
Microbial Surface Biofilm Responds to the Growth-Reproduction-Senescence Cycle of the Dominant Coral Reef Macroalgae Sargassum spp.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. 16S rRNA Gene Sequencing Data
2.3. Metagenome-Assembled Genomes (MAGs)
2.4. Statistical Analyses of Microbial Community Composition
3. Results
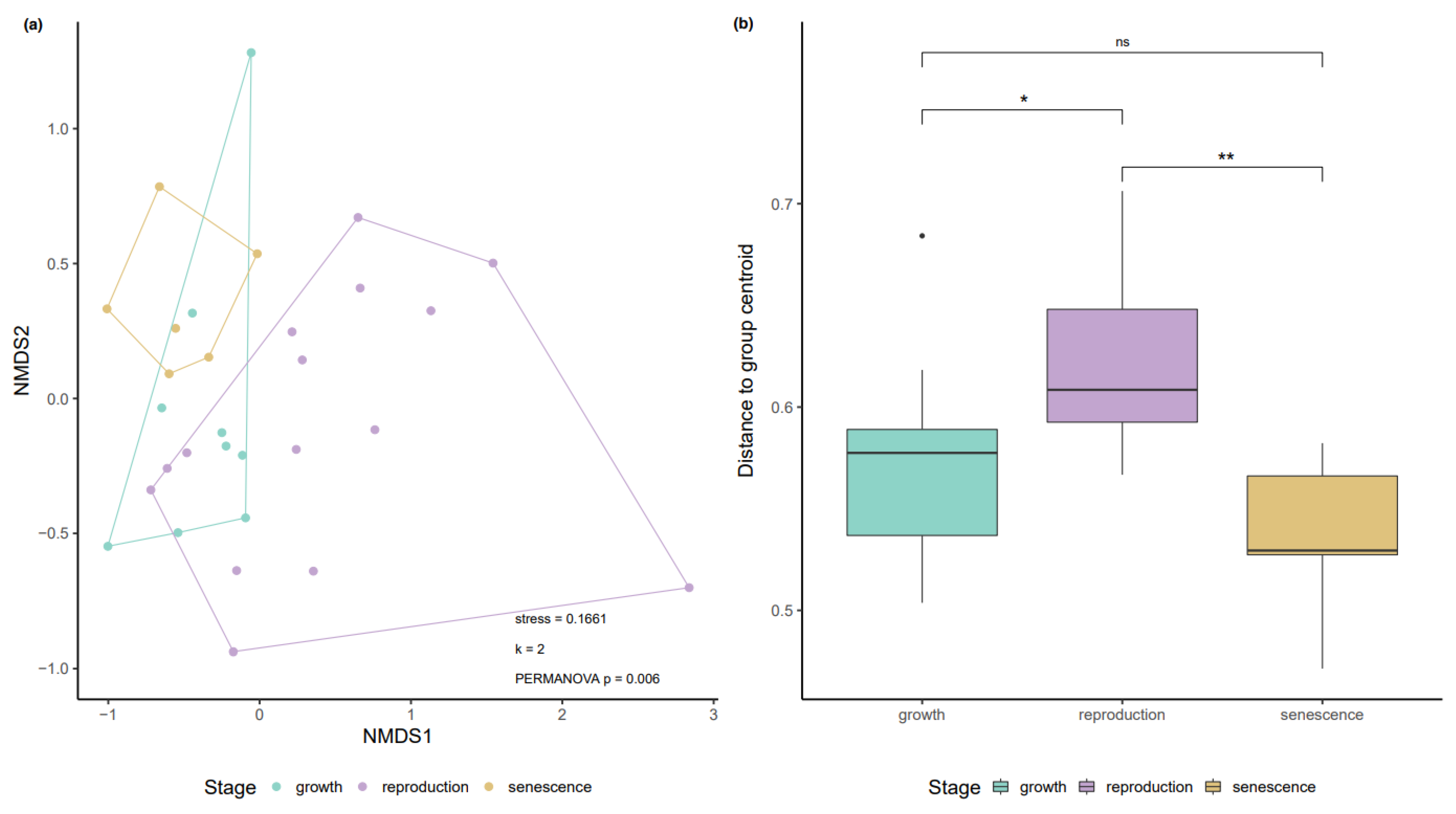
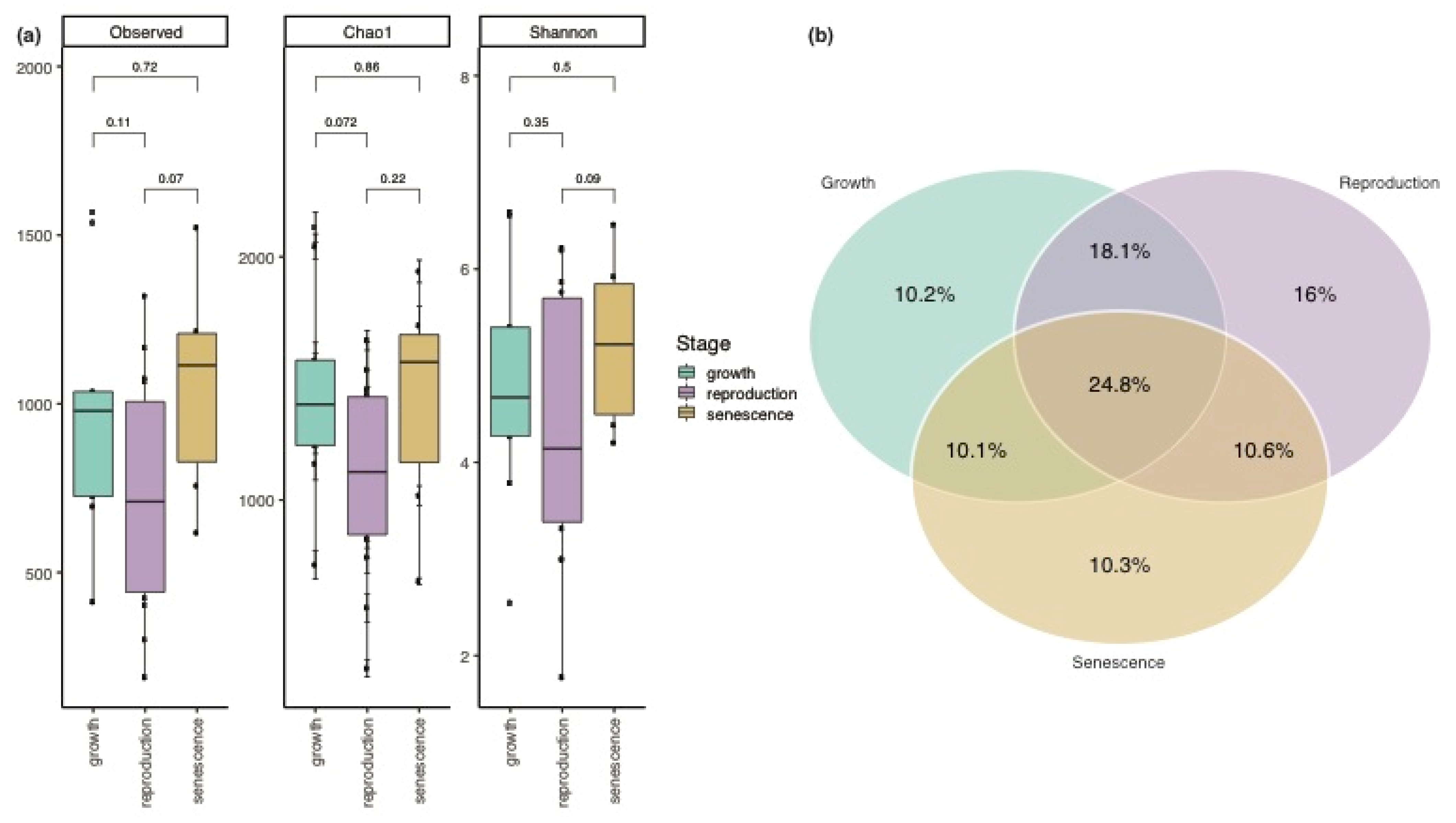
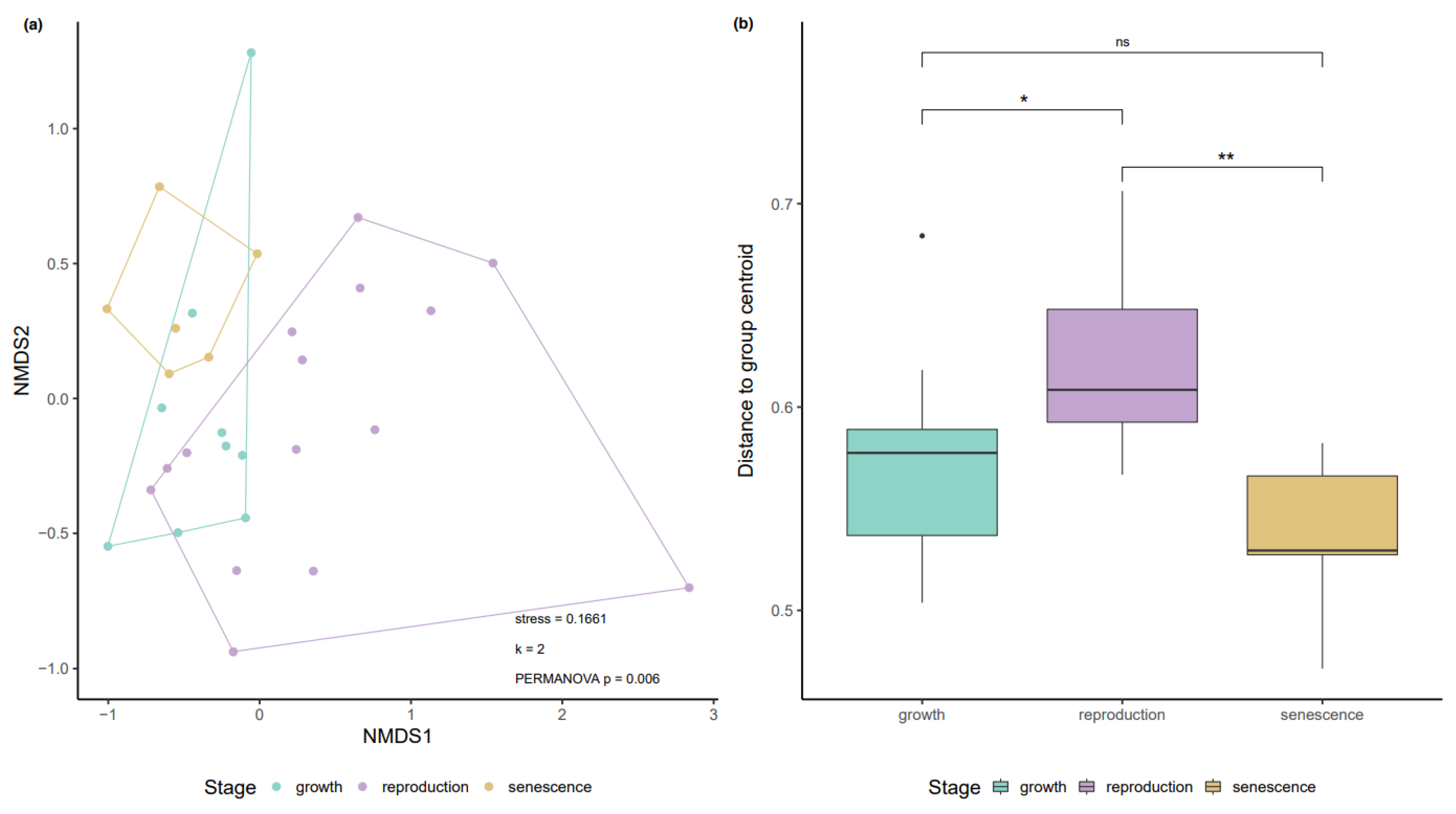
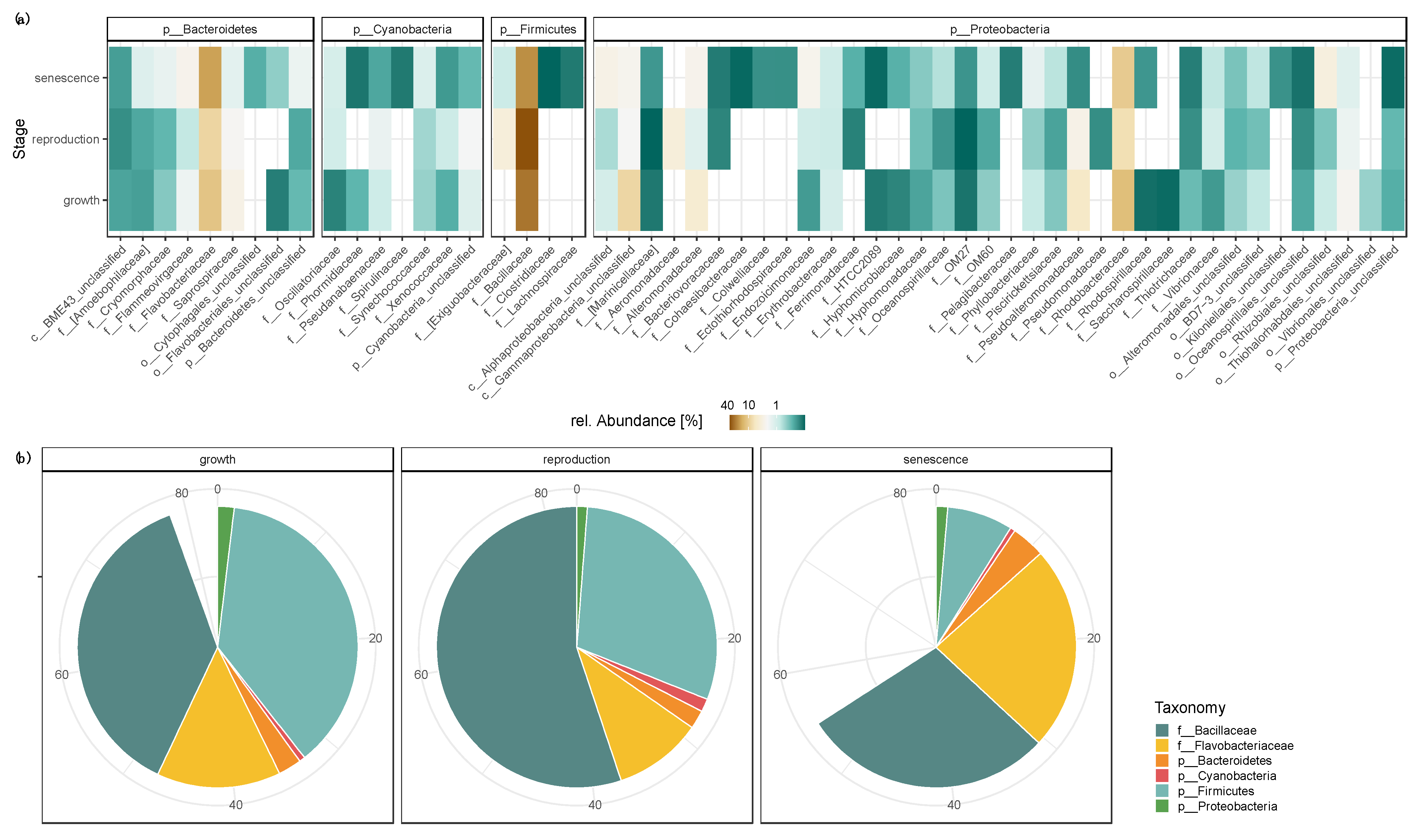
3.1. Variation in the Biofilm Community along the Growth-Reproduction-Senescence Cycle
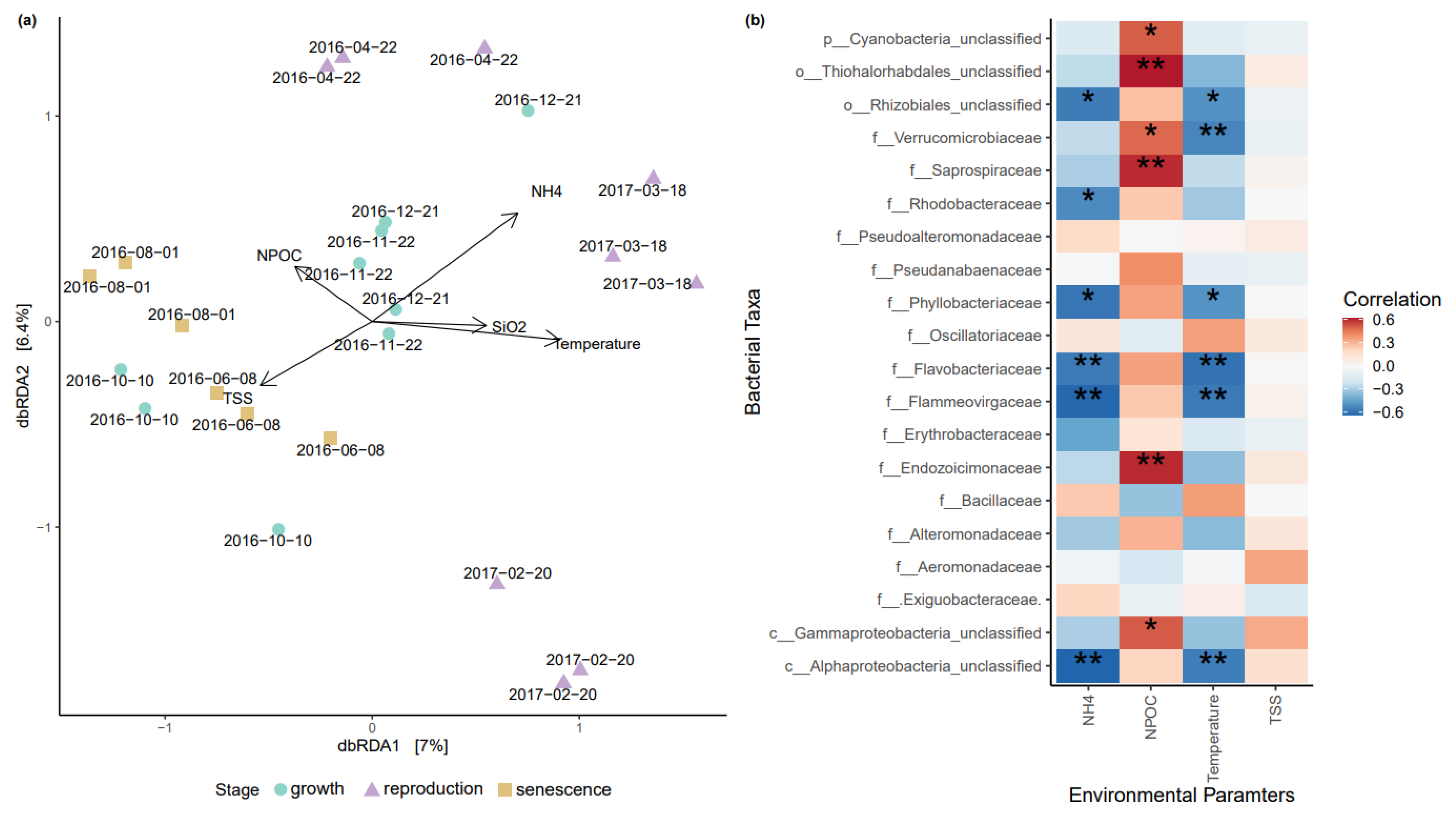
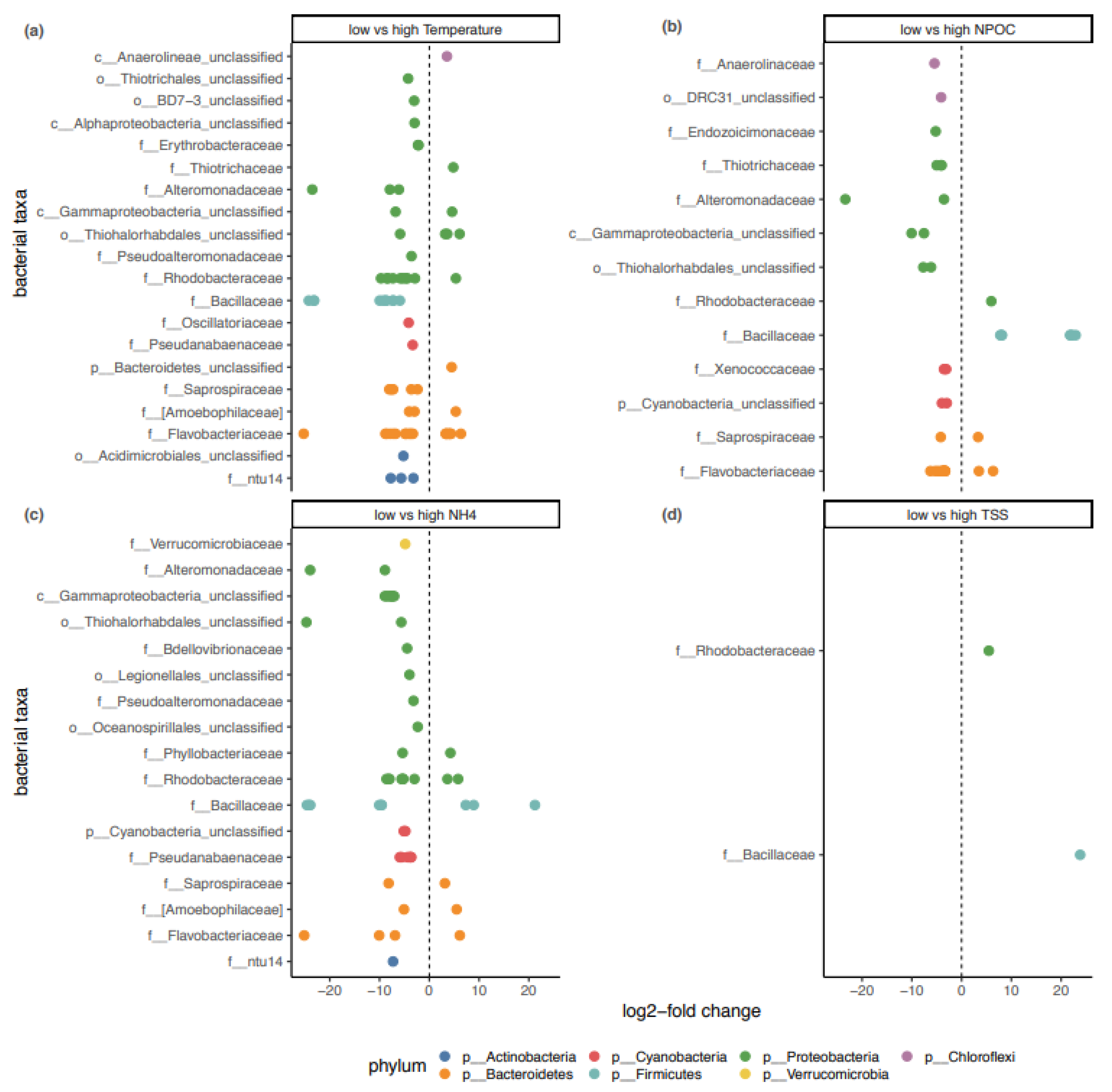
3.2. Influence of Abiotic Factors
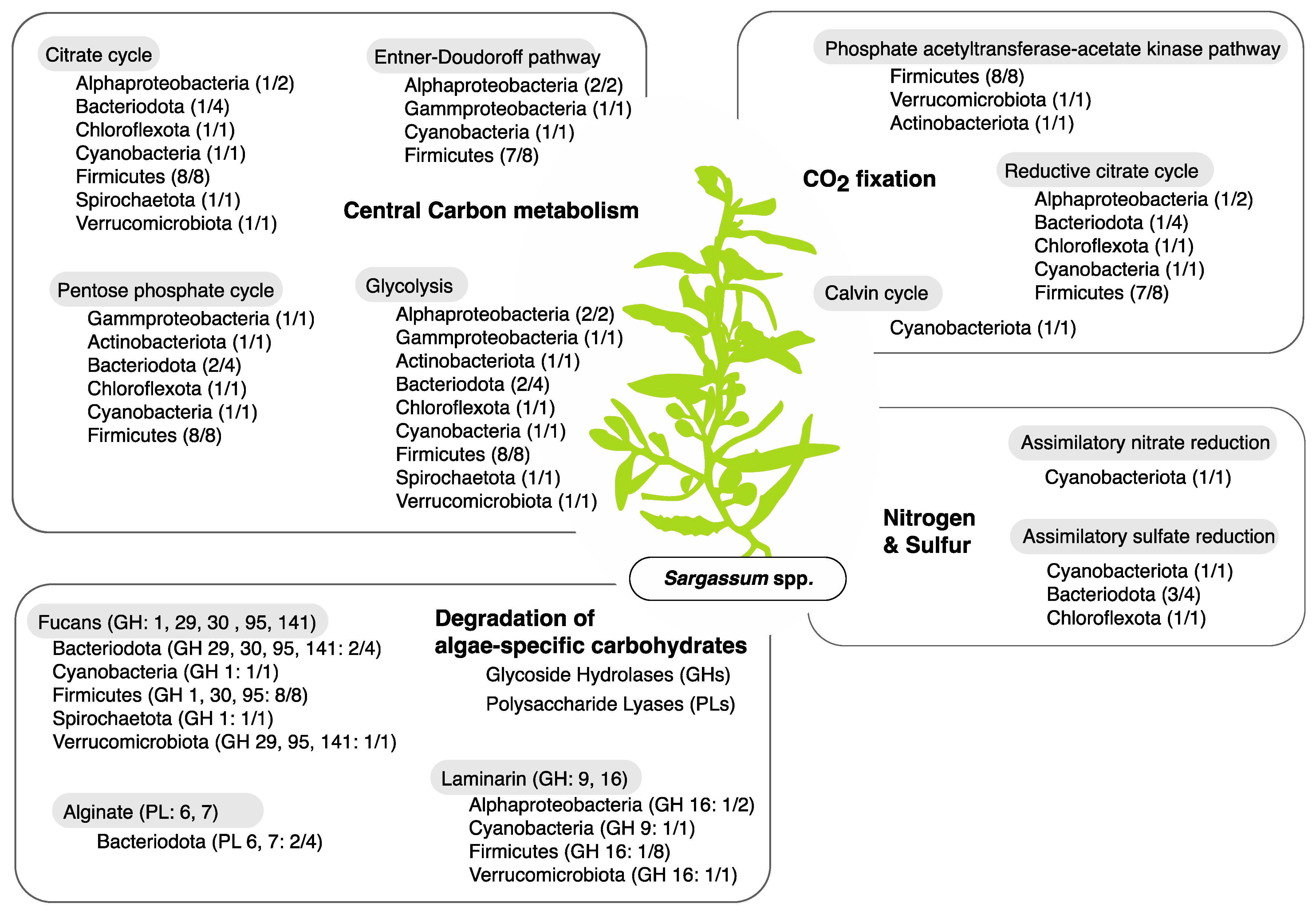
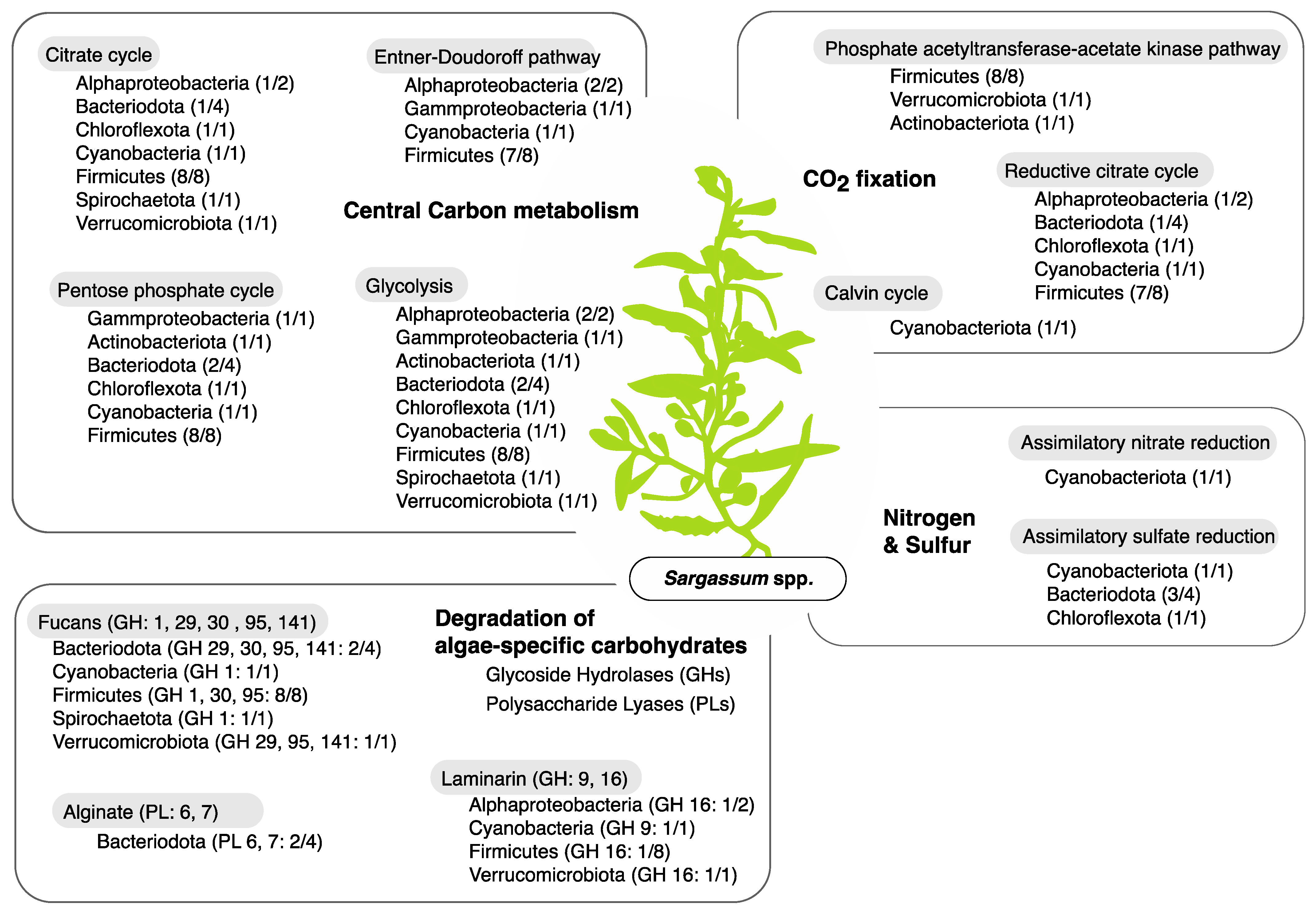
3.3. Metabolic Functions of Metagenome Assembled Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruno, J.F.; Sweatman, H.; Precht, W.F.; Selig, E.R.; Schutte, V.G. Assessing evidence of phase shifts from coral to macroalgal dominance on coral reefs. Ecology 2009, 90, 1478–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.P.; Rodrigues, M.J.; Bellwood, D.R.; Ceccarelli, D.; Hoegh-Guldberg, O.; McCook, L.; Moltschaniwskyj, N.; Pratchett, M.S.; Steneck, R.S.; Willis, B. Phase shifts, herbivory, and the resilience of coral reefs to climate change. Curr. Biol. 2007, 17, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Sandin, S.A.; Smith, J.E.; DeMartini, E.E.; Dinsdale, E.A.; Donner, S.D.; Friedlander, A.M.; Konotchick, T.; Malay, M.; Maragos, J.E.; Obura, D.; et al. Baselines and degradation of coral reefs in the Northern Line Islands. PLoS ONE 2008, 3, e1548. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.P.; Baird, A.H.; Bellwood, D.R.; Card, M.; Connolly, S.R.; Folke, C.; Grosberg, R.; Hoegh-Guldberg, O.; Jackson, J.B.C.; Kleypas, J.; et al. Climate change, human impacts, and the resilience of coral reefs. Science 2003, 301, 929–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.P.; Kerry, J.T.; Baird, A.H.; Connolly, S.R.; Dietzel, A.; Eakin, C.M.; Heron, S.F.; Hoey, A.S.; Hoogenboom, M.O.; Liu, G.; et al. Global warming transforms coral reef assemblages. Nature 2018, 556, 492–496. [Google Scholar] [CrossRef]
- Haas, A.F.; Nelson, C.E.; Kelly, L.W.; Carlson, C.A.; Rohwer, F.; Leichter, J.J.; Wyatt, A.; Smith, J.E. Effects of coral reef benthic primary producers on dissolved organic carbon and microbial activity. PLoS ONE 2011, 6, e27973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.E.; Goldberg, S.J.; Kelly, L.W.; Haas, A.F.; Smith, J.E.; Rohwer, F.; Carlson, C.A. Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J. 2013, 7, 962–979. [Google Scholar] [CrossRef]
- Silveira, C.B.; Cavalcanti, G.S.; Walter, J.M.; Silva-Lima, A.W.; Dinsdale, E.A.; Bourne, D.G.; Thompson, C.C.; Thompson, F.L. Microbial processes driving coral reef organic carbon flow. FEMS Microbiol. Rev. 2017, 41, 575–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A.F.; Fairoz, M.F.M.; Kelly, L.W.; Nelson, C.E.; Dinsdale, E.A.; Edwards, R.A.; Giles, S.; Hatay, M.; Hisakawa, N.; Knowles, B.; et al. Global microbialization of coral reefs. Nat. Microbiol. 2016, 1, 16042. [Google Scholar] [CrossRef] [PubMed]
- Barott, K.L.; Rohwer, F.L. Unseen players shape benthic competition on coral reefs. Trends Microbiol. 2012, 20, 621–628. [Google Scholar] [CrossRef] [PubMed]
- McDole, T.; Nulton, J.; Barott, K.L.; Felts, B.; Hand, C.; Hatay, M.; Lee, H.; Nadon, M.O.; Nosrat, B.; Salamon, P.; et al. Assessing coral reefs on a pacific-wide scale using the microbialization score. PLoS ONE 2012, 7, e43233. [Google Scholar] [CrossRef]
- Morrow, K.M.; Paul, V.J.; Liles, M.R.; Chadwick, N.E. Allelochemicals produced by Caribbean macroalgae and cyanobacteria have species-specific effects on reef coral microorganisms. Coral Reefs 2011, 30, 309–320. [Google Scholar] [CrossRef]
- Sweet, M.J.; Bythell, J.C.; Nugues, M.M. Algae as reservoirs for coral pathogens. PLoS ONE 2013, 8, e69717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohwer, F.; Seguritan, V.; Azam, F.; Knowlton, N. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 2002, 243, 1–10. [Google Scholar] [CrossRef] [Green Version]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [Green Version]
- Van Der Loos, L.M.; Eriksson, B.K.; Falcao Salles, J. The Macroalgal Holobiont in a Changing Sea. Trends Microbiol. 2019, 27, 635–650. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.D.; Clyne, J. Significance of algal excretory products for growth of epilimnetic bacteria. Appl. Environ. Microbiol. 1984, 47, 731–734. [Google Scholar] [CrossRef] [Green Version]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical interactions between marine macroalgae and bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Bengtsson, M.M.; Sjotun, K.; Storesund, J.E.; Ovreas, L. Utilization of kelp-derived carbon sources by kelp surface-associated bacteria. Aquat. Microb. Ecol. 2011, 62, 191–199. [Google Scholar] [CrossRef]
- Singh, R.P.; Mantri, V.A.; Reddy, C.R.K.; Jha, B. Isolation of seaweed-associated bacteria and their morphogenesis-inducing capability in axenic cultures of the green alga Ulva fasciata. Aquat Biol 2011, 12, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Reddy, C.R.K. Seaweed-microbial interactions: Key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 2014, 88, 213–230. [Google Scholar] [CrossRef]
- Mei, X.Y.; Wu, C.H.; Zhao, J.; Yan, T.; Jiang, P. Community structure of bacteria associated with drifting Sargassum horneri, the causative species of golden tide in the Yellow Sea. Front. Microbiol. 2019, 10, 1192. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Gardiner, M. Microbial Dysbiosis: Rethinking disease in marine ecosystems. Front. Microbiol 2016, 7, 991. [Google Scholar] [CrossRef] [PubMed]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial community patterns on marine macroalgae are host-specific but temporally variable. Environ. Microbiol. 2011, 13, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Nylund, G.M.; Persson, F.; Lindegarth, M.; Cervin, G.; Hermansson, M.; Pavia, H. The red alga Bonnemaisonia asparagoides regulates epiphytic bacterial abundance and community composition by chemical defence. FEMS Microbiol. Ecol. 2010, 71, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, M.M.; Sjotun, K.; Ovreas, L. Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat. Microb. Ecol. 2010, 60, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Aires, T.; Serrao, E.A.; Engelen, A.H. Host and environmental specificity in bacterial communities associated to two highly invasive marine species (genus Asparagopsis). Front. Microbiol. 2016, 7, 559. [Google Scholar] [CrossRef]
- Bengtsson, M.M.; Sjotun, K.; Lanzen, A.; Ovreas, L. Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. ISME J. 2012, 6, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, F.P.; D’Hondt, S.; Willems, A.; Airoldi, L.; De Clerck, O. Diversity and temporal dynamics of the epiphytic bacterial communities associated with the canopy-forming seaweed Cystoseira cornpressa (Esper) Gerloff and Nizamuddin. Front. Microbiol. 2016, 7, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serebryakova, A.; Aires, T.; Viard, F.; Serrao, E.A.; Engelen, A.H. Summer shifts of bacterial communities associated with the invasive brown seaweed Sargassum muticum are location and tissue dependent. PLoS ONE 2018, 13, e0206734. [Google Scholar] [CrossRef] [PubMed]
- Tuya, F.; Wernberg, T.; Thomsen, M.S. The spatial arrangement of reefs alters the ecological patterns of fauna between interspersed algal habitats. Estuar. Coast. Shelf Sci. 2008, 78, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Webster, F.J.; Babcock, R.C.; Van Keulen, M.; Loneragan, N.R. Macroalgae inhibits larval settlement and increases recruit mortality at Ningaloo Reef, Western Australia. PLoS ONE 2015, 10, e0124162. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.E. Competition between scleractinian corals and macroalgae—an experimental investigation of coral growth, survival and reproduction. J. Exp. Mar. Biol. Ecol. 1995, 190, 151–168. [Google Scholar] [CrossRef]
- van Woesik, R.; Ripple, K.; Miller, S.L. Macroalgae reduces survival of nursery-reared Acropora corals in the Florida reef tract. Restor. Ecol. 2018, 26, 563–569. [Google Scholar] [CrossRef]
- Smith, J.E.; Shaw, M.; Edwards, R.A.; Obura, D.; Pantos, O.; Sala, E.; Sandin, S.A.; Smriga, S.; Hatay, M.; Rohwer, F.L. Indirect effects of algae on coral: Algae-mediated, microbe-induced coral mortality. Ecol. Lett. 2006, 9, 835–845. [Google Scholar] [CrossRef]
- Birrell, C.L.; McCook, L.J.; Willis, B.L.; Harrington, L. Chemical effects of macroalgae on larval settlement of the broadcast spawning coral Acropora millepora. Mar. Ecol. Prog. Ser. 2008, 362, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Vega Thurber, R.; Burkepile, D.E.; Correa, A.M.; Thurber, A.R.; Shantz, A.A.; Welsh, R.; Pritchard, C.; Rosales, S. Macroalgae decrease growth and alter microbial community structure of the reef-building coral, Porites astreoides. PLoS ONE 2012, 7, e44246. [Google Scholar] [CrossRef] [Green Version]
- Nugues, M.M.; Smith, G.W.; Hooidonk, R.J.; Seabra, M.I.; Bak, R.P.M. Algal contact as a trigger for coral disease. Ecol. Lett. 2004, 7, 919–923. [Google Scholar] [CrossRef]
- De’ath, G.; Fabricius, K. Water quality as a regional driver of coral biodiversity and macroalgae on the Great Barrier Reef. Ecol. Appl. 2010, 20, 840–850. [Google Scholar] [CrossRef]
- Wismer, S.; Hoey, A.S.; Bellwood, D.R. Cross-shelf benthic community structure on the Great Barrier Reef: Relationships between macroalgal cover and herbivore biomass. Mar. Ecol. Prog. Ser. 2009, 376, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, D.M.; Evans, R.D.; Logan, M.; Mantel, P.; Puotinen, M.; Petus, C.; Russ, G.R.; Williamson, D.H. Long-term dynamics and drivers of coral and macroalgal cover on inshore reefs of the Great Barrier Reef Marine Park. Ecol. Appl. 2020, 30, e02008. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, C.D.; Bellwood, D.R. Seasonality and dynamics in coral reef macroalgae: Variation in condition and susceptibility to herbivory. Mar. Biol. 2010, 157, 955–965. [Google Scholar] [CrossRef]
- Schaffelke, B.; Klumpp, D.W. Biomass and productivity of tropical macroalgae on three nearshore fringing reefs in the central Great Barrier Reef, Australia. Bot. Mar. 1997, 40, 373–383. [Google Scholar] [CrossRef]
- Ceccarelli, D.M.; Loffler, Z.; Bourne, D.G.; Al Moajil-Cole, G.S.; Boström-Einarsson, L.; Evans-Illidge, E.; Fabricius, K.; Glasl, B.; Marshall, P.; McLeod, I.; et al. Rehabilitation of coral reefs through removal of macroalgae: State of knowledge and considerations for management and implementation. Restor. Ecol. 2018, 26, 827–838. [Google Scholar] [CrossRef]
- Martin-Smith, K.M. The phenology of four species of Sargassum at Magnetic Island, Australia. Bot. Mar. 1993, 327–334. [Google Scholar] [CrossRef]
- Loffler, Z.; Hoey, A.S.; Hughes, A.R. Canopy-forming macroalgal beds (Sargassum) on coral reefs are resilient to physical disturbance. J. Ecol. 2018, 106, 1164. [Google Scholar] [CrossRef]
- Walther, B.D.; Kingsford, M.J.; McCulloch, M.T. Environmental records from Great Barrier Reef corals: Inshore versus offshore drivers. PLoS ONE 2013, 8, e77091. [Google Scholar] [CrossRef]
- Glasl, B.; Bourne, D.G.; Frade, P.R.; Thomas, T.; Schaffelke, B.; Webster, N.S. Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 2019, 7, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasl, B.; Robbins, S.; Frade, P.R.; Marangon, E.; Laffy, P.W.; Bourne, D.G.; Webster, N.S. Comparative genome-centric analysis reveals seasonal variation in the function of coral reef microbiomes. ISME J. 2020, 14, 1435–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, M.J.; Lourey, M.J. Water Quality—Field and Analytical Procedures; Australian Institute of Marine Science: Townsville, Australia, 2000. [Google Scholar]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley and Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Turner, S.; Pryer, K.M.; Miao, V.P.; Palmer, J.D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryot. Microbiol. 1999, 46, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.V.; Kamp, J.v.d.; Ostrowski, M.; Seymour, J.R.; Ingleton, T.; Messer, L.F.; Jeffries, T.; Siboni, N.; Laverock, B.; Bibiloni-Isaksson, J.; et al. Systematic, continental scale temporal monitoring of marine pelagic microbiota by the Australian Marine Microbial Biodiversity Initiative. Sci. Data 2018, 5, 180130. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 1 November 2021).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-9; 2020. Available online: https://github.com/vegandevs/vegan (accessed on 1 November 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Barott, K.L.; Rodriguez-Brito, B.; Janouskovec, J.; Marhaver, K.L.; Smith, J.E.; Keeling, P.; Rohwer, F.L. Microbial diversity associated with four functional groups of benthic reef algae and the reef-building coral Montastraea annularis. Environ. Microbiol. 2011, 13, 1192–1204. [Google Scholar] [CrossRef]
- De Mesquita, M.M.F.; Crapez, M.A.C.; Teixeira, V.L.; Cavalcanti, D.N. Potential interactions bacteria-brown algae. J. Appl. Phycol. 2019, 31, 867–883. [Google Scholar] [CrossRef]
- Li, J.L.; Wang, T.; Yu, S.X.; Bai, J.; Qin, S. Community characteristics and ecological roles of bacterial biofilms associated with various algal settlements on coastal reefs. J. Environ. Manag. 2019, 250, 109459. [Google Scholar] [CrossRef] [PubMed]
- Susilowati, R.; Sabdono, A.; Widowati, I. Isolation and characterization of bacteria associated with brown algae Sargassum sp. from Panjang Island and their antibacterial activities. Procedia Environ. Sci 2015, 23, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, K.; Thilakan, B.; Chakraborty, R.D.; Raola, V.K.; Joy, M. O-heterocyclic derivatives with antibacterial properties from marine bacterium Bacillus subtilis associated with seaweed, Sargassum myriocystum. Appl. Microbiol. Biotechnol. 2017, 101, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Bondoso, J.; Balague, V.; Gasol, J.M.; Lage, O.M. Community composition of the Planctomycetes associated with different macroalgae. FEMS Microbiol. Ecol. 2014, 88, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaneveld, J.R.; Burkepile, D.E.; Shantz, A.A.; Pritchard, C.E.; McMinds, R.; Payet, J.P.; Welsh, R.; Correa, A.M.S.; Lemoine, N.P.; Rosales, S.; et al. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 2016, 7, 11833. [Google Scholar] [CrossRef] [PubMed]
- Frade, P.R.; Glasl, B.; Matthews, S.A.; Mellin, C.; Serrao, E.A.; Wolfe, K.; Mumby, P.J.; Webster, N.S.; Bourne, D.G. Spatial patterns of microbial communities across surface waters of the Great Barrier Reef. Commun. Biol. 2020, 3, 442. [Google Scholar] [CrossRef] [PubMed]
- Krüger, K.; Chafee, M.; Ben Francis, T.; Glavina del Rio, T.; Becher, D.; Schweder, T.; Amann, R.I.; Teeling, H. In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 2019, 13, 2800–2816. [Google Scholar] [CrossRef] [Green Version]
- Dethier, M.N.; Williams, S.L. Seasonal stresses shift optimal intertidal algal habitats. Mar. Biol. 2009, 156, 555–567. [Google Scholar] [CrossRef]
- Gorham, J.; Lewey, S.A. Seasonal-changes in the chemical-composition of Sargassum muticum. Mar. Biol. 1984, 80, 103–107. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glasl, B.; Haskell, J.B.; Aires, T.; Serrão, E.A.; Bourne, D.G.; Webster, N.S.; Frade, P.R. Microbial Surface Biofilm Responds to the Growth-Reproduction-Senescence Cycle of the Dominant Coral Reef Macroalgae Sargassum spp. Life 2021, 11, 1199. https://doi.org/10.3390/life11111199
Glasl B, Haskell JB, Aires T, Serrão EA, Bourne DG, Webster NS, Frade PR. Microbial Surface Biofilm Responds to the Growth-Reproduction-Senescence Cycle of the Dominant Coral Reef Macroalgae Sargassum spp. Life. 2021; 11(11):1199. https://doi.org/10.3390/life11111199
Chicago/Turabian StyleGlasl, Bettina, Jasmine B. Haskell, Tania Aires, Ester A. Serrão, David G. Bourne, Nicole S. Webster, and Pedro R. Frade. 2021. "Microbial Surface Biofilm Responds to the Growth-Reproduction-Senescence Cycle of the Dominant Coral Reef Macroalgae Sargassum spp." Life 11, no. 11: 1199. https://doi.org/10.3390/life11111199
APA StyleGlasl, B., Haskell, J. B., Aires, T., Serrão, E. A., Bourne, D. G., Webster, N. S., & Frade, P. R. (2021). Microbial Surface Biofilm Responds to the Growth-Reproduction-Senescence Cycle of the Dominant Coral Reef Macroalgae Sargassum spp. Life, 11(11), 1199. https://doi.org/10.3390/life11111199