Struvite Crystallisation and the Effect of Co2+ Ions

 , , , and
, , , and
Abstract
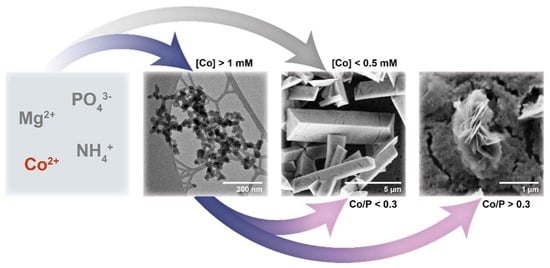
:
1. Introduction
2. Materials and Methods
2.1. Crystallisation Experiments
2.2. Analytical Methods
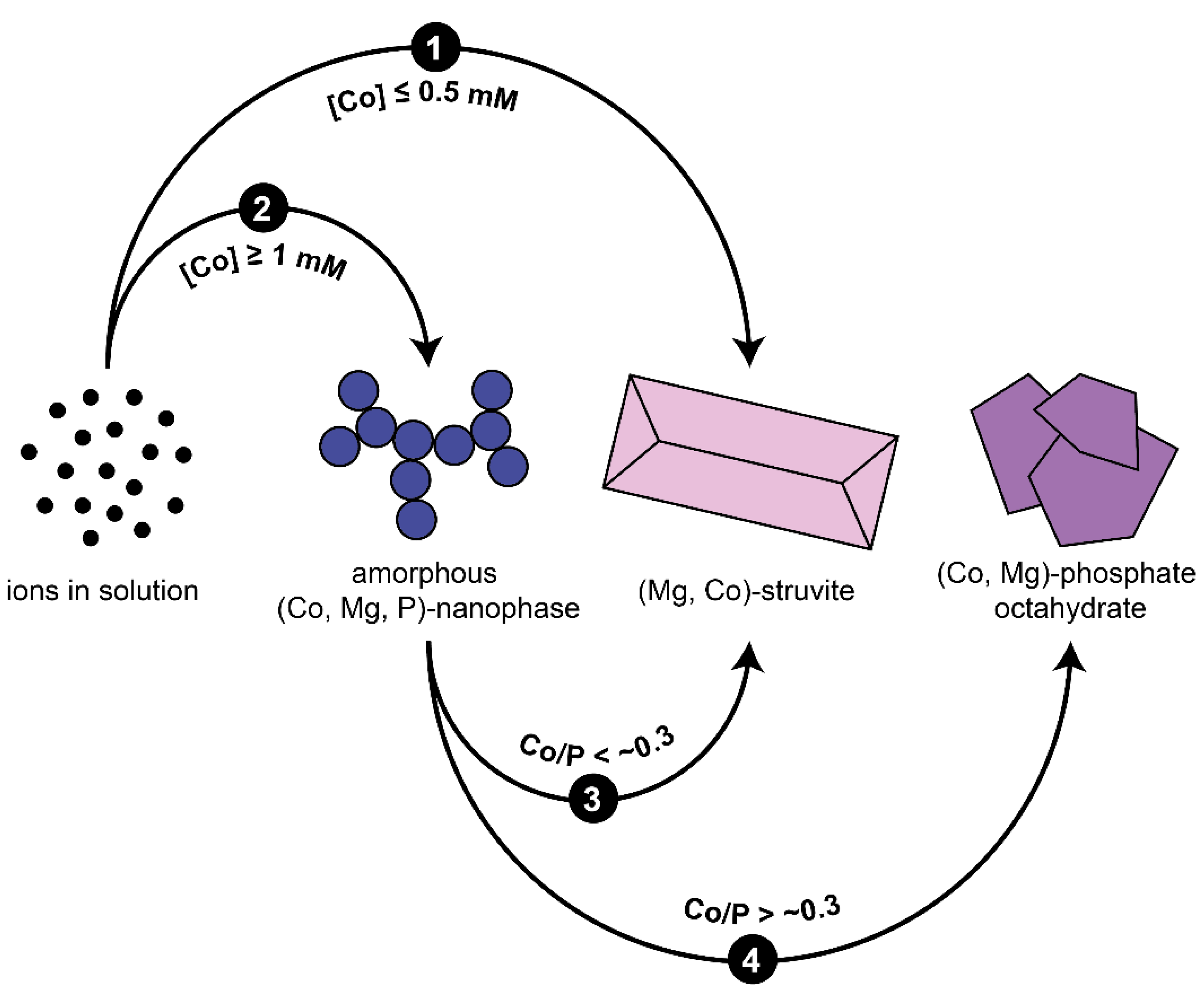
3. Results and Discussion
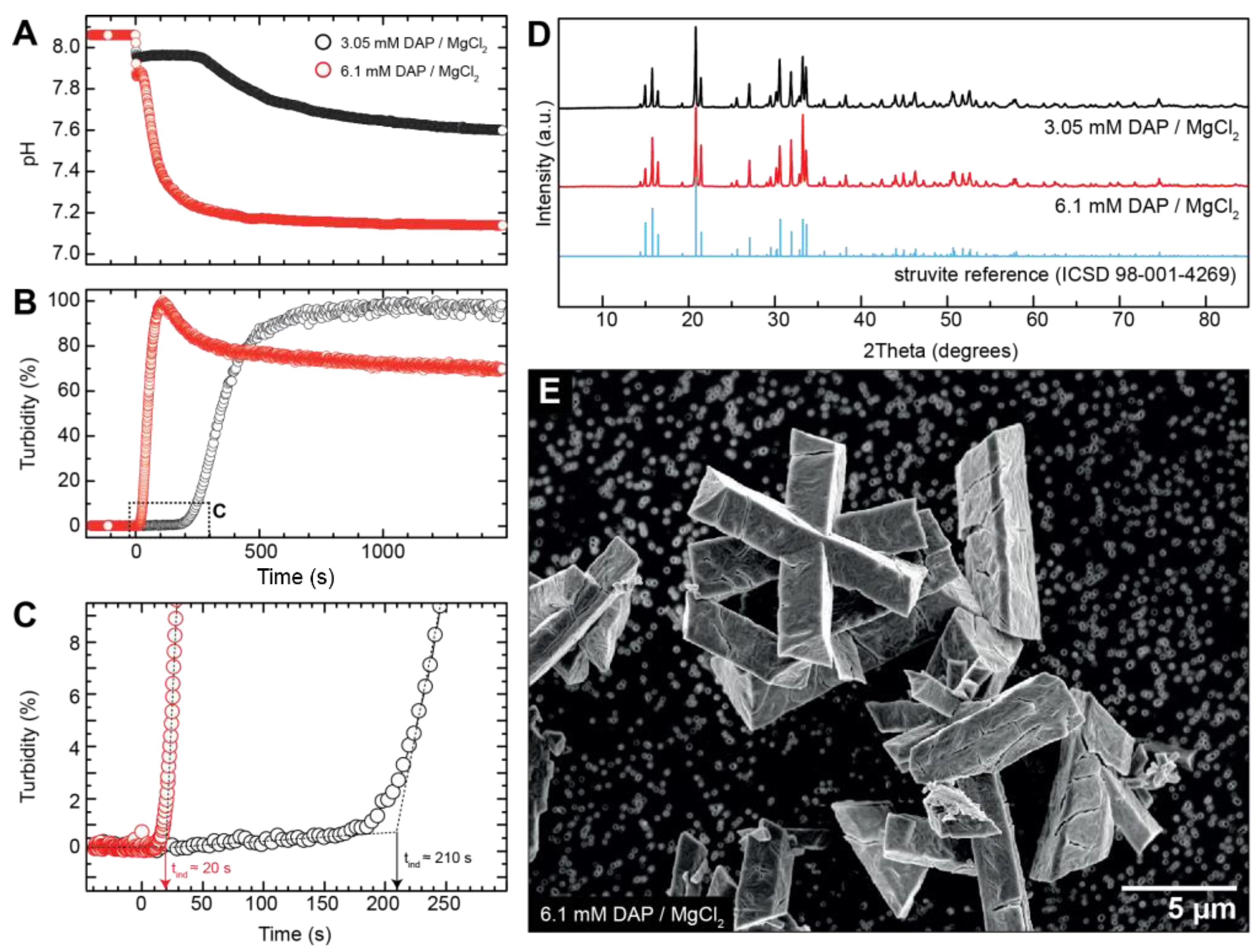
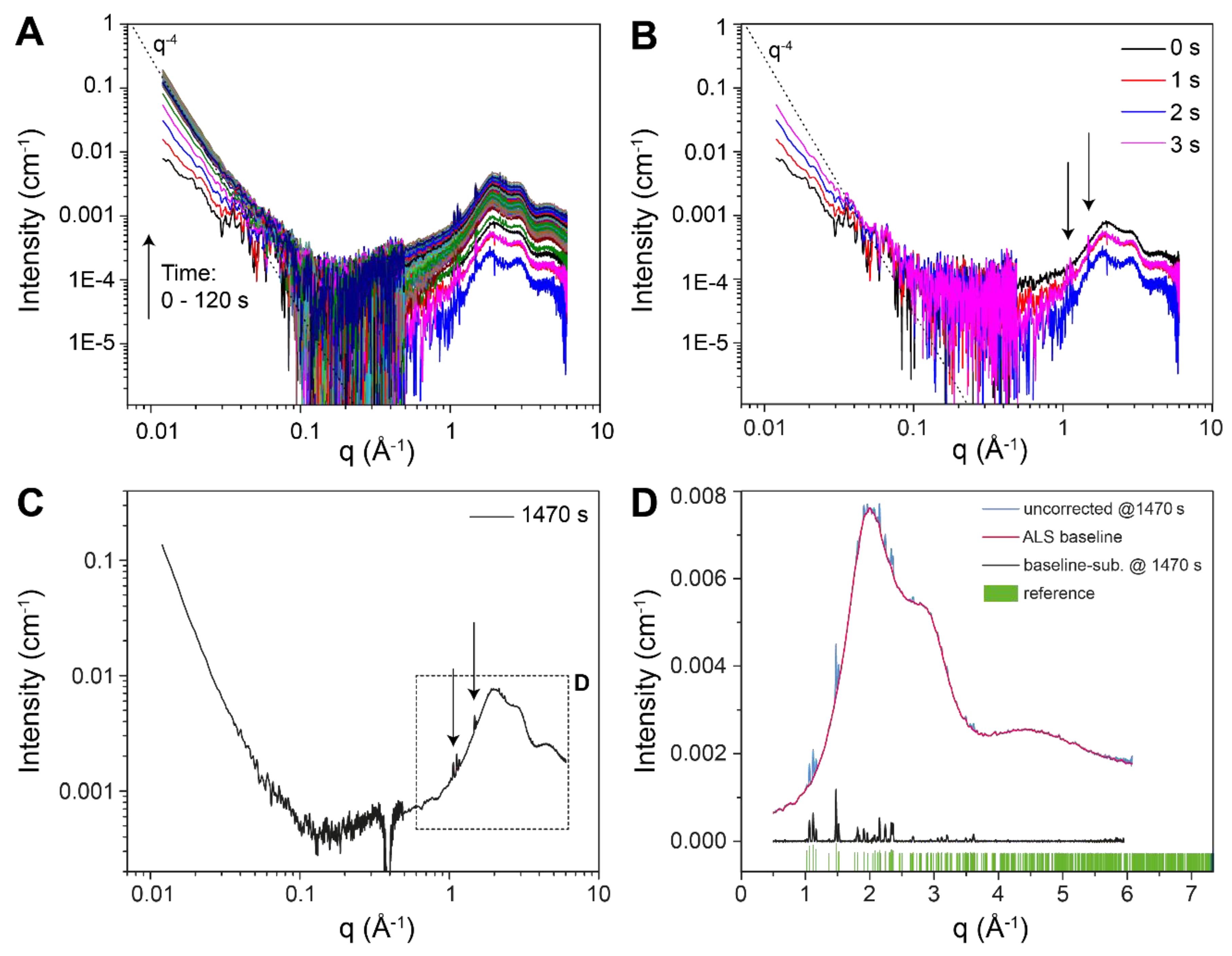
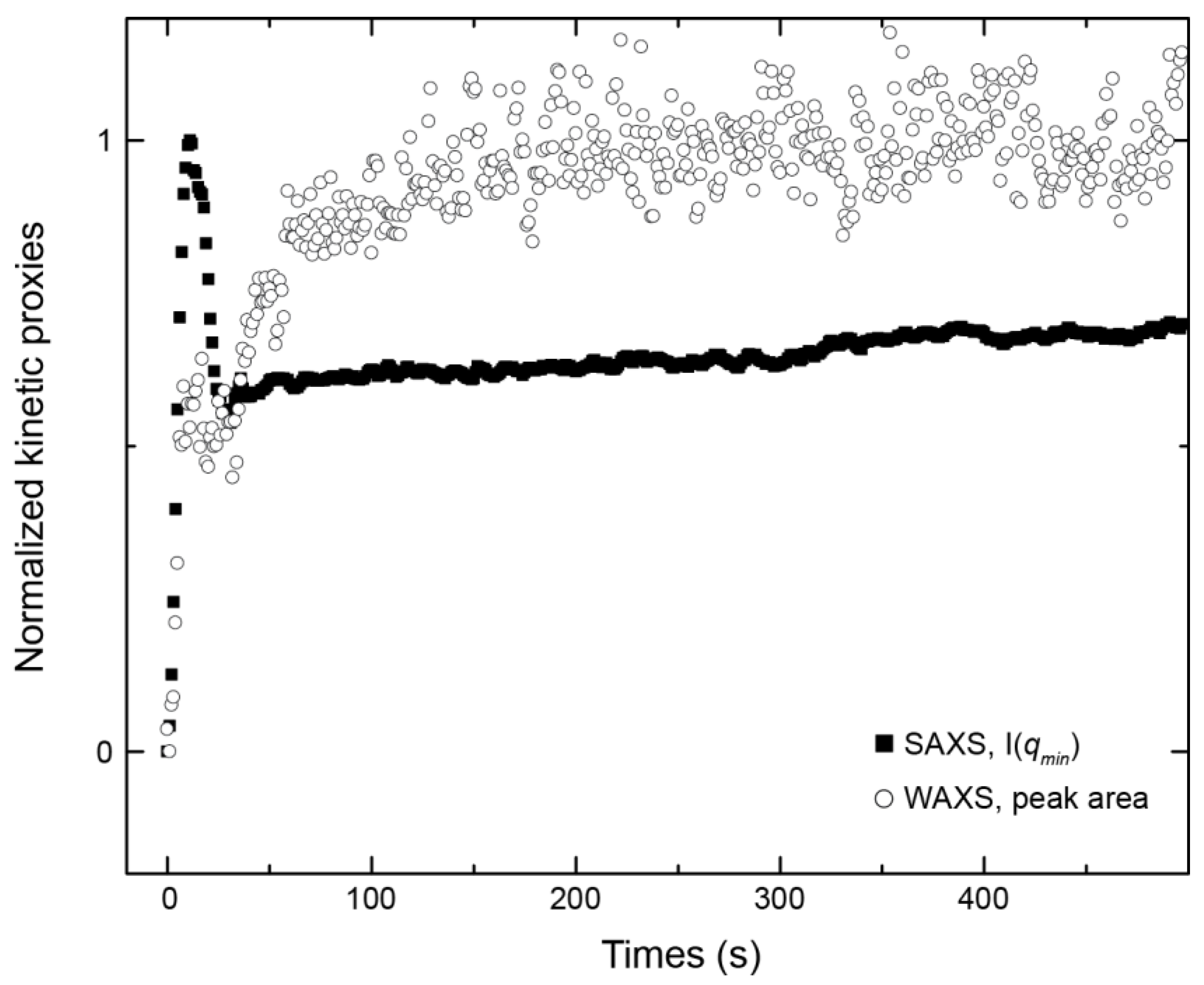
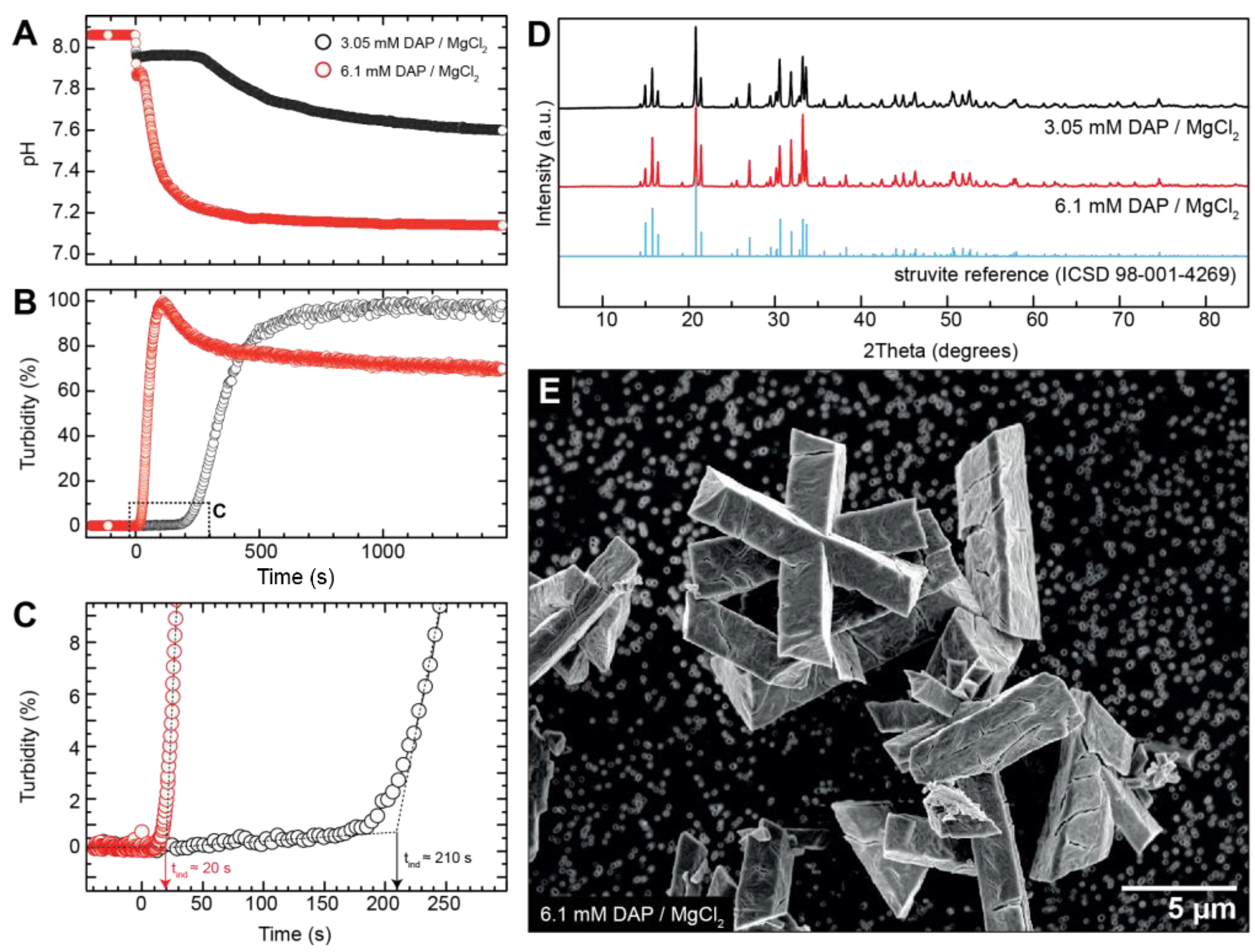
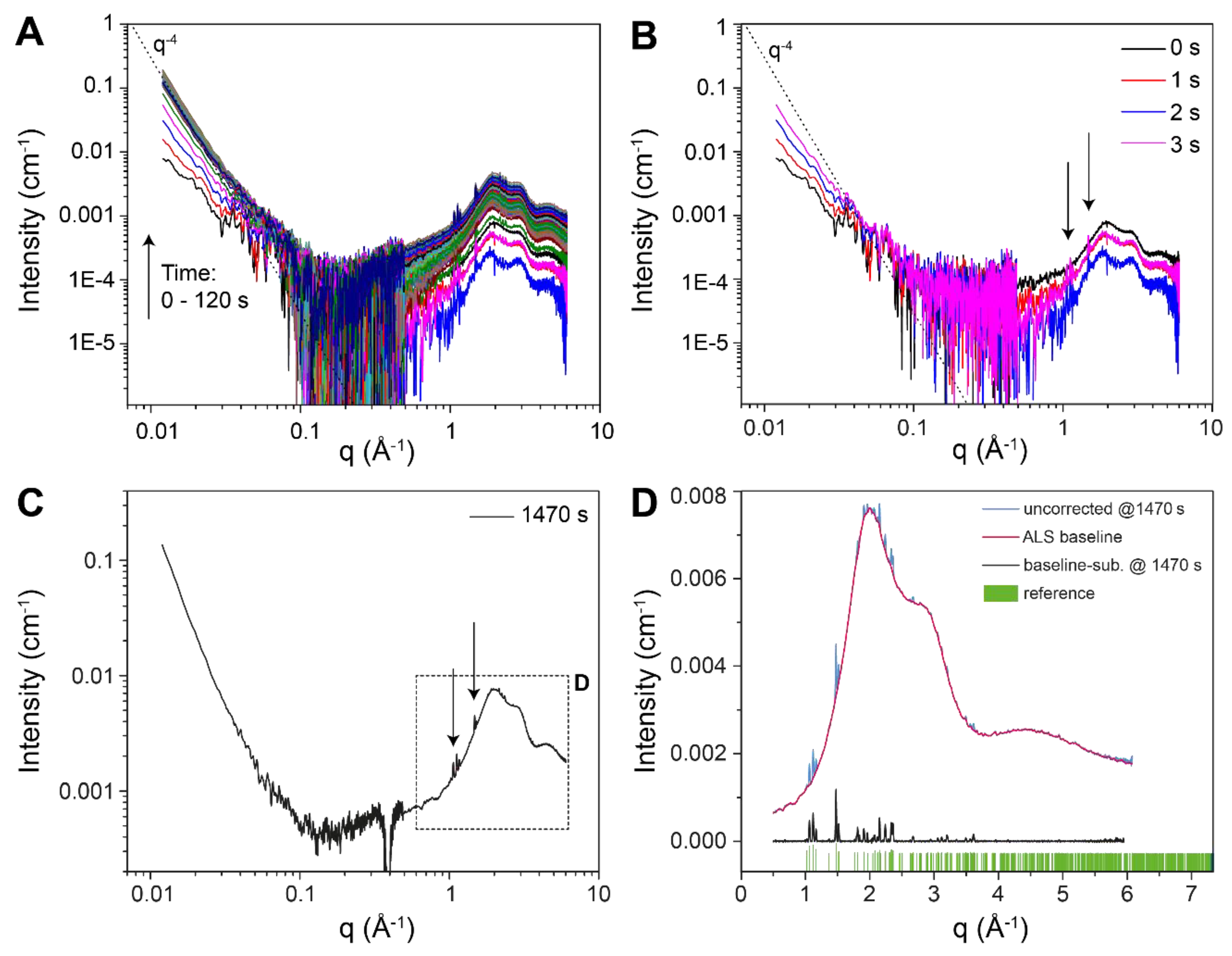
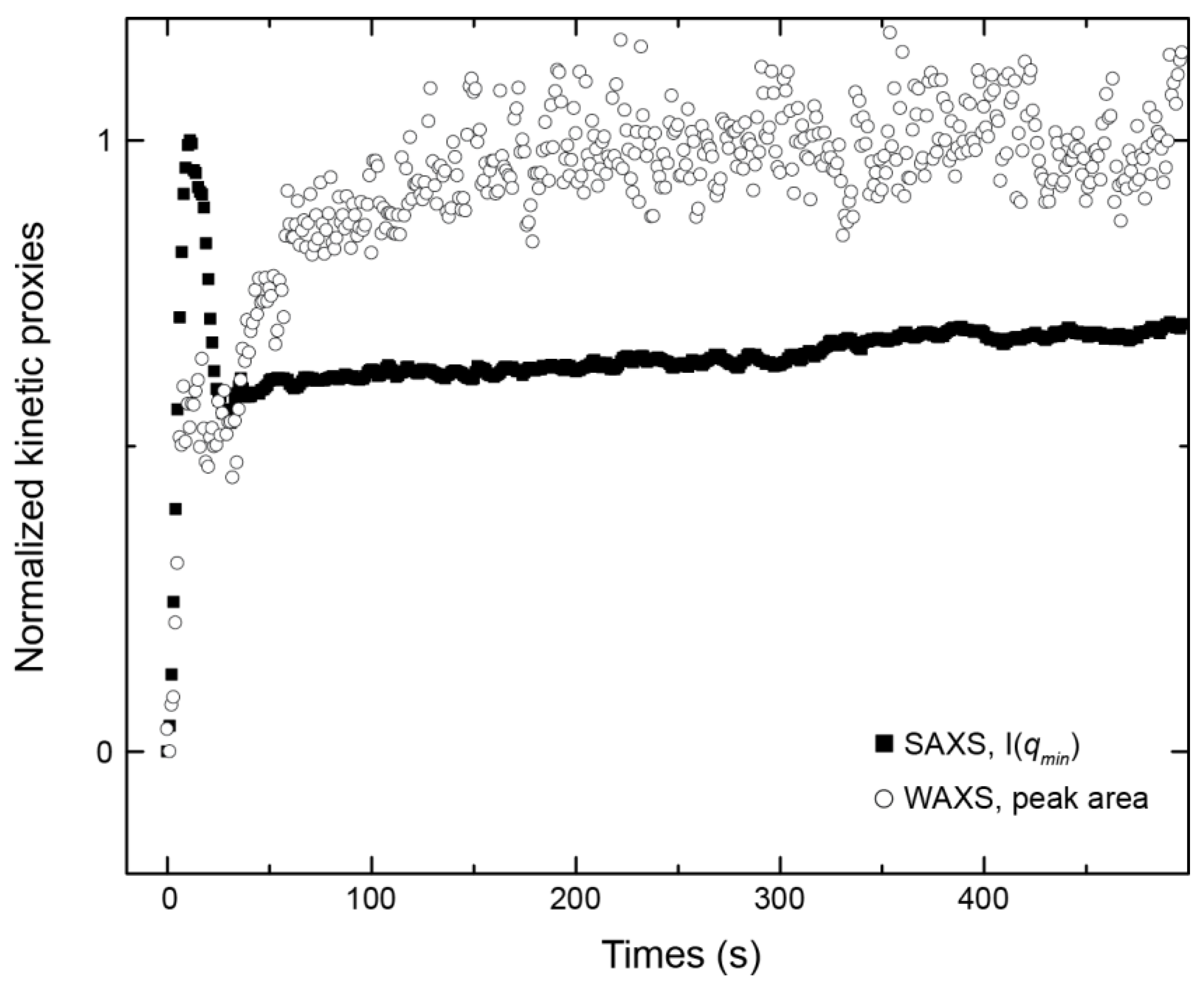
3.1. Crystallisation in the Absence of Co
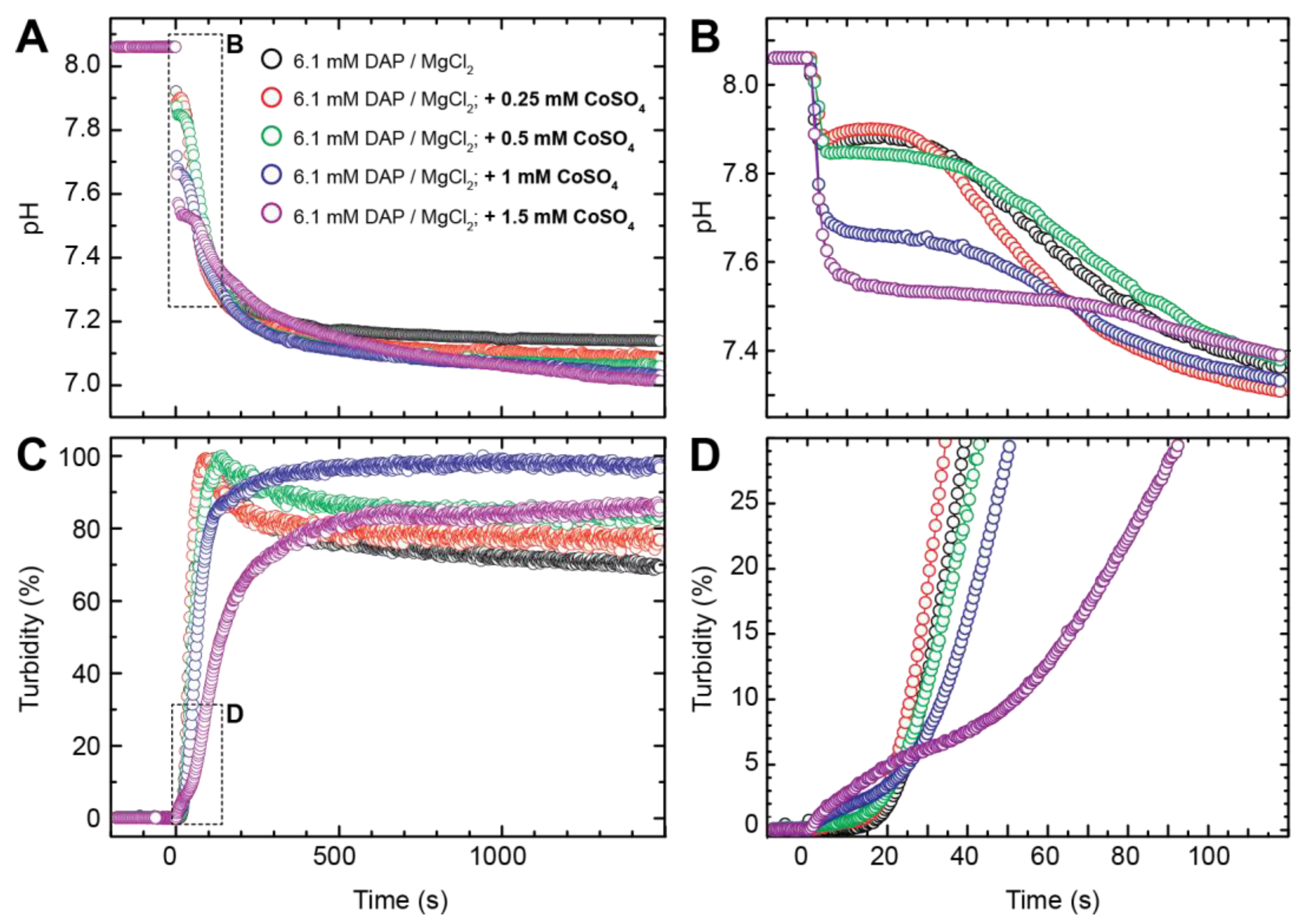
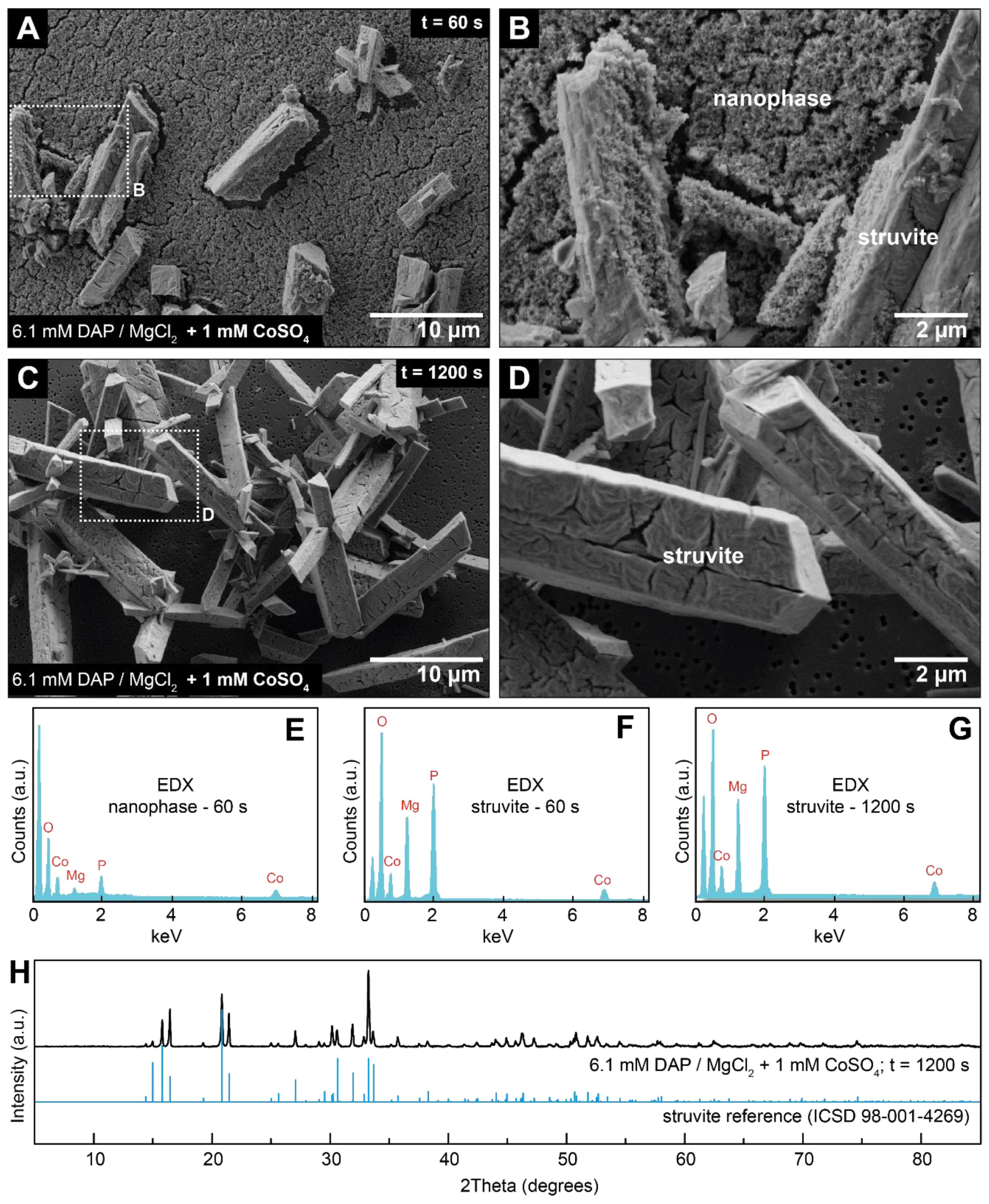
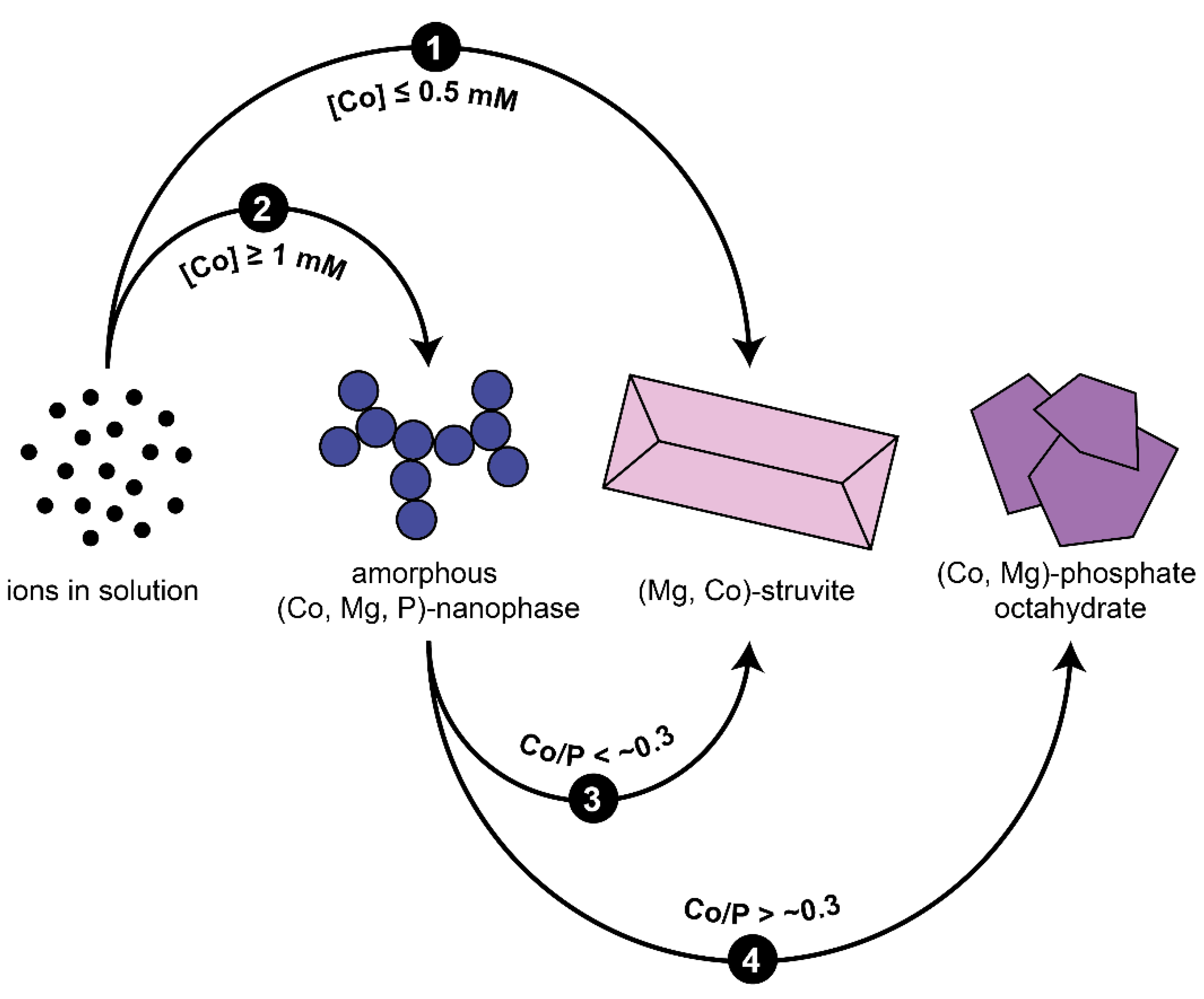
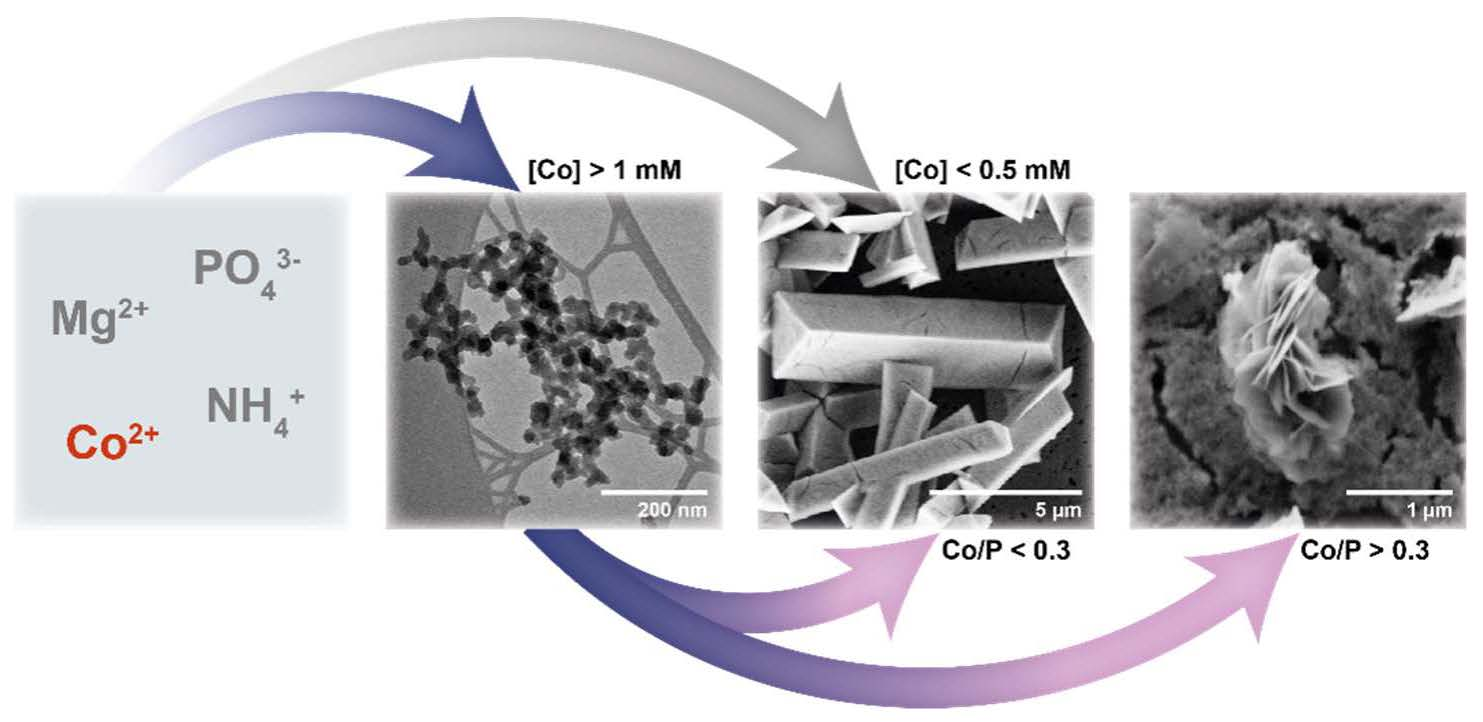
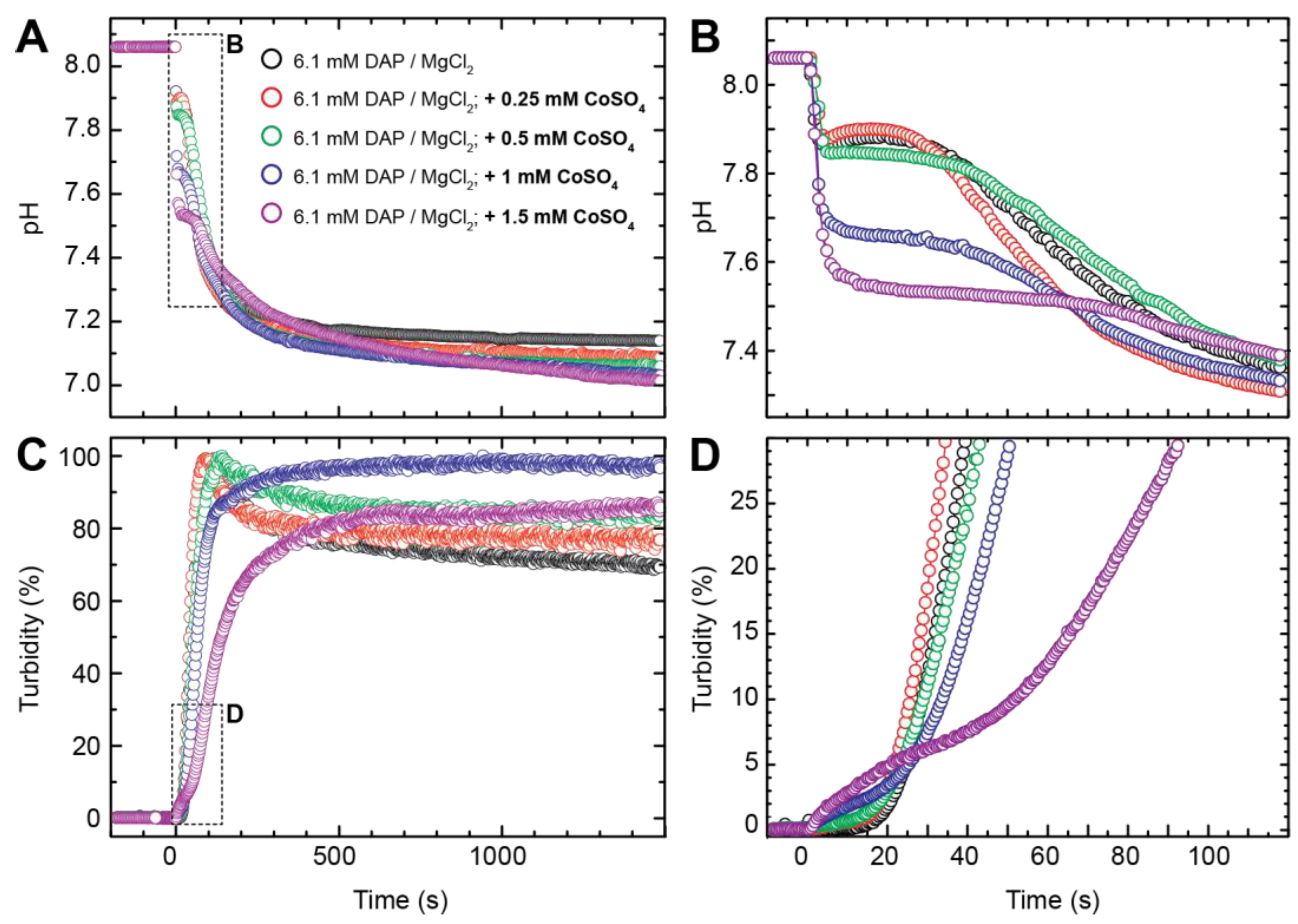
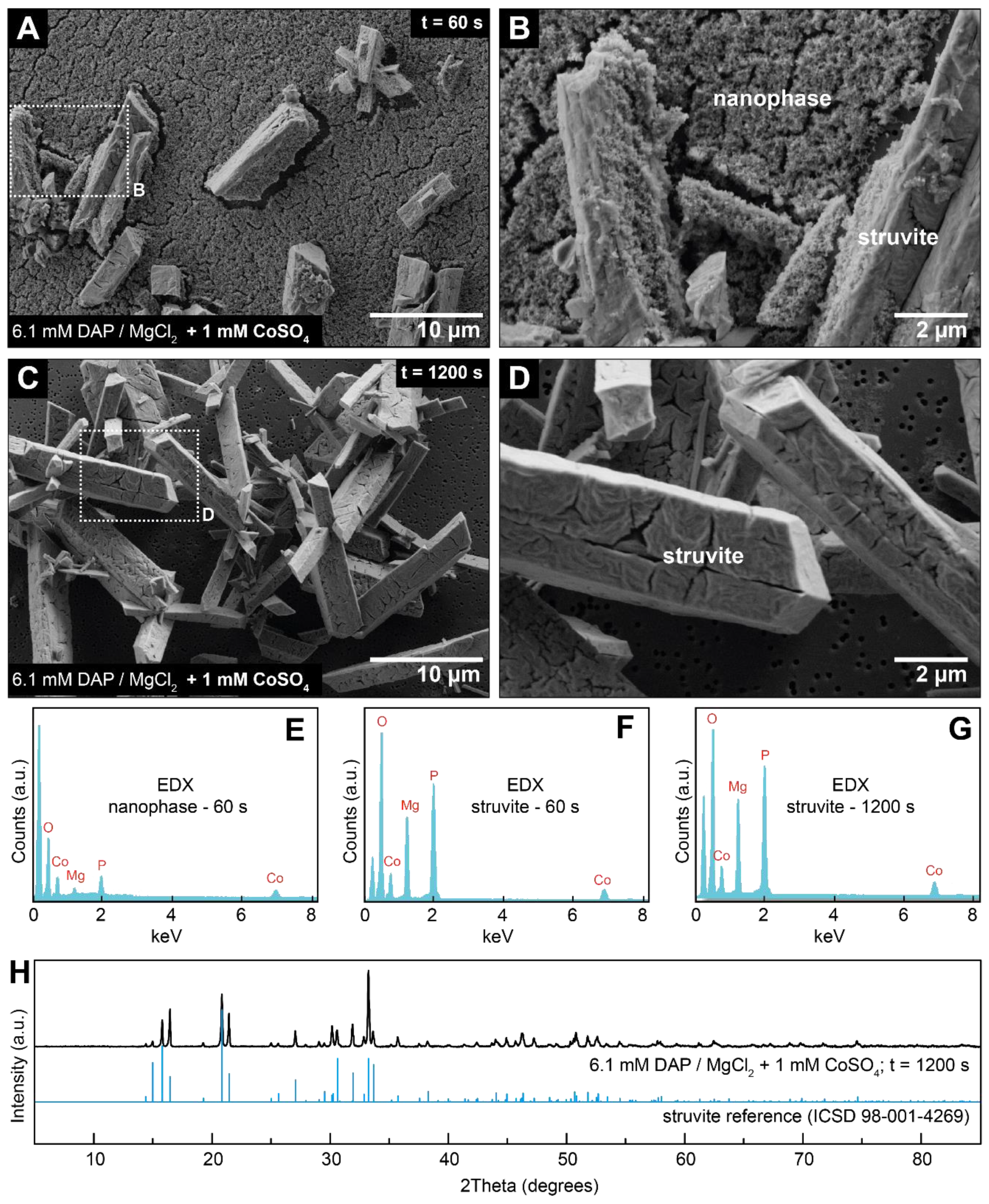
3.2. Crystallisation in the Presence of Low (0.25–1.5 mM) Co Concentrations
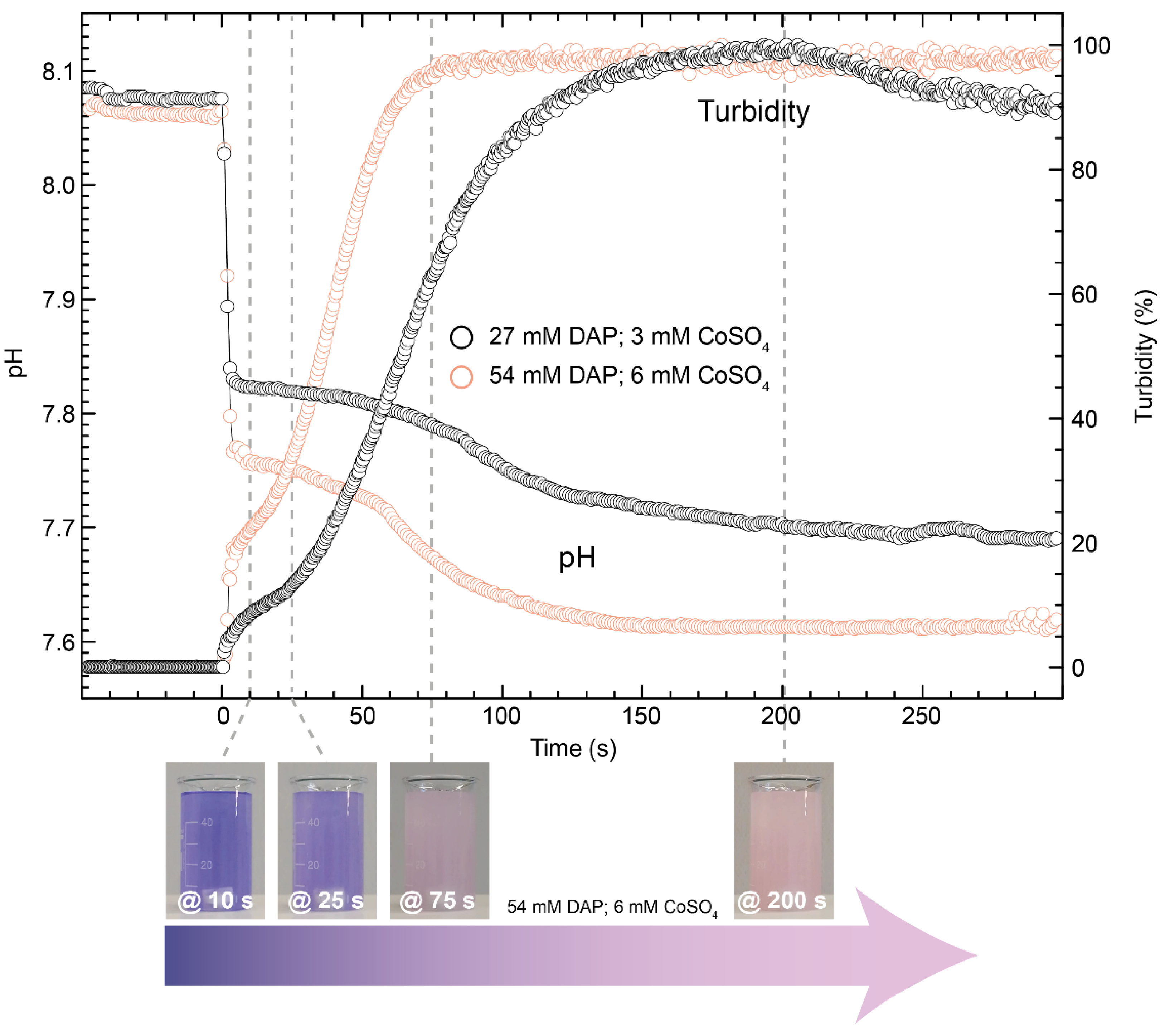
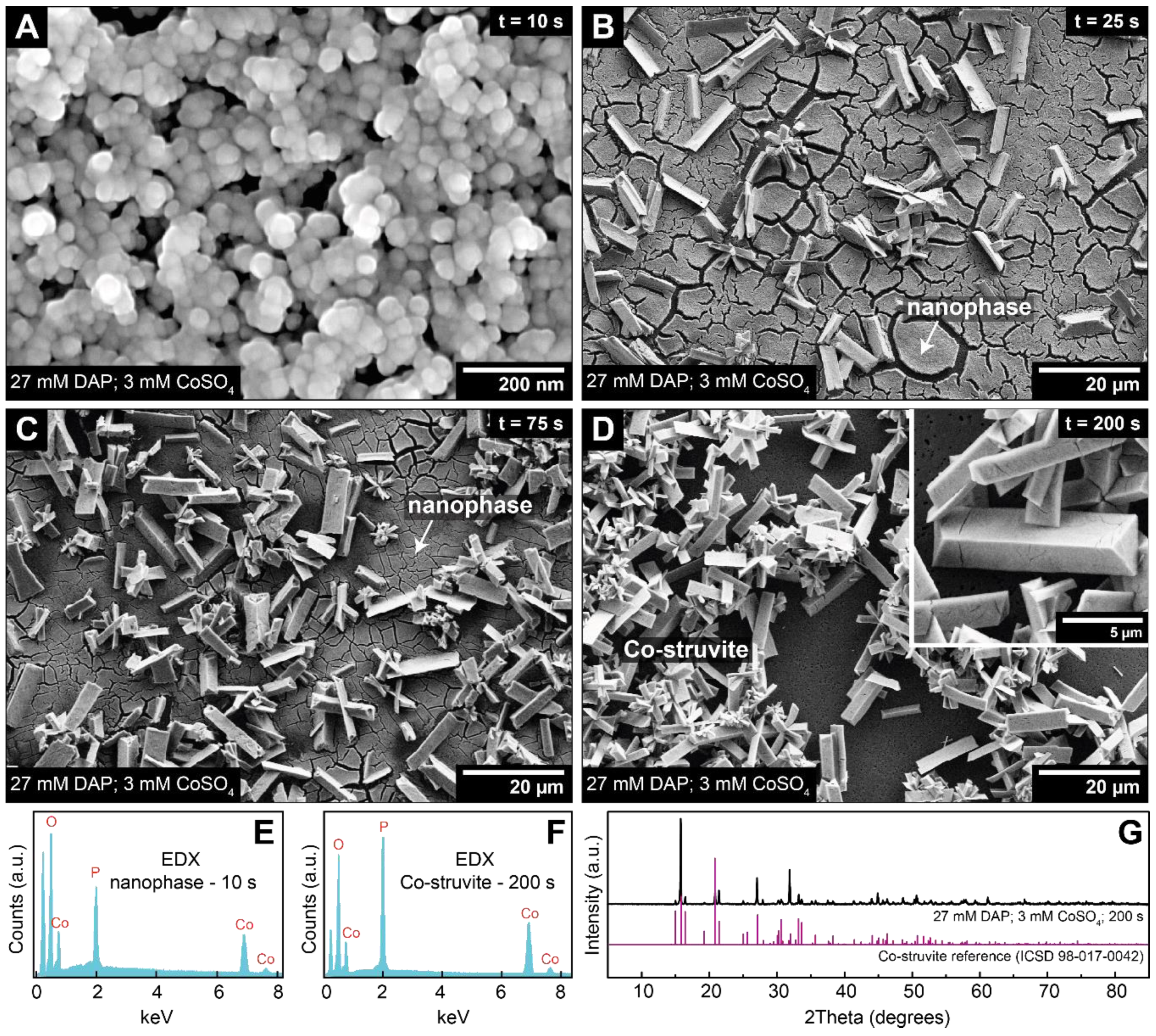
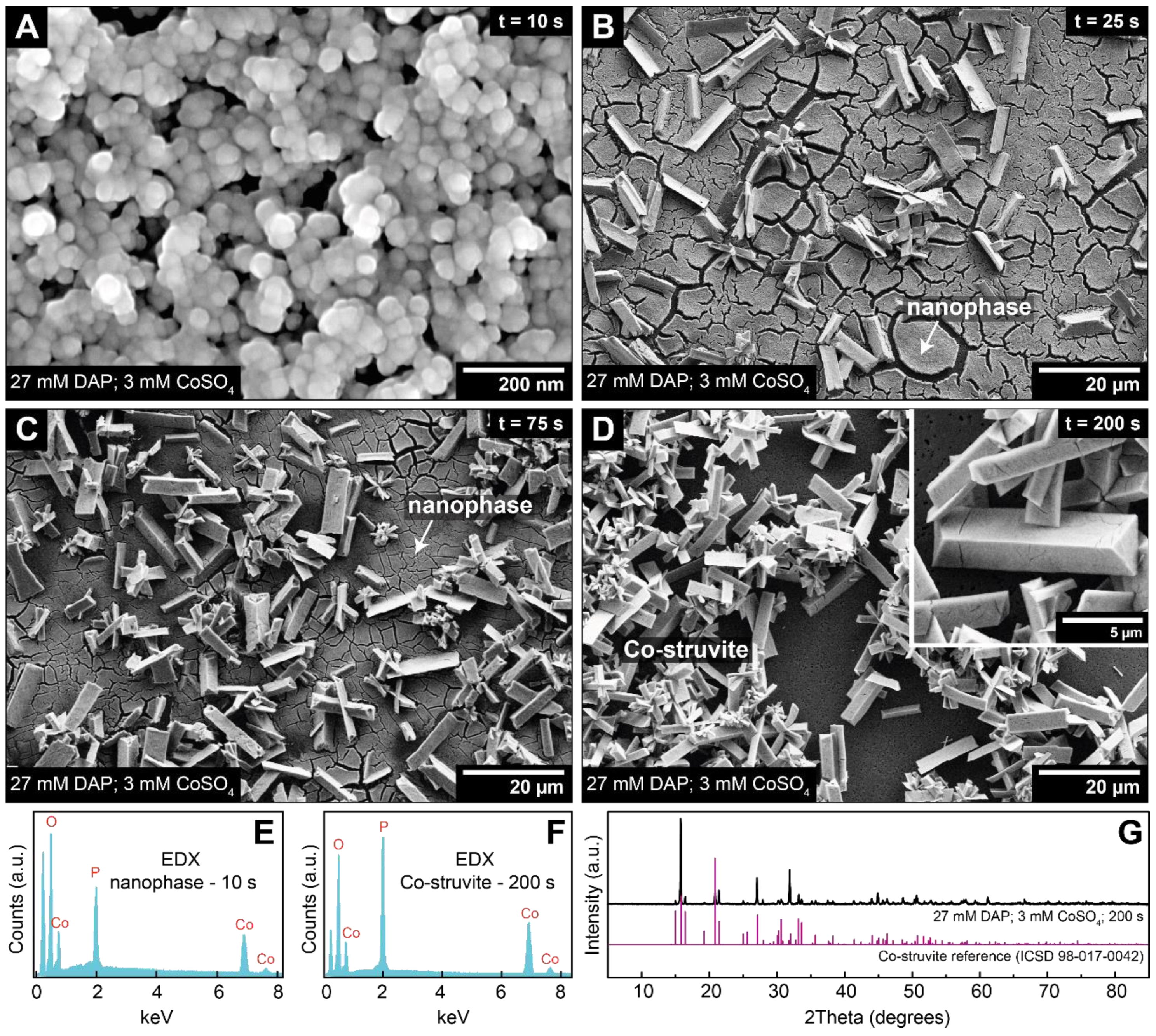
3.3. Crystallisation in the Presence of High (2–6 mM) Co Concentrations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cordell, D.; Drangert, J.O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar] [CrossRef]
- El Diwani, G.; El Rafie, S.; El Ibiari, N.N.; El-Aila, H.I. Recovery of ammonia nitrogen from industrial wastewater treatment as struvite slow releasing fertilizer. Desalination 2007, 214, 200–214. [Google Scholar] [CrossRef]
- Uysal, A.; Yilmazel, Y.D.; Demirer, G.N. The determination of fertilizer quality of the formed struvite from effluent of a sewage sludge anaerobic digester. J. Hazard. Mater. 2010, 181, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Kataki, S.; West, H.; Clarke, M.; Baruah, D.C. Phosphorus recovery as struvite from farm, municipal and industrial waste: Feedstock suitability, methods and pre-treatments. Waste Manag. 2016, 49, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergström, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.J.M.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, aaa6760. [Google Scholar] [CrossRef] [PubMed]
- Ariyanto, E.; Ang, H.M.; Sen, T.K. Impact of various physico-chemical parameters on spontaneous nucleation of struvite (MgNH4PO4·6H2O) formation in a wastewater treatment plant: Kinetic and nucleation mechanism. Desalin. Water Treat. 2013, 52, 6620–6631. [Google Scholar] [CrossRef]
- Mehta, C.M.; Batstone, D.J. Nucleation and growth kinetics of struvite crystallization. Water Res. 2013, 47, 2890–2900. [Google Scholar] [CrossRef] [PubMed]
- Rouff, A.A.; Juarez, K.M. Zinc interaction with struvite during and after mineral formation. Environ. Sci. Technol. 2014, 48, 6342–6349. [Google Scholar] [CrossRef]
- Rouff, A.A.; Ramlogan, M.V.; Rabinovich, A. Synergistic removal of zinc and copper in greenhouse waste effluent by struvite. ACS Sustain. Chem. Eng. 2016, 4, 1319–1327. [Google Scholar] [CrossRef]
- Ma, N.; Rouff, A.A. Influence of pH and oxidation state on the interaction of arsenic with struvite during mineral formation. Environ. Sci. Technol. 2012, 46, 8791–8798. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, R.; Honkanen, M.; Kolppo, K.; Lepistö, R. Nutrient management via struvite precipitation and recovery from various agroindustrial wastewaters: Process feasibility and struvite quality. J. Environ. Manag. 2018, 212, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Rouff, A.A. Sorption of chromium with struvite during phosphorus recovery. Environ. Sci. Technol. 2012, 46, 12493–12501. [Google Scholar] [CrossRef] [PubMed]
- Basset, H.; Bedwell, W.L. Studies of phosphates. Part I. ammonium magnesium phosphate and related compounds. J. Chem. Soc. 1933, 1933, 854–871. [Google Scholar] [CrossRef]
- Ravikumar, R.V.S.S.N.; Rao, S.N.; Reddy, B.J.; Reddy, Y.P. Electronic spectra of hexa aqua coordinated transition metal doped zinc struvite. Ferroelectrics 1996, 189, 139–147. [Google Scholar] [CrossRef]
- Ravikumar, R.V.S.S.N.; Chandrasekhar, A.V.; Reddy, B.J.; Reddy, Y.P.; Ikeda, K. X-ray powder diffraction, DTA and vibrational studies of CdNH4PO4·6H2O crystals. Cryst. Res. Technol. 2002, 37, 1127–1132. [Google Scholar] [CrossRef]
- Chand, P.; Agarwal, O.P. Electron paramagnetic resonance study of doped synthetic crystals of struvite and its zinc analogue. Spectrochim. Acta Part Mol. Spectrosc. 1991, 47, 775–783. [Google Scholar] [CrossRef]
- Fytili, D.; Zabaniotou, A. Utilization of sewage sludge in EU application of old and new methods—A review. Renew. Sustain. Energy Rev. 2008, 12, 116–140. [Google Scholar] [CrossRef]
- European Commission Report on Critical Raw Materials for the EU—Report of the Ad Hoc Working Group on Defining Critical Raw Materials. Available online: https://ec.europa.eu/growth/tools-databases/eip-raw-materials/en/system/files/ged/79%20report-b_en.pdf (accessed on 4 March 2019).
- Perez, J.P.H.; Folens, K.; Leus, K.; Vanhaecke, F.; Van Der Voort, P.; Du Laing, G. Progress in hydrometallurgical technologies to recover critical raw materials and precious metals from low-concentrated streams. Resour. Conserv. Recycl. 2019, 142, 177–188. [Google Scholar] [CrossRef]
- Reid, R.L.; Horvath, D.J. Soil chemistry and mineral problems in farm livestock. A review. Anim. Feed Sci. Technol. 1980, 5, 95–167. [Google Scholar] [CrossRef]
- Gray, C.; Zhao, F.J.; Li, H.F.; McGrath, S.P.; Mico, C. Phytotoxicity and bioavailability of cobalt to plants in a range of soils. Chemosphere 2009, 75, 979–986. [Google Scholar]
- Simonsen, L.O.; Harbak, H.; Bennekou, P. Cobalt metabolism and toxicology—A brief update. Sci. Total Environ. 2012, 432, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Pauw, B.R.; Smith, A.J.; Snow, T.; Terrill, N.J.; Thünemann, A.F. The modular small-angle X-ray scattering data correction sequence. J. Appl. Crystallogr. 2017, 50, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Hövelmann, J.; Stawski, T.M.; Besselink, R.; Freeman, H.M.; Dietmann, K.M.; Mayanna, S.; Pauw, B.R.; Benning, L.G. A template-free and low temperature method for the synthesis of mesoporous magnesium phosphate with uniform pore structure and high surface area. Nanoscale 2019, 11, 6939–6951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaddel, S.; Ucar, S.; Andreassen, J.P.; Sterhus, S.W. Engineering of struvite crystals by regulating supersaturation—Correlation with phosphorus recovery, crystal morphology and process efficiency. J. Environ. Chem. Eng. 2019, 7, 102918. [Google Scholar] [CrossRef]
- Eilers, P.H.; Boelens, H.F. Baseline correction with asymmetric least squares smoothing. Leiden Univ. Med. Cent. Rep. 2005, 1, 5. [Google Scholar]
- Stawski, T.M.; Van Driessche, A.E.S.; Ossorio, M.; Diego Rodriguez-Blanco, J.; Besselink, R.; Benning, L.G. Formation of calcium sulfate through the aggregation of sub-3 nanometre primary species. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Panthöfer, M.; Bienert, R.; Buzanich, A.D.O.G.; Emmerling, F.; Tremel, W. Role of water during crystallization of amorphous cobalt phosphate nanoparticles. Cryst. Growth Des. 2016, 16, 4232–4239. [Google Scholar] [CrossRef]
- Pouget, E.M.; Bomans, P.H.H.; Goos, J.A.C.M.; Frederik, P.M.; de With, G.; Sommerdijk, N.A.J.M. The initial stages of template-controlled CaCO3 formation revealed by Cryo-TEM. Science 2009, 323, 1455–1458. [Google Scholar] [CrossRef]
- Hövelmann, J.; Putnis, C.V. In situ nanoscale imaging of struvite formation during the dissolution of natural brucite: Implications for phosphorus recovery from wastewaters. Environ. Sci. Technol. 2016, 50, 13032–13041. [Google Scholar] [CrossRef]
- Liu, L.; Song, W.; Guo, M. Remediation techniques for heavy metal-contaminated soils: Principles and applicability. Sci. Total Environ. 2018, 633, 206–219. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAP | MgCl2 | CoSO4 | Co/P | Precipitates |
|---|---|---|---|---|
| [mM] | [mM] | [mM] | ||
| 3.05 | 3.05 | - | - | Mg-struvite |
| 6.1 | 6.1 | - | - | Mg-struvite |
| 27 | 3 | - | - | Mg-struvite |
| 54 | 6 | - | - | Mg-struvite |
| 6.1 | 6.1 | 0.25 | 0.04 | Mg-struvite |
| 6.1 | 6.1 | 0.5 | 0.08 | Mg-struvite |
| 6.1 | 6.1 | 1 | 0.16 | (Co, Mg, P)-NP * → (Mg, Co)-struvite |
| 6.1 | 6.1 | 1.5 | 0.25 | (Co, Mg, P)-NP * → (Mg, Co)-struvite |
| 6.1 | 6.1 | 2 | 0.33 | (Co, Mg, P)-NP * → (Mg, Co)-struvite + (Co, Mg)-phosphate octahydrate |
| 6.1 | 6.1 | 3 | 0.5 | (Co, Mg, P)-NP * → (Co, Mg)-phosphate octahydrate |
| 6.1 | 6.1 | 4 | 0.66 | (Co, Mg, P)-NP * → (Co, Mg)-phosphate octahydrate |
| 6.1 | 6.1 | 6 | 0.98 | (Co, Mg, P)-NP * → (Co, Mg)-phosphate octahydrate |
| 6.1 | - | 6 | 0.98 | (Co, P)-NP * → Co-phosphate octahydrate |
| 27 | - | 3 | 0.11 | (Co, P)-NP * → Co-struvite |
| 54 | - | 6 | 0.11 | (Co, P)-NP * → Co-struvite |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hövelmann, J.; Stawski, T.M.; Freeman, H.M.; Besselink, R.; Mayanna, S.; Perez, J.P.H.; Hondow, N.S.; Benning, L.G. Struvite Crystallisation and the Effect of Co2+ Ions. Minerals 2019, 9, 503. https://doi.org/10.3390/min9090503
Hövelmann J, Stawski TM, Freeman HM, Besselink R, Mayanna S, Perez JPH, Hondow NS, Benning LG. Struvite Crystallisation and the Effect of Co2+ Ions. Minerals. 2019; 9(9):503. https://doi.org/10.3390/min9090503
Chicago/Turabian StyleHövelmann, Jörn, Tomasz M. Stawski, Helen M. Freeman, Rogier Besselink, Sathish Mayanna, Jeffrey Paulo H. Perez, Nicole S. Hondow, and Liane G. Benning. 2019. "Struvite Crystallisation and the Effect of Co2+ Ions" Minerals 9, no. 9: 503. https://doi.org/10.3390/min9090503
APA StyleHövelmann, J., Stawski, T. M., Freeman, H. M., Besselink, R., Mayanna, S., Perez, J. P. H., Hondow, N. S., & Benning, L. G. (2019). Struvite Crystallisation and the Effect of Co2+ Ions. Minerals, 9(9), 503. https://doi.org/10.3390/min9090503