Identification of New, Functionally Relevant Mutations in the Coding Regions of the Human Fos and Jun Proto-Oncogenes in Rheumatoid Arthritis Synovial Tissue

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Detection of Fos and Jun Single Nucleotide Polymorphisms (SNPs) and Mutations in Synovial Membrane (SM) Samples from Rheumatoid Arthritis (RA), Osteoarthritis (OA), and Normal Control (NC) Individuals
2.2. Functional Analyses
2.3. DNA Binding Capacity of Expressed cFos and cJun Mutants
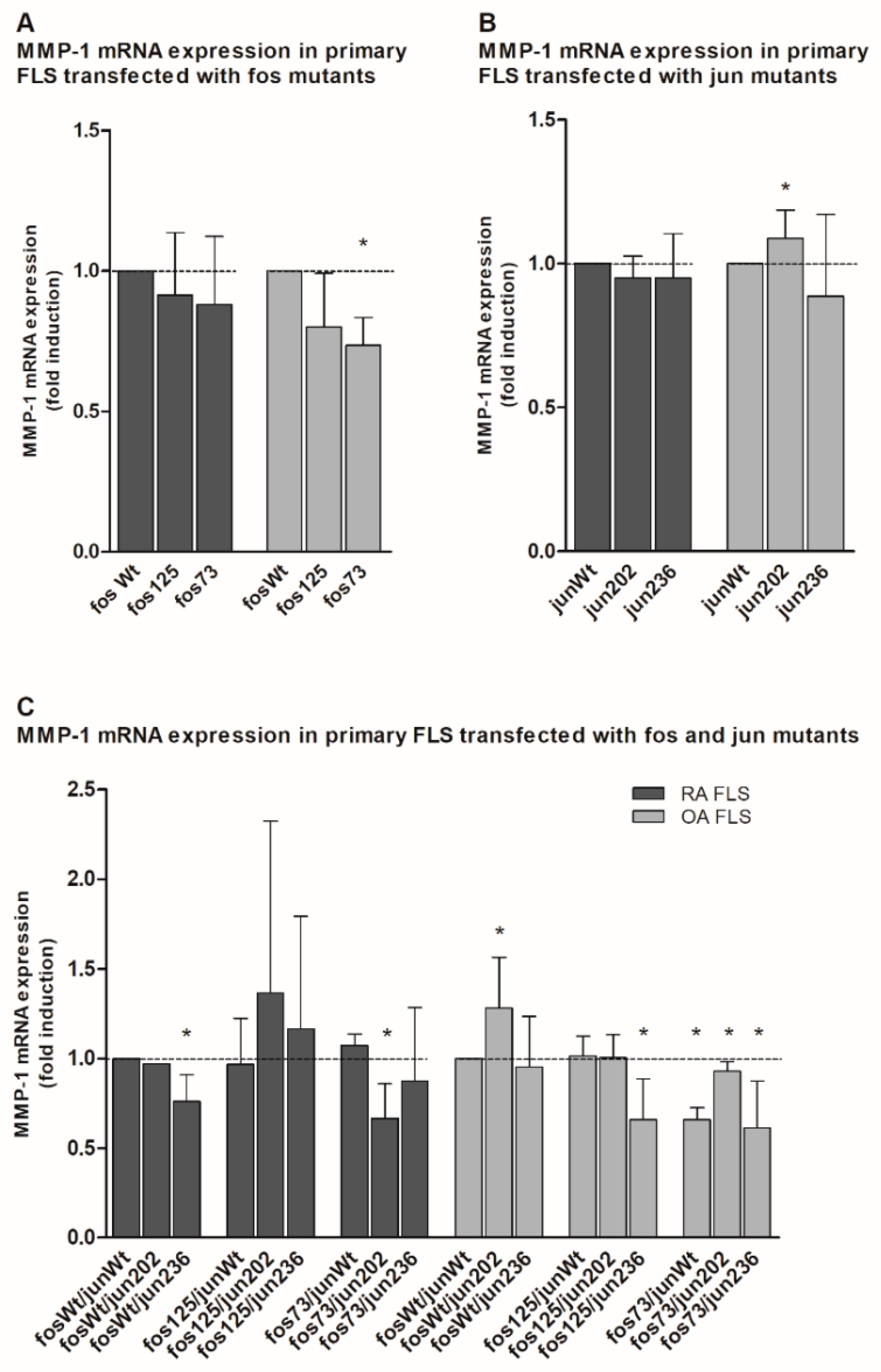
2.4. Analysis of Matrix Metalloproteinase 1 (MMP-1) and Interleukin 6 (IL-6) Gene Expression in the Presence of cFos and cJjun Mutants
2.5. Allelic Distribution of Mutations
3. Discussion
4. Materials and Methods
4.1. Patients, Tissue Samples, and Isolation of Primary Fibroblast-Like Synoviocytes (FLS)
4.2. DNA Preparation
4.3. RNA Extraction, cDNA Synthesis, and Conventional Polymerase Chain Reaction (PCR)
4.4. Cloning of DNA Fragments
4.5. Non-Isotopic RNase Cleavage Assay
4.6. Functional Analyses
4.7. Quantitative Polymerase Chain Reaction (qPCR)
4.8. Preparation of Nuclear and Whole Cell Extracts
4.9. Electrophoretic Mobility Shift Assay
4.10. Antibodies, Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), and Western Blot
4.11. Genotyping and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid |
| AP | activator protein |
| del, | deletion |
| dup | duplication |
| EMSA | electrophoretic mobility shift assay |
| FLS | fibroblast-like synoviocytes |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| gnomAD | genome aggregation database |
| IL | interleukin |
| Jak | Janus kinase |
| JNK | cJun N-terminal kinase |
| MALDI-TOF | matrix assisted laser desorption ionization - time of flight |
| MMP | matrix metalloproteinase |
| NC | normal control |
| n.d. | not detected |
| NIRCA | Non-isotopic RNAse Cleavage Assay |
| OA | osteoarthritis |
| qPCR | quantitative real time polymerase chain reaction |
| RA | rheumatoid arthritis |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SM | synovial membrane |
| SNP | single nucleotide polymorphism |
| STAT | signal transducer and activator of transcription |
| wt | Wildtype |
References
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef]
- Pap, T.; Dankbar, B.; Wehmeyer, C.; Korb-Pap, A.; Sherwood, J. Synovial fibroblasts and articular tissue remodelling: Role and mechanisms. Semin. Cell Dev. Biol. 2020, 101, 140–145. [Google Scholar] [CrossRef]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Wollbold, J.; Huber, R.; Pohlers, D.; Koczan, D.; Guthke, R.; Kinne, R.W.; Gausmann, U. Adapted Boolean network models for extracellular matrix formation. BMC Syst. Biol. 2009, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, S.; Tsumiyama, K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle 2009, 8, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F. Bone development and inflammatory disease is regulated by AP-1 (Fos/Jun). Ann. Rheum. Dis. 2009, 69, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef]
- Okamoto, H.; Cujec, T.P.; Yamanaka, H.; Kamatani, N. Molecular aspects of rheumatoid arthritis: Role of transcription factors. FEBS J. 2008, 275, 4463–4470. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Kinne, R.W.; Boehm, S.; Iftner, T.; Aigner, T.; Vornehm, S.; Weseloh, G.; Bravo, R.; Emmrich, F.; Kroczek, R.A. Synovial Fibroblast-Like Cells Strongly Express Jun-B and C-Fos Proto-Oncogenes in Rheumatoid- and Osteoarthritis. Scand. J. Rheumatol. 1995, 24, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2007, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Papoudou-Bai, A.; Hatzimichael, E.; Barbouti, A.; Kanavaros, P. Expression patterns of the activator protein-1 (AP-1) family members in lymphoid neoplasms. Clin. Exp. Med. 2016, 17, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.-M.; Lee, S.-Y.; Kwok, S.-K.; Lee, S.H.; Kim, D.; Kim, W.K.; Her, Y.-M.; Son, H.-J.; Kim, E.-K.; Ryu, J.-G.; et al. The Fos-Related Antigen 1–JUNB/Activator Protein 1 Transcription Complex, a Downstream Target of Signal Transducer and Activator of Transcription 3, Induces T Helper 17 Differentiation and Promotes Experimental Autoimmune Arthritis. Front. Immunol. 2017, 8, 1793. [Google Scholar] [CrossRef]
- Behmoaras, J.; Bhangal, G.; Smith, J.; McDonald, K.; Mutch, B.; Lai, P.C.; Domin, J.; Game, L.; Salama, A.; Foxwell, B.M.; et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat. Genet. 2008, 40, 553–559. [Google Scholar] [CrossRef]
- Brown, P.H.; Kim, S.H.; Wise, S.C.; Sabichi, A.L.; Birrer, M.J. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 1996, 7, 1013–1021. [Google Scholar]
- Knapp, J.I.; Heppner, C.; Hickman, A.B.; Burns, A.L.; Chandrasekharappa, S.C.; Collins, F.S.; Marx, S.J.; Spiegel, A.M.; Agarwal, S.K. Identification and characterization of JunD missense mutants that lack menin binding. Oncogene 2000, 19, 4706–4712. [Google Scholar] [CrossRef][Green Version]
- Knebel, B.; Kotzka, J.; Knebel, B.; Hartwig, S.; Avci, H.; Jacob, S.; Nitzgen, U.; Schiller, M.; März, W.; Hoffmann, M.M.; et al. A mutation in the c-Fos gene associated with congenital generalized lipodystrophy. Orphanet J. Rare Dis. 2013, 8, 119. [Google Scholar] [CrossRef]
- Huber, R.; Kirsten, H.; Näkki, A.; Pohlers, D.; Thude, H.; Eidner, T.; Heinig, M.; Brand, K.; Ahnert, P.; Kinne, R.W. Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study. Int. J. Mol. Sci. 2019, 20, 1382. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Bang, S.-Y.; Lee, H.-S.; Bae, S.-Y.B.H.-S.L.S.-C. Update on the genetic architecture of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.; Kollnberger, S.; Mellins, E.D. HLA associations in inflammatory arthritis: emerging mechanisms and clinical implications. Nat. Rev. Rheumatol. 2019, 15, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Database of Single Nucleotide Polymorphisms (dbSNP). Available online: https://www.ncbi.nlm.nih.gov/snp/ (accessed on 20 October 2020).
- Karczewski, K.J.; Francioli, L.C.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Yearb. Paediatr. Endocrinol. 2020, C, 434–443. [Google Scholar] [CrossRef]
- The Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 20 August 2020).
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef]
- Supek, F. The Code of Silence: Widespread Associations Between Synonymous Codon Biases and Gene Function. J. Mol. Evol. 2015, 82, 65–73. [Google Scholar] [CrossRef]
- Quax, T.E.; Claassens, N.J.; Söll, D.; Van Der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef]
- Gingold, H.; Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011, 7, 481. [Google Scholar] [CrossRef]
- Kunisch, E.; Pohlers, D.; Dunger, S.; Huber, R.; Kreusch, A.; Wiederanders, B.; Kinne, R.W. What can experimental research offer to rheumatology today—The viewpoint of molecular biology? Contribution of molecular biology to pathogenesis research in rheumatology using the example of rheumatoid arthritis. Z. Rheumatol. 2002, 61. [Google Scholar] [CrossRef]
- Chung, S.A.; Shum, A.K. Rare variants, autoimmune disease, and arthritis. Curr. Opin. Rheumatol. 2016, 28, 346–351. [Google Scholar] [CrossRef]
- Kinne, R.W.; Liehr, T.; Beensen, V.; Kunisch, E.; Zimmermann, T.; Holland, H.; Pfeiffer, R.; Stahl, H.-D.; Lungershausen, W.; Hein, G.; et al. Mosaic chromosomal aberrations in synovial fibroblasts of patients with rheumatoid arthritis, osteoarthritis, and other inflammatory joint diseases. Arthritis Res. 2001, 3, 319–330. [Google Scholar] [CrossRef]
- Kinne, R.W.; Kunisch, E.; Beensen, V.; Zimmermann, T.; Emmrich, F.; Petrow, P.; Lungershausen, W.; Hein, G.; Braun, R.K.; Foerster, M.; et al. Synovial fibroblasts and synovial macrophages from patients with rheumatoid arthritis and other inflammatory joint diseases show chromosomal aberrations. Genes Chromosom. Cancer 2003, 38, 53–67. [Google Scholar] [CrossRef]
- May, G.H.; Allen, K.E.; Clark, W.; Funk, M.; Gillespie, D.A. Analysis of the interaction between c-Jun and c-Jun N-terminal kinase in vivo. J. Biol. Chem. 1998, 273, 33429–33435. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Characteristics of amino acids. Curr. Protoc. Protein. Sci. 2001. [Google Scholar] [CrossRef]
- Schauer, M.; Kottek, T.; Schönherr, M.; Bhattacharya, A.; Ibrahim, S.M.; Hirose, M.; Köhling, R.; Fuellen, G.; Schmitz, U.; Kunz, M. A mutation in the NADH-dehydrogenase subunit 2 suppresses fibroblast aging. Oncotarget 2015, 6, 8552–8566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ginter, T.; Fahrer, J.; Kröhnert, U.; Fetz, V.; Garrone, A.; Stauber, R.H.; Reichardt, W.; Müller-Newen, G.; Kosan, C.; Heinzel, T.; et al. Arginine residues within the DNA binding domain of STAT3 promote intracellular shuttling and phosphorylation of STAT3. Cell. Signal. 2014, 26, 1698–1706. [Google Scholar] [CrossRef]
- Bossis, G.; Malnou, C.E.; Farras, R.; Andermarcher, E.; Hipskind, R.; Rodriguez, M.; Schmidt, D.; Muller, S.; Jariel-Encontre, I.; Piechaczyk, M. Down-Regulation of c-Fos/c-Jun AP-1 Dimer Activity by Sumoylation. Mol. Cell. Biol. 2005, 25, 6964–6979. [Google Scholar] [CrossRef]
- Ryseck, R.P.; Bravo, R. c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: Effect of FOS proteins. Oncogene 1991, 6, 533–542. [Google Scholar]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Bejjani, F.; Evanno, E.; Zibara, K.; Piechaczyk, M.; Jariel-Encontre, I. The AP-1 transcriptional complex: Local switch or remote command? Biochim. Biophys. Acta 2019, 1872, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Georgopoulos, K.; Greenberg, M.E.; Leder, P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 1988, 55, 917–924. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Intracellular Signaling Pathways in Rheumatoid Arthritis. J. Clin. Cell. Immunol. 2013, 4, 160. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Zhou, C.; Nandakumar, K.S. Molecular and Cellular Pathways Contributing to Joint Damage in Rheumatoid Arthritis. Mediat. Inflamm. 2020, 2020, 1–20. [Google Scholar] [CrossRef]
- Aud, D.; Peng, S.L. Mechanisms of Disease: Transcription factors in inflammatory arthritis. Nat. Clin. Pr. Rheumatol. 2006, 2, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Alonso, I.G.D.L.F.; Liang, H.-C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93. [Google Scholar] [CrossRef]
- Hamaratoglu, F.; Atkins, M. Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 4580. [Google Scholar] [CrossRef]
- Giuliani, C.; Bucci, I.; Napolitano, G.; Giuliani, C.; Bucci, I.; Napolitano, G. The Role of the Transcription Factor Nuclear Factor-kappa B in Thyroid Autoimmunity and Cancer. Front. Endocrinol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Huber, R.; Pietsch, D.; Panterodt, T.; Brand, K. Regulation of C/EBPbeta and resulting functions in cells of the monocytic lineage. Cell Signal. 2012, 24, 1287–1296. [Google Scholar] [CrossRef]
- Wei, W.; Jin, J.; Schlisio, S.; Harper, J.W.; Kaelin, W.G., Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005, 8, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Stuhlmüller, B.; Kunisch, E.; Kinne, R.W. Discrepancy between Jun/Fos Proto-Oncogene mRNA and Protein Expression in the Rheumatoid Arthritis Synovial Membrane. J 2020, 3, 15. [Google Scholar] [CrossRef]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Boyle, D.L.; Shi, Y.; Green, D.R.; Firestein, G.S. Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1088–1092. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef]
- McGonagle, D.; Watad, A.; Savic, S. Mechanistic immunological based classification of rheumatoid arthritis. Autoimmun. Rev. 2018, 17, 1115–1123. [Google Scholar] [CrossRef]
- Uspenskaya, N.Y.; Akopov, S.B.; Snezhkov, E.V.; Sverdlov, E.D. The Rate of Human Germline Mutations—Variable Factor of Evolution and Diseases. Russ. J. Genet. 2019, 55, 523–534. [Google Scholar] [CrossRef]
- Ferguson, L.R. Chronic inflammation and mutagenesis. Mutat. Res. Mol. Mech. Mutagen. 2010, 690, 3–11. [Google Scholar] [CrossRef]
- Carlson, J.; Locke, A.E.; Flickinger, M.; Zawistowski, M.; Levy, S.; Myers, R.M.; Boehnke, M.; Kang, H.M.; Scott, L.J.; Jun, Z.L.; et al. Extremely rare variants reveal patterns of germline mutation rate heterogeneity in humans. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Huber, R.; Hummert, C.; Gausmann, U.; Pohlers, D.; Koczan, D.; Guthke, R.; Kinne, R.W. Identification of intra-group, inter-individual, and gene-specific variances in mRNA expression profiles in the rheumatoid arthritis synovial membrane. Arthritis Res. Ther. 2008, 10, R98. [Google Scholar] [CrossRef]
- Mankia, K.; Emery, P. Review: Preclinical Rheumatoid Arthritis: Progress Toward Prevention. Arthritis Rheumatol. 2016, 68, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Kunisch, E.; Pfeiffer, R.; Hirth, A.; Stahl, H.-D.; Sack, U.; Laube, A.; Liesaus, E.; Roth, A.; Palombo-Kinne, E.; et al. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture—Primary culture cells markedly differ from fourth-passage cells. Arthritis Res. 2000, 3, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The american rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Asch, E.; Bloch, D.; Bole, G.; Borenstein, D.; Brandt, K.; Christy, W.; Cooke, T.D.; Greenwald, R.; Hochberg, M.; et al. Development of criteria for the classification and reporting of osteoarthritis: Classification of osteoarthritis of the knee. Arthritis Rheum. 1986, 29, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Kunisch, E.; Gluck, B.; Egerer, R.; Sickinger, S.; Kinne, R.W. Comparison of conventional and real-time RT-PCR for the quantitation of jun protooncogene mRNA and analysis of junB mRNA expression in synovial membranes and isolated synovial fibroblasts from rheumatoid arthritis patients. Z. Rheumatol. 2003, 62, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.C.; Huber, R.; Gutsch, R.; Kandemir, J.D.; Cappello, C.; Krauter, J.; Duyster, J.; Ganser, A.; Brand, K. ITD- and FL-induced FLT3 signal transduction leads to increased C/EBPbeta-LIP expression and LIP/LAP ratio by different signalling modules. Br. J. Haematol. 2010, 148, 777–790. [Google Scholar] [CrossRef]
- Umesono, K.; Murakami, K.K.; Thompson, C.C.; Evans, R.M. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991, 65, 1255–1266. [Google Scholar] [CrossRef]
- De Martin, R.; Strasswimmer, J.; Philipson, L. A new luciferase promoter insertion vector for the analysis of weak transcriptional activities. Gene 1993, 124, 137–138. [Google Scholar] [CrossRef]
- White, L.A.; E Brinckerhoff, C. Two activator protein-1 elements in the matrix metalloproteinase-1 promoter have different effects on transcription and bind Jun D, c-Fos, and Fra-2. Matrix Biol. 1995, 14, 715–725. [Google Scholar] [CrossRef]
- Dungera, S.; Neumannb, S.; Zellc, R.; Hirschfeldc, E.B.-; Stelznerc, A.; Paschkeb, R.; Kinne, R.W.; Sickingera, S. Mutation Detection in Mosaic Situations: RNA Mismatch Assay and Denaturing Gradient Gel Electrophoresis Are More Sensitive Than Conventional Cycle Sequencing. Anal. Biochem. 2001, 294, 89–93. [Google Scholar] [CrossRef]
- Poxon, S.W.; Hughes, J.A. A biofunctional assay to study pRL-CMV plasmid DNA formulation stability. PDA. J. Pharm. Sci. Technol. 1999, 53, 314–317. [Google Scholar]
- Kunisch, E.; Jansen, A.; Kojima, F.; Löffler, I.; Kapoor, M.; Kawai, S.; Rubio, I.; Crofford, L.J.; Kinne, R.W. Prostaglandin E2 Differentially Modulates Proinflammatory/Prodestructive Effects of TNF-α on Synovial Fibroblasts via Specific E Prostanoid Receptors/cAMP. J. Immunol. 2009, 183, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Aicher, W.K.; Dinkel, A.; Eibel, H.; Peter, H.H. Characterization of SV40 T antigen immortalized human synovial fibroblasts: Maintained expression patterns of EGR-i, HLA-DR and some surface receptors. Rheumatol. Int. 1997, 16, 241–247. [Google Scholar] [CrossRef]
- Pohlers, D.; Schmidt-Weber, C.B.; Franch, A.; Kuhlmann, J.; Brauer, R.; Emmrich, F.; Kinne, R.W. Differential clinical efficacy of anti-CD4 monoclonal antibodies in rat adjuvant arthritis is paralleled by differential influence on NF-kappaB binding activity and TNF-alpha secretion of T cells. Arthritis Res. 2002, 4, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Panterodt, T.; Welz, B.; Christmann, M.; Friesenhagen, J.; Westphal, A.; Pietsch, D.; Brand, K. C/EBPbeta-LAP*/LAP Expression Is Mediated by RSK/eIF4B-Dependent Signalling and Boosted by Increased Protein Stability in Models of Monocytic Differentiation. PLoS ONE 2015, 10, e0144338. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, H.; Dienst, S.; Emmrich, F.; Ahnert, P. CalcDalton: A tool for multiplex genotyping primer design for single-base extension reactions using cleavable primers. Biotechniques 2006, 40, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, H.; Teupser, D.; Weissfuss, J.; Wolfram, G.; Emmrich, F.; Ahnert, P. Robustness of single-base extension against mismatches at the site of primer attachment in a clinical assay. J. Mol. Med. 2006, 85, 361–369. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | Incidence of Mutations | |||||
|---|---|---|---|---|---|---|
| Gene | Description | nt Exchange | aa Exchange | RA | OA | NC |
| cfos | fos84 | T252C * | none | 7/11 | 3/10 | 1/5 |
| fos121/123 | C361G A367G | Gln121Glu Ile123Val | 0 | 1/10 | 0 | |
| fos125 | G374A | Arg125Lys | 4/11 | 0 | 0 | |
| fos73/125 | C217A G374A | Leu73Met Arg125Lys | 1/11 | 0 | 0 | |
| cjun | jun202 | 604–606ΔCAG | 202ΔGln | 2/10 | 0 | 0 |
| jun236 | C706T | Pro236Ser | 1/10 | 0 | 0 | |
| jun250 | G750A | none | 3/10 | 0 | 0 | |
| Observed Mutations | GnomAD [25,26] at aa Site | |||
|---|---|---|---|---|
| Gene | Description | Position hg19 (aa Exchange) | ID | Frequency (n-Minor Alleles) |
| cfos | fos73/ | chr14:75746655 (Leu73Met) | 14-75746684-C-T | L73F: 3 × 10−5 (1) |
| fos125 | chr14:75746812 (Arg125Lys) | n.d. | n.d. | |
| cjun | jun202 | chr1:59248125-59248133 (202ΔGln) | 1-59248123-GGCT-G | Q206del: 9 × 10−4 (204) |
| 1-59248123-G-GGCT | Q206dup: 2 × 10−4 (39) | |||
| 1-59248123-G-GGCTGCT | Q205_206dup: 2 × 10−5 (5) | |||
| 1-59248123-GGCT-G | Q205_206del: 5 × 10−6 (1) | |||
| jun236 | chr1:59248037 (Pro236Ser) | n.d. | n.d. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huber, R.; Augsten, S.; Kirsten, H.; Zell, R.; Stelzner, A.; Thude, H.; Eidner, T.; Stuhlmüller, B.; Ahnert, P.; Kinne, R.W. Identification of New, Functionally Relevant Mutations in the Coding Regions of the Human Fos and Jun Proto-Oncogenes in Rheumatoid Arthritis Synovial Tissue. Life 2021, 11, 5. https://doi.org/10.3390/life11010005
Huber R, Augsten S, Kirsten H, Zell R, Stelzner A, Thude H, Eidner T, Stuhlmüller B, Ahnert P, Kinne RW. Identification of New, Functionally Relevant Mutations in the Coding Regions of the Human Fos and Jun Proto-Oncogenes in Rheumatoid Arthritis Synovial Tissue. Life. 2021; 11(1):5. https://doi.org/10.3390/life11010005
Chicago/Turabian StyleHuber, René, Sandra Augsten, Holger Kirsten, Roland Zell, Axel Stelzner, Hansjörg Thude, Thorsten Eidner, Bruno Stuhlmüller, Peter Ahnert, and Raimund W. Kinne. 2021. "Identification of New, Functionally Relevant Mutations in the Coding Regions of the Human Fos and Jun Proto-Oncogenes in Rheumatoid Arthritis Synovial Tissue" Life 11, no. 1: 5. https://doi.org/10.3390/life11010005
APA StyleHuber, R., Augsten, S., Kirsten, H., Zell, R., Stelzner, A., Thude, H., Eidner, T., Stuhlmüller, B., Ahnert, P., & Kinne, R. W. (2021). Identification of New, Functionally Relevant Mutations in the Coding Regions of the Human Fos and Jun Proto-Oncogenes in Rheumatoid Arthritis Synovial Tissue. Life, 11(1), 5. https://doi.org/10.3390/life11010005