Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania during the First Months of the Pandemic

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Whole-Genome Sequencing (WGS) of SARS-CoV-2 Strains
2.3. WGS Assembling and Reference Mapping
2.4. Phylogenetic Analysis
2.5. Lineage and Mutations Prediction
2.6. Variability Profiling
2.7. Graphic Representations
2.8. Nucleotide Sequence Accession Numbers
3. Results
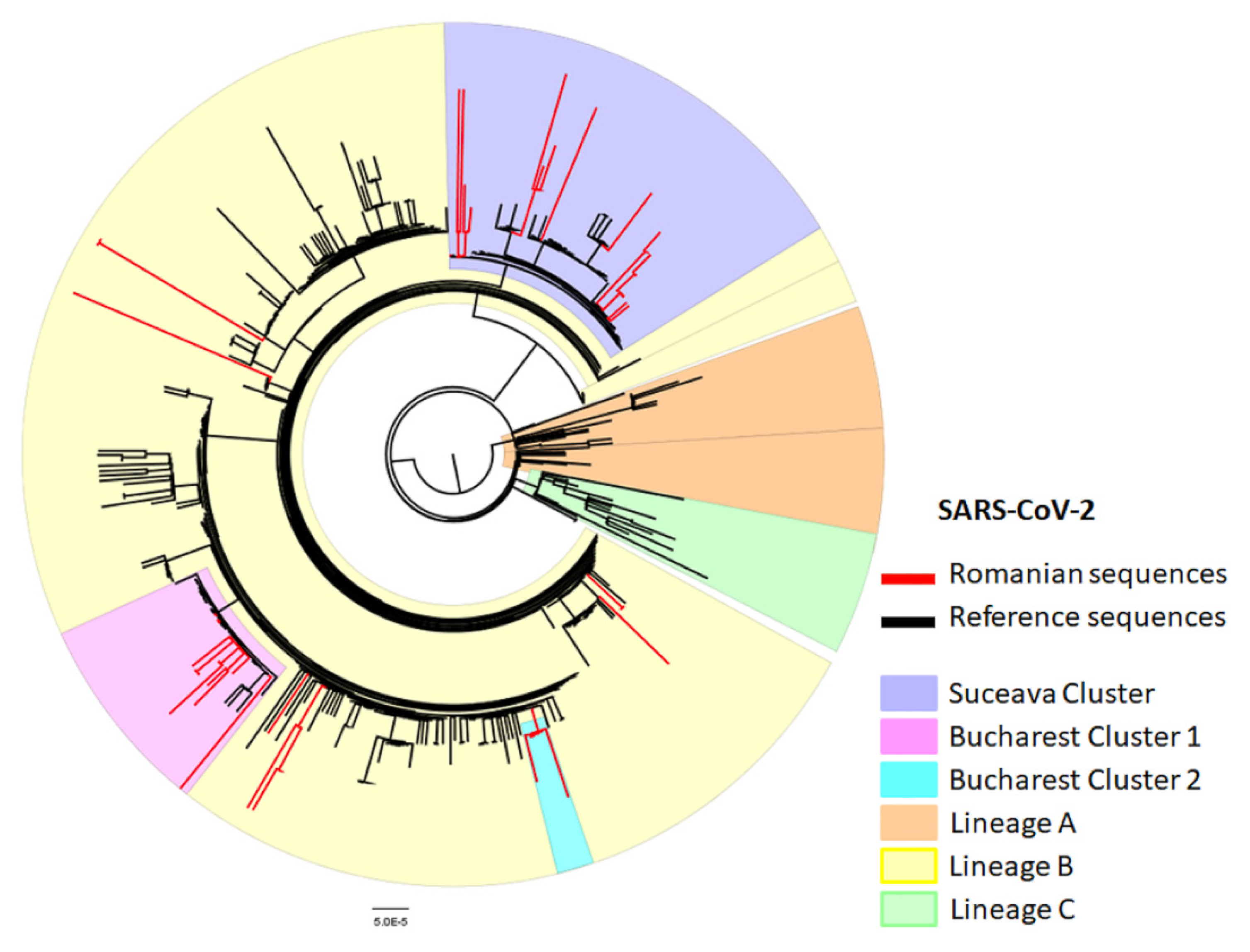
3.1. Phylogenetic and Subtyping Analysis
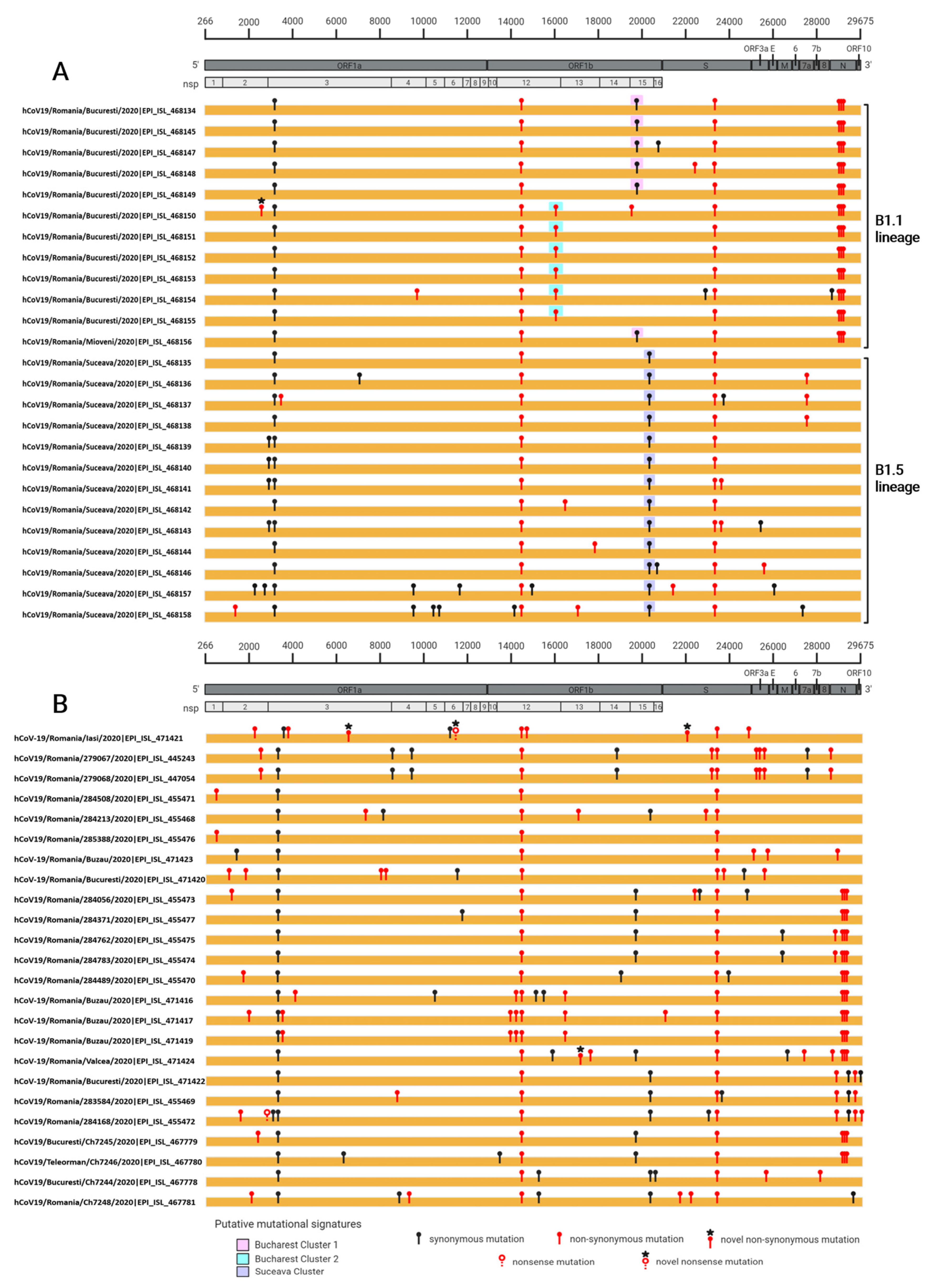
3.2. Mutations Analysis
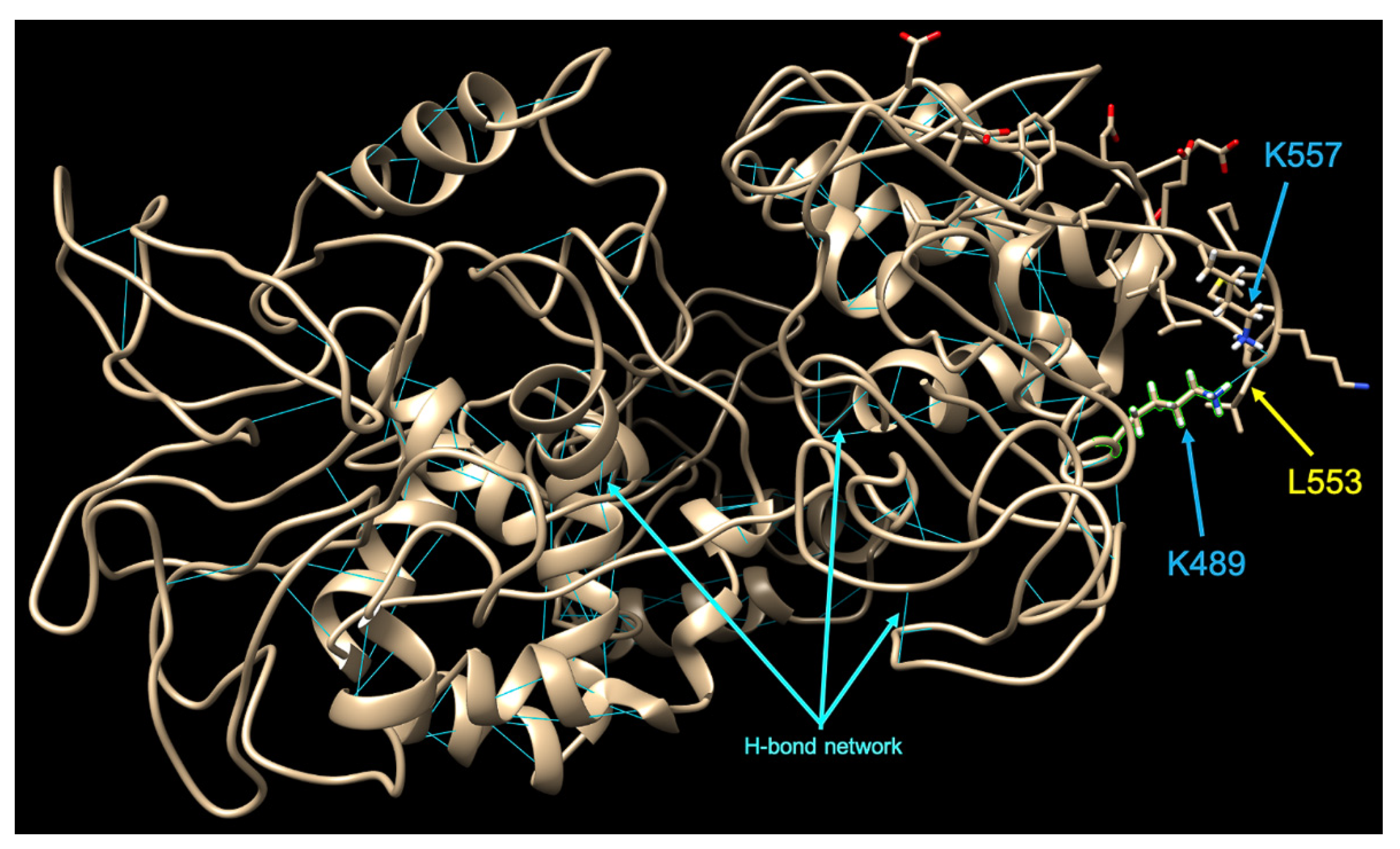
3.3. Variability Profile
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petersen, E.; Petrosillo, N.; Koopmans, M. ESCMID Emerging Infections Task Force Expert Panel. Emerging infections—an increasingly important topic: Review by the Emerging Infections Task Force. Clin. Microbiol. Infect. 2018, 24, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Rothe, C.; Schunk, M.; Sothmann, P.; Bretzel, G.; Froeschl, G.; Wallrauch, C.; Zimmer, T.; Thiel, V.; Janke, C.; Guggemos, W.; et al. Transmission of 2019-NCOV infection from an asymptomatic contact in Germany. N. Engl. J. Med. 2020, 382, 970–971. [Google Scholar] [CrossRef]
- Prather, K.A.; Wang, C.C.; Schooley, R.T. Reducing transmission of SARS-CoV-2. Science 2020, 368, 1422–1424. [Google Scholar] [CrossRef]
- Streinu-Cercel, A. SARS-CoV-2 in Romania—Situation Update and Containment Strategies. Germs 2020, 10, 8. [Google Scholar] [CrossRef]
- Asadi, S.; Bouvier, N.; Wexler, A.S.; Ristenpart, W.D. The coronavirus pandemic and aerosols: Does COVID-19 transmit via expiratory particles? Aerosol Sci. Tech. 2020, 54, 635–638. [Google Scholar] [CrossRef]
- Lemey, P.; Pybus, O.G.; Bin, W.; Saksena, N.K.; Salemi, M.; Vandamme, A.M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. USA 2003, 100, 6588–6592. [Google Scholar] [CrossRef]
- Holmes, E.C.; Dudas, G.; Rambaut, A.; Andersen, K.G. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature 2016, 538, 193–200. [Google Scholar] [CrossRef]
- Yang, C.F.; Chang, S.F.; Hsu, T.C.; Su, C.L.; Wang, T.C.; Lin, S.H.; Yang, S.L.; Lin, C.C.; Shu, P.Y. Molecular characterization and phylogenetic analysis of dengue viruses imported into Taiwan during 2011–2016. PLoS Negl. Trop Dis. 2018, 12, 9. [Google Scholar] [CrossRef]
- Lam, T.T.Y.; Jia, N.; Zhang, Y.W.; Shum, M.H.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020. [Google Scholar] [CrossRef]
- Paraskevis, D.; Kostaki, E.G.; Magiorkinis, G.; Panayiotakopoulos, G.; Sourvinos, G.; Tsiodras, S. Full-genome evolutionary analysis of the novel coronavirus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect. Genet. Evol. 2020, 79, 104212. [Google Scholar] [CrossRef] [PubMed]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 17, 9241–9243. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; Hill, V.; OToole, A.; McCrone, J.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints 2020. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Agerpres. Video Conference Held by Ludovic Orban, the Romanian PM. Available online: https://www.agerpres.ro/politica/2020/05/04/video-conferinta-de-presa-sustinuta-de-prim-ministrul-ludovic-orban-la-ora-20-00--498320 (accessed on 6 July 2020).
- CNSCBT. Analysis of Confirmed COVID-19 Cases in Romania. Available online: https://www.cnscbt.ro/index.php/analiza-cazuri-confirmate-covid19/1643-analiza-cazuri-confirmate-pana-la-5-04-2020/file (accessed on 6 July 2020).
- MAI.GOV.RO. Briefing by the National Institute of Public Health. 25 May 2020. Available online: https://www.mai.gov.ro/informare-covid-19-grupul-de-comunicare-strategica-25-mai-2020-ora-13-00/ (accessed on 6 July 2020).
- CNSCBT. COVID-19 Surveillance Methodology, Actualised 16 March 2020. Available online: http://cnscbt.ro/index.php/metodologii/infectia-2019-cu-ncov/1535-metodologia-de-supraveghere-a-covid-19-actualizare-16-03-2020-1/file (accessed on 6 July 2020).
- CNSCBT. COVID-19 Surveillance Methodology, Actualised 23 March 2020. Available online: http://cnscbt.ro/index.php/metodologii/infectia-2019-cu-ncov/1570-metodologia-de-supraveghere-a-covid-19-actualizare-23-03-2020-1/file (accessed on 6 July 2020).
- Castells, M.; Lopez-Tort, F.; Colina, R.; Cristina, J. Evidence of Increasing Diversification of Emerging SARS-CoV-2 Strains. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, D.; Lon, J.R.; Chen, X.; Hu, M.; Lin, S.; Chen, Z.; Wang, X.; Meng, Y.; Du, H. Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes revealed its epidemic trend and possible origins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making sense of mutation: What D614G means for the COVID-19 pandemic remains unclear. Cell 2020. [Google Scholar] [CrossRef]
- Sakai, Y.; Kawachi, K.; Terada, Y.; Omori, H.; Matsuura, Y.; Kamitani, W. Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 2017, 510, 165–174. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Graham, R.L.; Sims, A.C.; Brockway, S.M.; Baric, R.S.; Denison, M.R. The nsp2 Replicase Proteins of Murine Hepatitis Virus and Severe Acute Respiratory Syndrome Coronavirus Are Dispensable for Viral Replication. J. Virol. 2005, 79, 13399–13411. [Google Scholar] [CrossRef] [PubMed]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R.; Kuhn, P.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 2 Interacts with a Host Protein Complex Involved in Mitochondrial Biogenesis and Intracellular Signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surleac, M.; Banica, L.; Casangiu, C.; Cotic, M.; Florea, D.; Sandulescu, O.; Milu, P.; Streinu-Cercel, A.; Vlaicu, O.; Paraskevis, D.; et al. Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania during the First Months of the Pandemic. Life 2020, 10, 152. https://doi.org/10.3390/life10080152
Surleac M, Banica L, Casangiu C, Cotic M, Florea D, Sandulescu O, Milu P, Streinu-Cercel A, Vlaicu O, Paraskevis D, et al. Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania during the First Months of the Pandemic. Life. 2020; 10(8):152. https://doi.org/10.3390/life10080152
Chicago/Turabian StyleSurleac, Marius, Leontina Banica, Corina Casangiu, Marius Cotic, Dragos Florea, Oana Sandulescu, Petre Milu, Anca Streinu-Cercel, Ovidiu Vlaicu, Dimitrios Paraskevis, and et al. 2020. "Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania during the First Months of the Pandemic" Life 10, no. 8: 152. https://doi.org/10.3390/life10080152
APA StyleSurleac, M., Banica, L., Casangiu, C., Cotic, M., Florea, D., Sandulescu, O., Milu, P., Streinu-Cercel, A., Vlaicu, O., Paraskevis, D., Paraschiv, S., & Otelea, D. (2020). Molecular Epidemiology Analysis of SARS-CoV-2 Strains Circulating in Romania during the First Months of the Pandemic. Life, 10(8), 152. https://doi.org/10.3390/life10080152