Inferring the Phylogenetic Positions of Two Fig Wasp Subfamilies of Epichrysomallinae and Sycophaginae Using Transcriptomes and Mitochondrial Data


Abstract
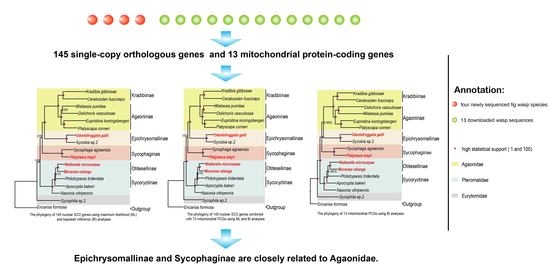
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Data Collection
2.2. RNA Extraction, Transcriptome Sequencing and Assembly
2.3. Prediction of CDS and Identification of SCO Genes
2.4. DNA Extraction, Library Construction and Sequencing; Mitogenome Assembly and Annotation of 13 PCGs
2.5. Multiple Sequences Alignment, Model Selection and Construction of Phylogeny
3. Results
3.1. Transcriptomes and Mitogenomes of the Four Newly Sequenced Fig Wasp Species Obtained from High-Throughput Sequencing
3.2. Identification of SCO Genes and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weiblen, G.D. How to be a fig wasp. Annu. Rev. Entomol. 2002, 47, 299–330. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.A.; Jousselin, E.; Kjellberg, F.; Compton, S.G.; Herre, E.A. Phylogenetic relationships, historical biogeography and character evolution of fig-pollinating wasps. Proc. Biol. Sci. 2001, 268, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.A.; Robbins, N.; Gilbert, M.T.P.; Herre, E.A. Critical review of host specificity and its coevolutionary implications in the fig/fig-wasp mutualism. Proc. Natl. Acad. Sci. USA 2005, 102 (Suppl. S1), 6558–6565. [Google Scholar] [CrossRef]
- Heraty, J.M.; Burks, R.A.; Cruaud, A.; Gibson, G.A.P.; Liljeblad, J.; Munro, J.; Rasplus, J.-Y.; Delvare, G.; Janšta, P.; Gumovsky, A.; et al. A phylogenetic analysis of the megadiverse chalcidoida. Cladistics 2013, 29, 466–542. [Google Scholar] [CrossRef]
- Hill, D.S. Figs (Ficus spp.) and fig-wasps (Chalcidoidea). J. Nat. Hist. 1967, 1, 413–434. [Google Scholar] [CrossRef]
- Bouček, Z.; Watsham, A.; Wiebes, J.T. The fig wasp fauna of the receptacles of Ficus thonningii (Hymenoptera, Chalcidoidea). Tijdschr. Entomol. 1981, 124, 149–233. [Google Scholar]
- Bouček, Z. Australasian Chalcidoidea (Hymenoptera): A Biosystematic Revision of Genera of Fourteen Families, with a Reclassification of Species; CAB International: Wallingford, UK, 1988. [Google Scholar]
- Rasplus, J.-Y.; Kerdelhue, C.; Le Clainche, I.; Mondor, G. Molecular phylogeny of fig wasps. Agaonidae are not monophyletic. C. R. Acad. Sci. III 1998, 321, 517–526. [Google Scholar] [CrossRef]
- Walker, F. Descriptions of new gengera and species of parasites, belonging to the families Proctotrupidae and Chalcididae, which attack insects destructive to the fig in India. Entomologist 1875, 8, 15–18. [Google Scholar]
- Joseph, K.J. A proposed revision of the classification of the fig insects of the families Agaonidae and Torymidae (Hymenoptera). Proc. R. Entomol. Soc. Lon. B 1964, 33, 63–66. [Google Scholar]
- Peters, R.S.; Niehuis, O.; Gunkel, S.; Blaser, M.; Mayer, C.; Podsiadlowski, L.; Kozlov, A.; Donath, A.; van Noort, S.; Liu, S.; et al. Transcriptome sequence-based phylogeny of chalcidoid wasps (Hymenoptera: Chalcidoidea) reveals a history of rapid radiations, convergence, and evolutionary success. Mol. Phylogenet. Evol. 2018, 120, 286–296. [Google Scholar] [CrossRef]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Cao, Y.; Wang, J.; Xiong, Z. Transcriptome sequencing of Pinus kesiya var. langbianensis and comparative analysis in the Pinus phylogeny. BMC Genom. 2018, 19, 725. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Wang, R.Q.; Wang, D.Z.; Li, C.T.; Yang, X.R. Mitochondrial genome of the shorthead catfish (Pelteobagrus eupogon): Structure, phylogeny, and intraspecific variation. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Bouček, Z. The genera of chalcidoid wasps from Ficus fruit in the New World. J. Nat. Hist. 1993, 27, 173–217. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using Trace signals and additional sequence Information. J. Comput. Sci. Syst. Biol. 1999, 99, 45–56. [Google Scholar]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Altekar, G.; Dwarkadas, S.; Huelsenbeck, J.P.; Ronquist, F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 2004, 20, 407–415. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J. Tracer v1.4. Encyclopedia Atmos. Sci. 2007, 141, 2297–2305. [Google Scholar] [CrossRef]
- Philippe, H.; Delsuc, F.; Brinkmann, H.; Lartillot, N. Phylogenomics. Annu. Rev. Ecol. Evol. S. 2005, 36, 541–562. [Google Scholar] [CrossRef]
- Dong, S.; Xiao, Y.; Kong, H.; Feng, C.; Harris, A.J.; Yan, Y.; Kang, M. Nuclear loci developed from multiple transcriptomes yield high resolution in phylogeny of scaly tree ferns (Cyatheaceae) from China and Vietnam. Mol. Phylogenet. Evol. 2019, 139, 106567. [Google Scholar] [CrossRef]
- Wang, G.; Lin, J.; Shi, Y.; Chang, X.; Wang, Y.; Guo, L.; Wang, W.; Dou, M.; Deng, Y.; Ming, R.; et al. Mitochondrial genome in Hypsizygus marmoreus and its evolution in Dikarya. BMC Genom. 2019, 20, 765. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Maddison, W.P. Molecular approaches and the growth of phylogenetic biology. In Molecular Zoology: Advances, Strategies, and Protocols; Ferraris, J.D., Palumbi, S.R., Eds.; Wiley-Liss: New York, NY, USA, 1996; pp. 47–63. [Google Scholar]
- Lin, G.-H.; Wang, K.; Deng, X.-G.; Nevo, E.; Zhao, F.; Su, J.-P.; Guo, S.-C.; Zhang, T.-Z.; Zhao, H. Transcriptome sequencing and phylogenomic resolution within Spalacidae (Rodentia). BMC Genom. 2014, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hong, W.; Jiao, H.; Zhao, H. Transcriptome sequencing and phylogenetic analysis of four species of luminescent beetles. Sci. Rep. 2017, 7, 1814. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Subfamily | Family | Accession No. (Mitochondrial Genome) | Accession No. (Genomes or Transcriptomes) |
|---|---|---|---|---|
| Platyneura mayri * | Sycophaginae * | MW167114 | PRJNA672045 | |
| Odontofroggatia galili * | Epichrysomallinae * | MW167113 | PRJNA671819 | |
| Walkerella microcarpae * | Otitesellinae * | Pteromalidae | MW167116 | PRJNA672219 |
| Micranisa ralianga * | Otitesellinae * | Pteromalidae | MW167115 | PRJNA672141 |
| Euprisitina koningsbergeri # | Agaoninae # | Agaonidae | MT947597 | PRJNA641212 |
| Platyscapa corneri # | Agaoninae # | Agaonidae | MT947604 | PRJNA641212 |
| Dolichoris vasculosae # | Agaoninae # | Agaonidae | MT947596 | PRJNA641212 |
| Wiebesia pumilae # | Agaoninae # | Agaonidae | MT947601 | PRJNA641212 |
| Kradibia gibbosae # | Kradibiinae # | Agaonidae | MT947598 | PRJNA641212 |
| Ceratosolen fusciceps # | Kradibiinae # | Agaonidae | MT916179 | PRJNA494992 |
| Sycophaga agreansis * | Sycophaginae * | MT947599 | PRJNA641212 | |
| Sycophila sp.2 * | - | Eurytomidae | MT947603 | PRJNA641212 |
| Sycobia sp.2 * | Epichrysomallinae * | MT947600 | PRJNA641212 | |
| Apocrypta bakeri * | Sycoryctinae * | Pteromalidae | MT906648 | PRJNA641212 |
| Philotrypesis tridentata * | Sycoryctinae * | Pteromalidae | MT947602 | PRJNA641212 |
| Nasonia vitripennis | Pteromalinae | Pteromalidae | EU746609.1, EU746613.1 | PRJNA594415 |
| Encarisa formosa | Coccophaginae | Aphelinidae | MG813797.1 | PRJNA252167 |
| Species | Primers | Sequence (5′–3’) | Annealing Temperature | Targeted CDS Region |
|---|---|---|---|---|
| Platyneura mayri | PF1 | ctatataaatttatgaaactatgattaatatctactaatcataaatatattgg | 54 °C | The middle part of the cox1 |
| PR1 | gataatctaggaggtaataatcaaaatcttatattatttattcgtgg | |||
| Micranisa ralianga | MF1 | caattaaagttaaacaaattaataagtaaataattgaaattaatattg | 50 °C | The middle part of the nad6 |
| MR1 | caatttaataataatcattgattttcttatattatatttttaatcatagtag |
| Platyneura Mayri | Odontofroggatia Galili | Walkerella Microcarpae | Micranisa Ralianga | |||||
|---|---|---|---|---|---|---|---|---|
| Length Range | Transcript | Unigene | Transcript | Unigene | Transcript | Unigene | Transcript | Unigene |
| 200 bp–500 bp | 14,720 | 4950 | 38,315 | 13,104 | 21,455 | 6249 | 24,187 | 7148 |
| 500 bp–1000 bp | 11,155 | 6015 | 27,822 | 14,868 | 13,405 | 8393 | 13,659 | 8435 |
| 1000 bp–2000 bp | 9368 | 3799 | 20,849 | 8211 | 11,040 | 4716 | 9517 | 4391 |
| >2000 bp | 16,581 | 5705 | 19,191 | 6501 | 22,073 | 6541 | 19,922 | 6411 |
| Total Number | 51,824 | 20,469 | 106,177 | 42,684 | 67,973 | 25,899 | 67,285 | 26,385 |
| Total Length | 99,849,096 | 35,330,681 | 130,935,638 | 49,857,034 | 133,605,390 | 42,636,711 | 127,749,958 | 42,751,535 |
| Mean Length | 1927 | 1726 | 1233 | 1168 | 1966 | 1646 | 1899 | 1620 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Xin, Z.; Hou, H.; Zhou, Y.; Wang, J.; Xiao, J.; Huang, D. Inferring the Phylogenetic Positions of Two Fig Wasp Subfamilies of Epichrysomallinae and Sycophaginae Using Transcriptomes and Mitochondrial Data. Life 2021, 11, 40. https://doi.org/10.3390/life11010040
Zhao D, Xin Z, Hou H, Zhou Y, Wang J, Xiao J, Huang D. Inferring the Phylogenetic Positions of Two Fig Wasp Subfamilies of Epichrysomallinae and Sycophaginae Using Transcriptomes and Mitochondrial Data. Life. 2021; 11(1):40. https://doi.org/10.3390/life11010040
Chicago/Turabian StyleZhao, Dan, Zhaozhe Xin, Hongxia Hou, Yi Zhou, Jianxia Wang, Jinhua Xiao, and Dawei Huang. 2021. "Inferring the Phylogenetic Positions of Two Fig Wasp Subfamilies of Epichrysomallinae and Sycophaginae Using Transcriptomes and Mitochondrial Data" Life 11, no. 1: 40. https://doi.org/10.3390/life11010040
APA StyleZhao, D., Xin, Z., Hou, H., Zhou, Y., Wang, J., Xiao, J., & Huang, D. (2021). Inferring the Phylogenetic Positions of Two Fig Wasp Subfamilies of Epichrysomallinae and Sycophaginae Using Transcriptomes and Mitochondrial Data. Life, 11(1), 40. https://doi.org/10.3390/life11010040