkova, N.V.; Brodsky, E.; Sharova, A.V.; Sharov, V.V.; Panov, J.; Kuzmin, D.A.; et al. Comparative Genomics Analysis of Repetitive Elements in Ten Gymnosperm Species: “Dark Repeatome” and Its Abundance in Conifer and Gnetum Species. Life 2021, 11, 1234. https://doi.org/10.3390/life11111234
Titievsky A, Putintseva YA, Taranenko EA, Baskin S, Oreshkova NV, Brodsky E, Sharova AV, Sharov VV, Panov J, Kuzmin DA, et al. Comparative Genomics Analysis of Repetitive Elements in Ten Gymnosperm Species: “Dark Repeatome” and Its Abundance in Conifer and Gnetum Species. Life. 2021; 11(11):1234. https://doi.org/10.3390/life11111234
Chicago/Turabian StyleTitievsky, Avi, Yuliya A. Putintseva, Elizaveta A. Taranenko, Sofya Baskin, Natalia V. Oreshkova, Elia Brodsky, Alexandra V. Sharova, Vadim V. Sharov, Julia Panov, Dmitry A. Kuzmin, and et al. 2021. "Comparative Genomics Analysis of Repetitive Elements in Ten Gymnosperm Species: “Dark Repeatome” and Its Abundance in Conifer and Gnetum Species" Life 11, no. 11: 1234. https://doi.org/10.3390/life11111234
APA StyleTitievsky, A., Putintseva, Y. A., Taranenko, E. A., Baskin, S., Oreshkova, N. V., Brodsky, E., Sharova, A. V., Sharov, V. V., Panov, J., Kuzmin, D. A., Brodsky, L., & Krutovsky, K. V. (2021). Comparative Genomics Analysis of Repetitive Elements in Ten Gymnosperm Species: “Dark Repeatome” and Its Abundance in Conifer and Gnetum Species. Life, 11(11), 1234. https://doi.org/10.3390/life11111234