Femtosecond Laser Ablation-ICP-Mass Spectrometry and CHNS Elemental Analyzer Reveal Trace Element Characteristics of Danburite from Mexico, Tanzania, and Vietnam

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Material
2.2. Analytical Methods
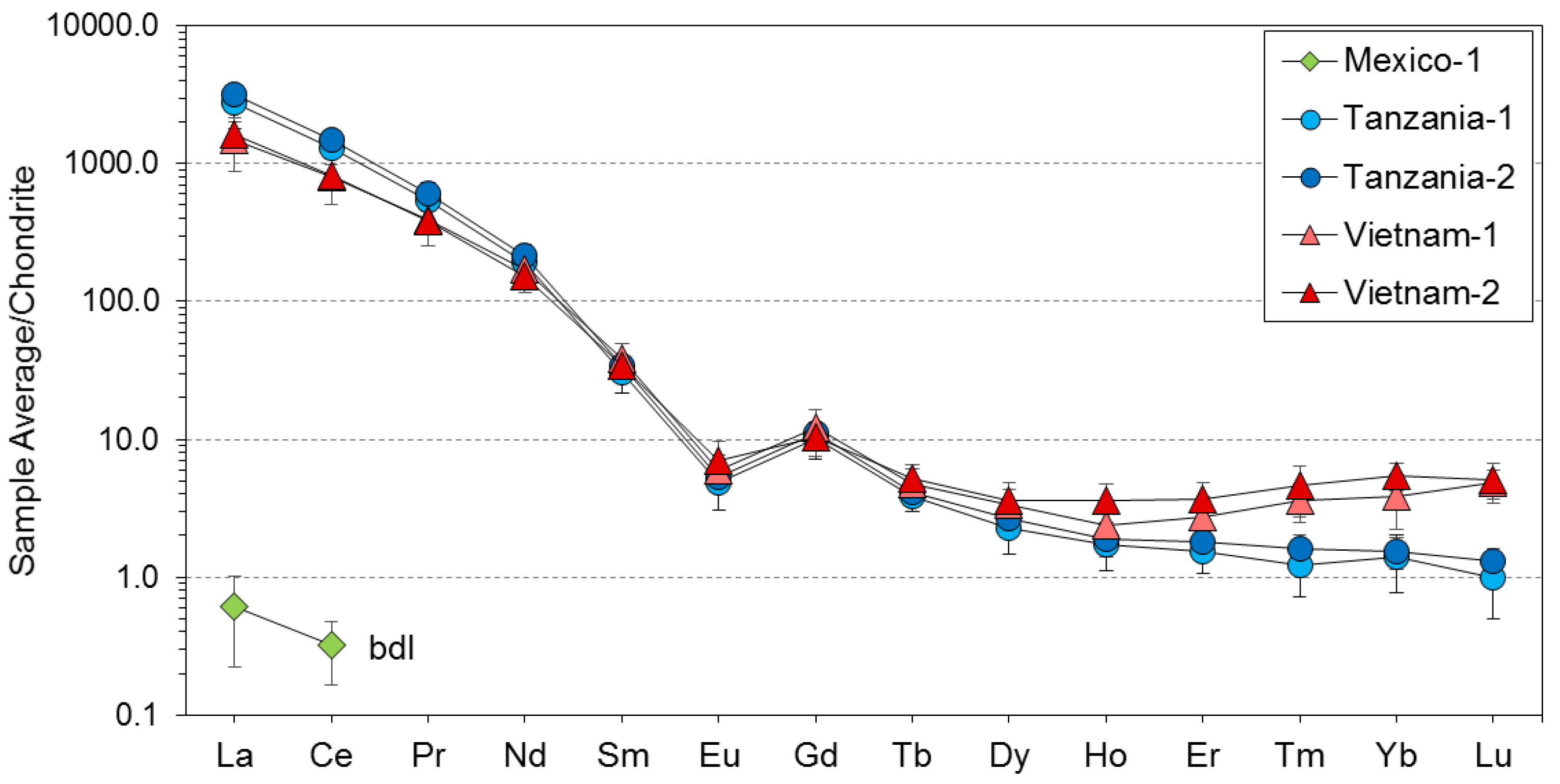
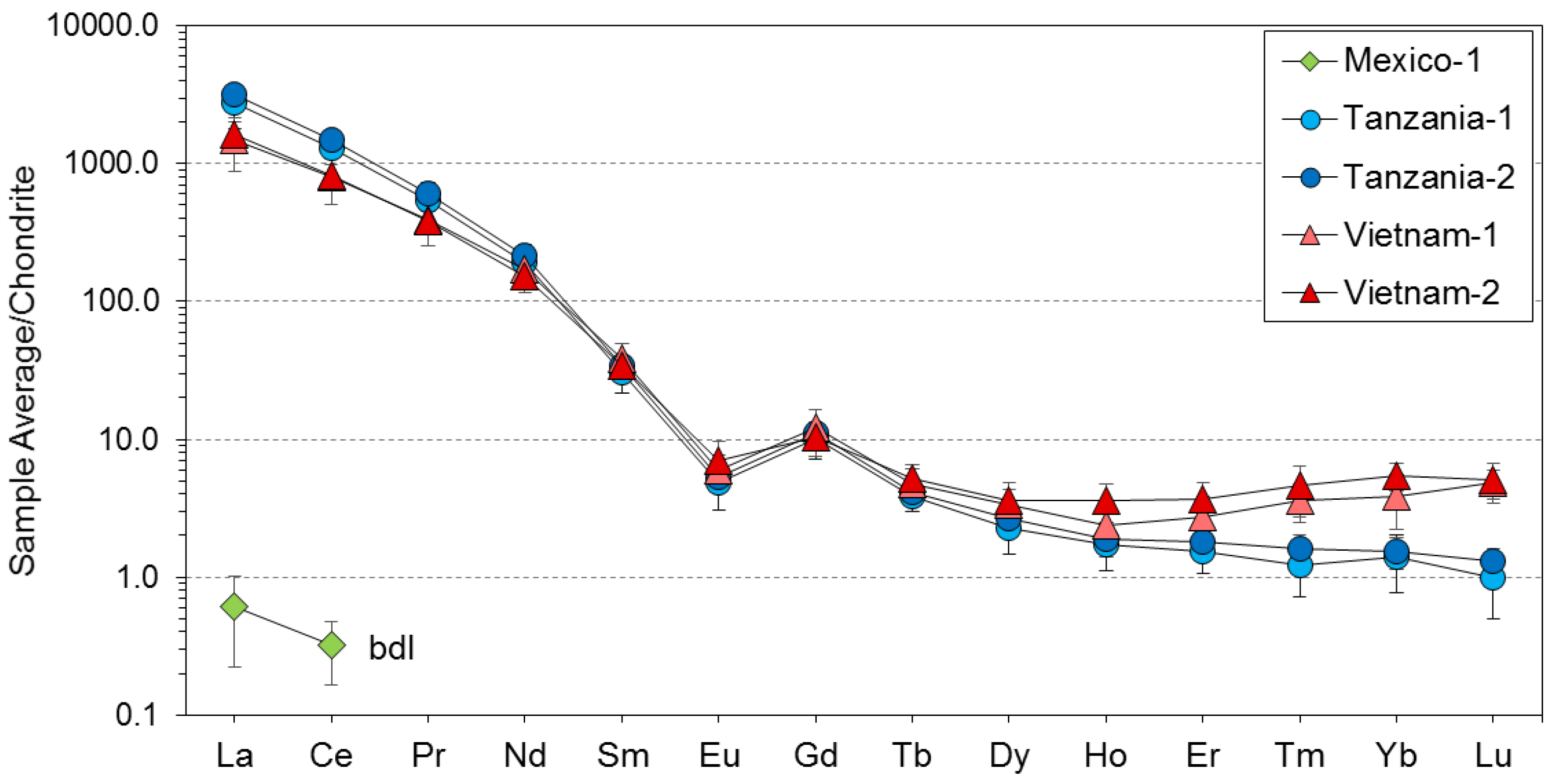
3. Results
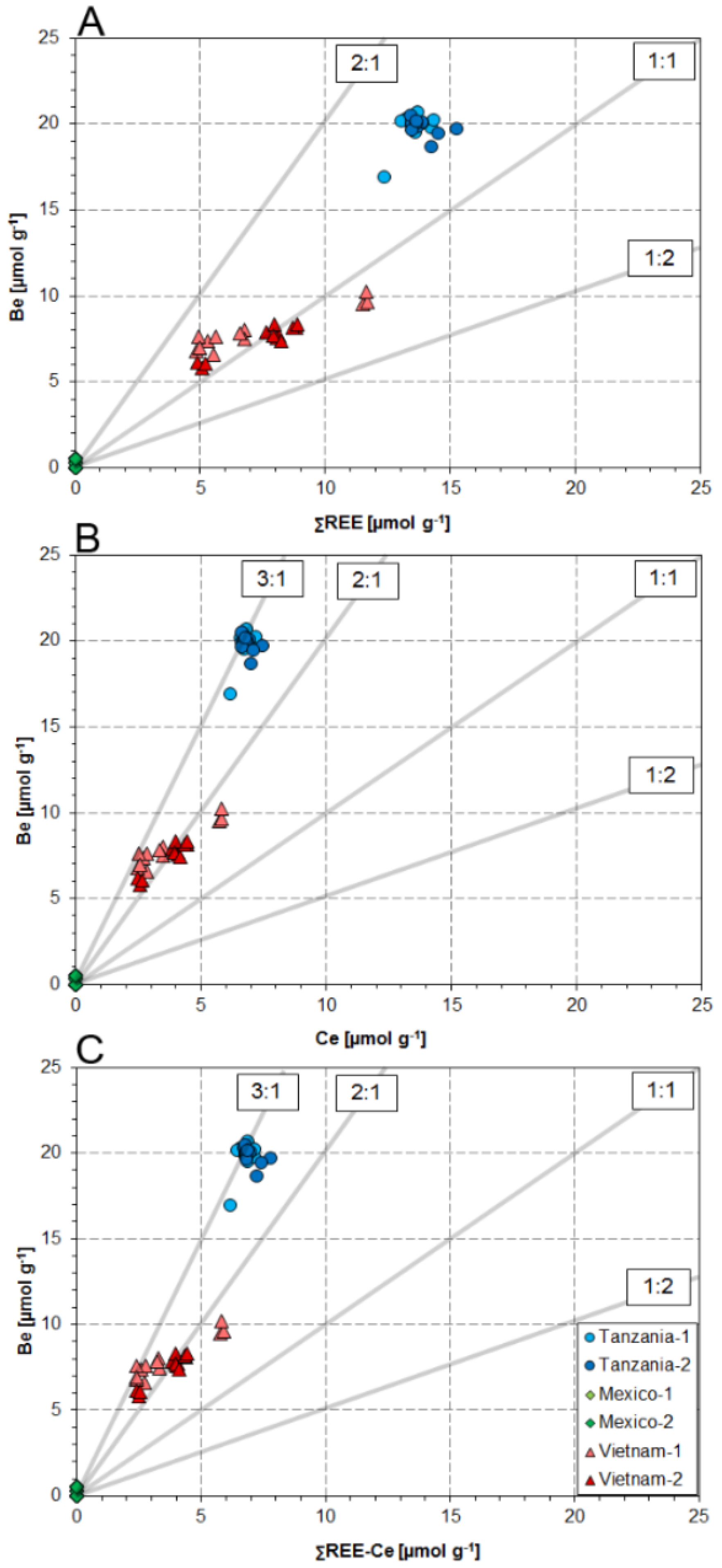
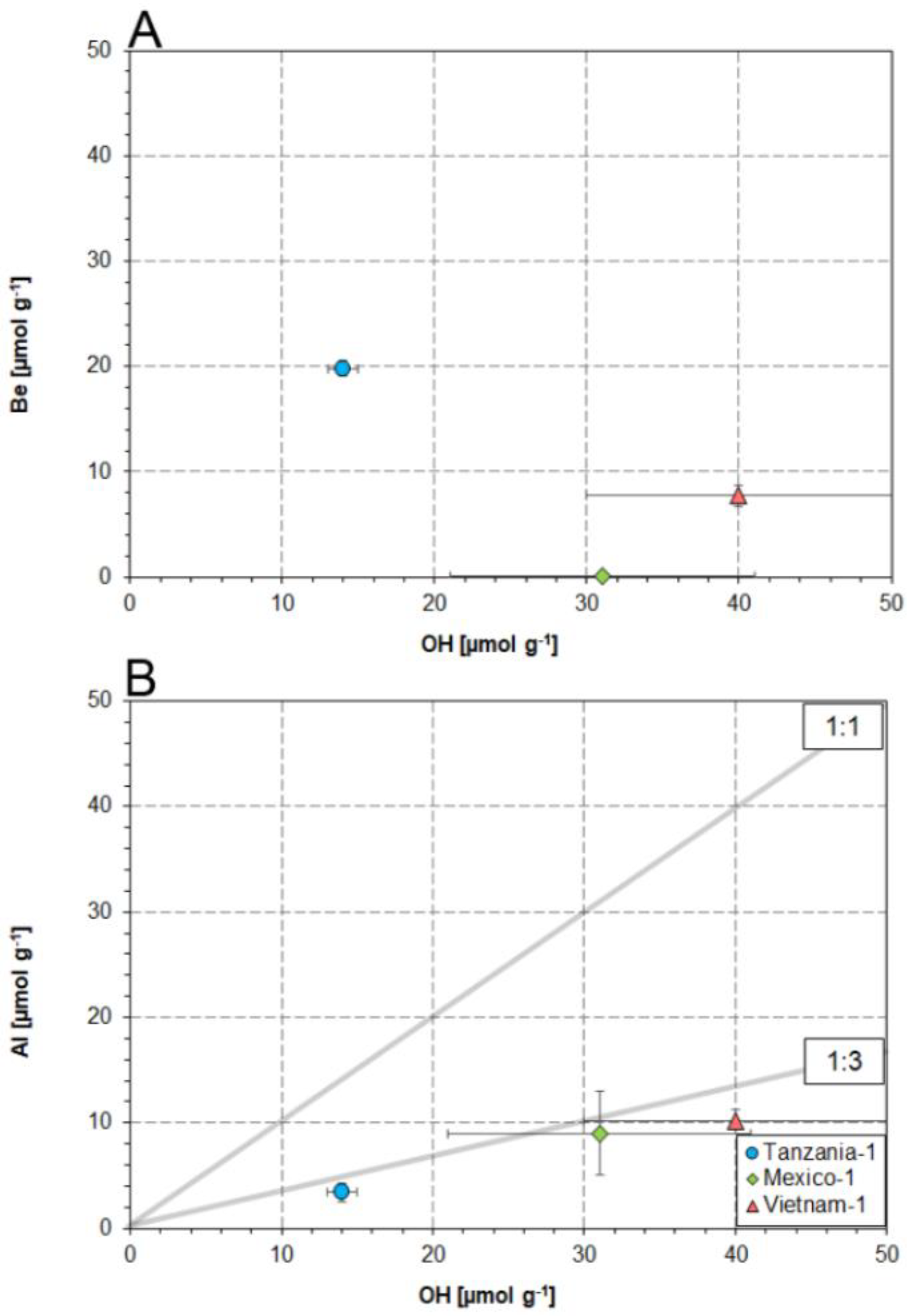
4. Discussion
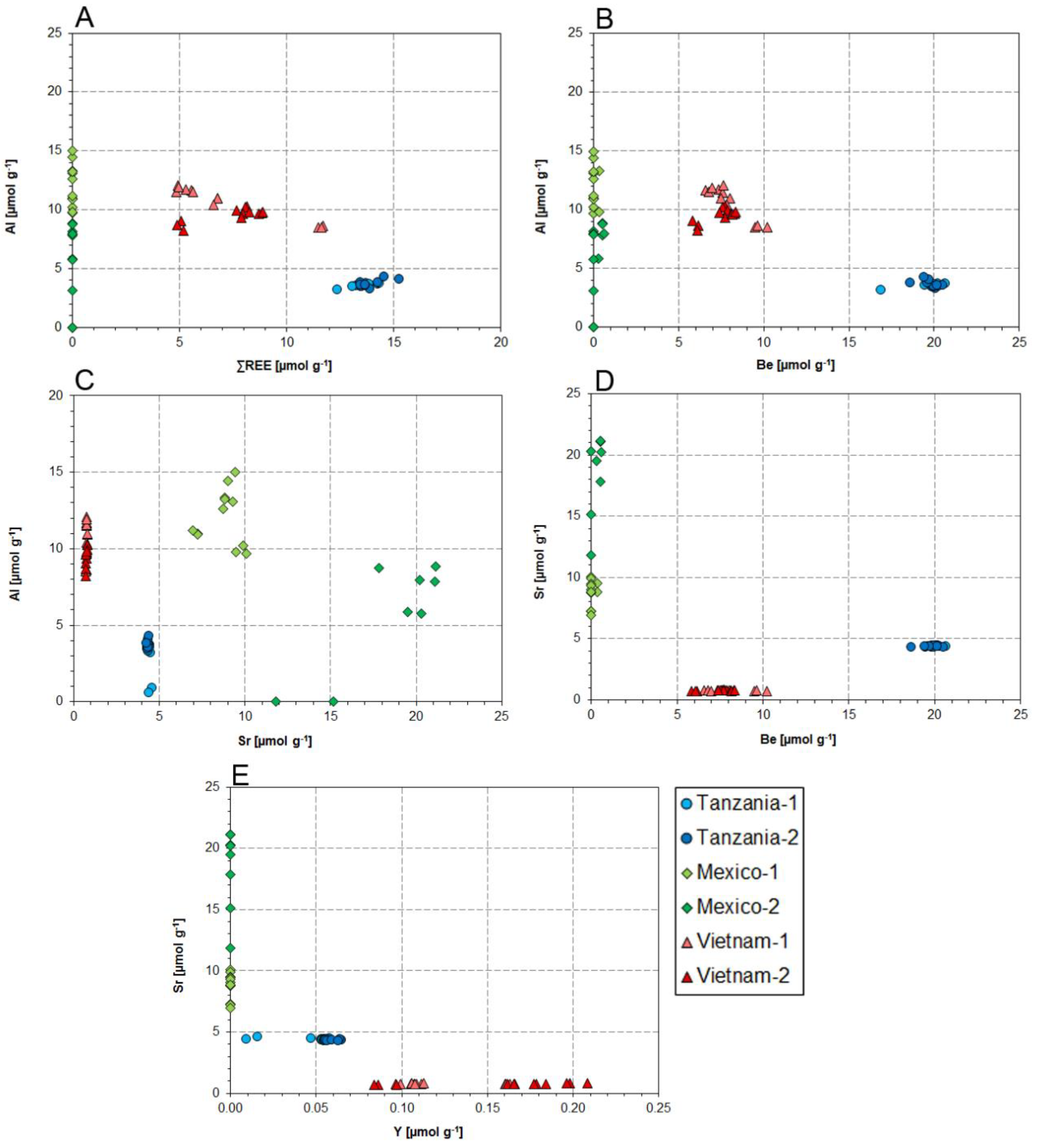
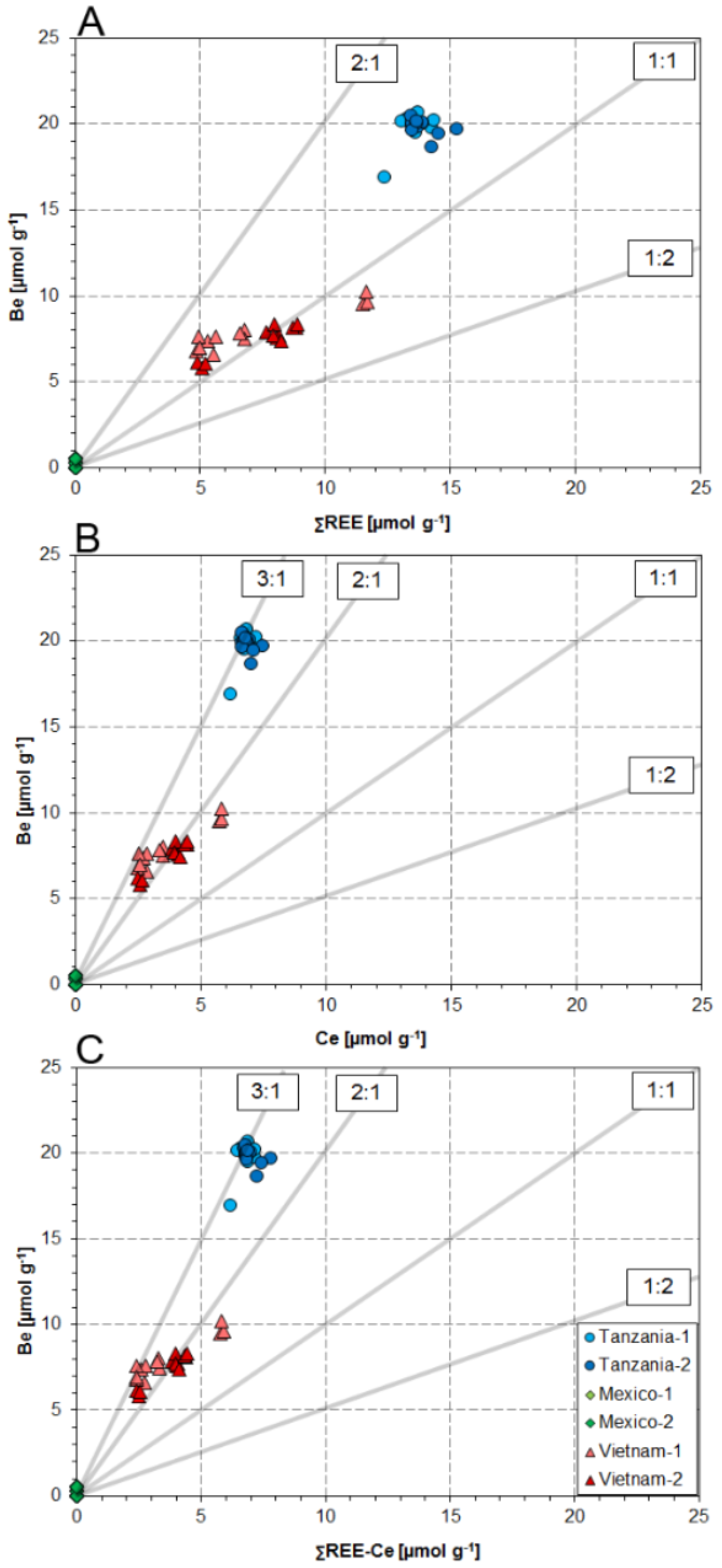
4.1. Substitution Mechanisms of REEs, Be, and Sr in Danburite Structure
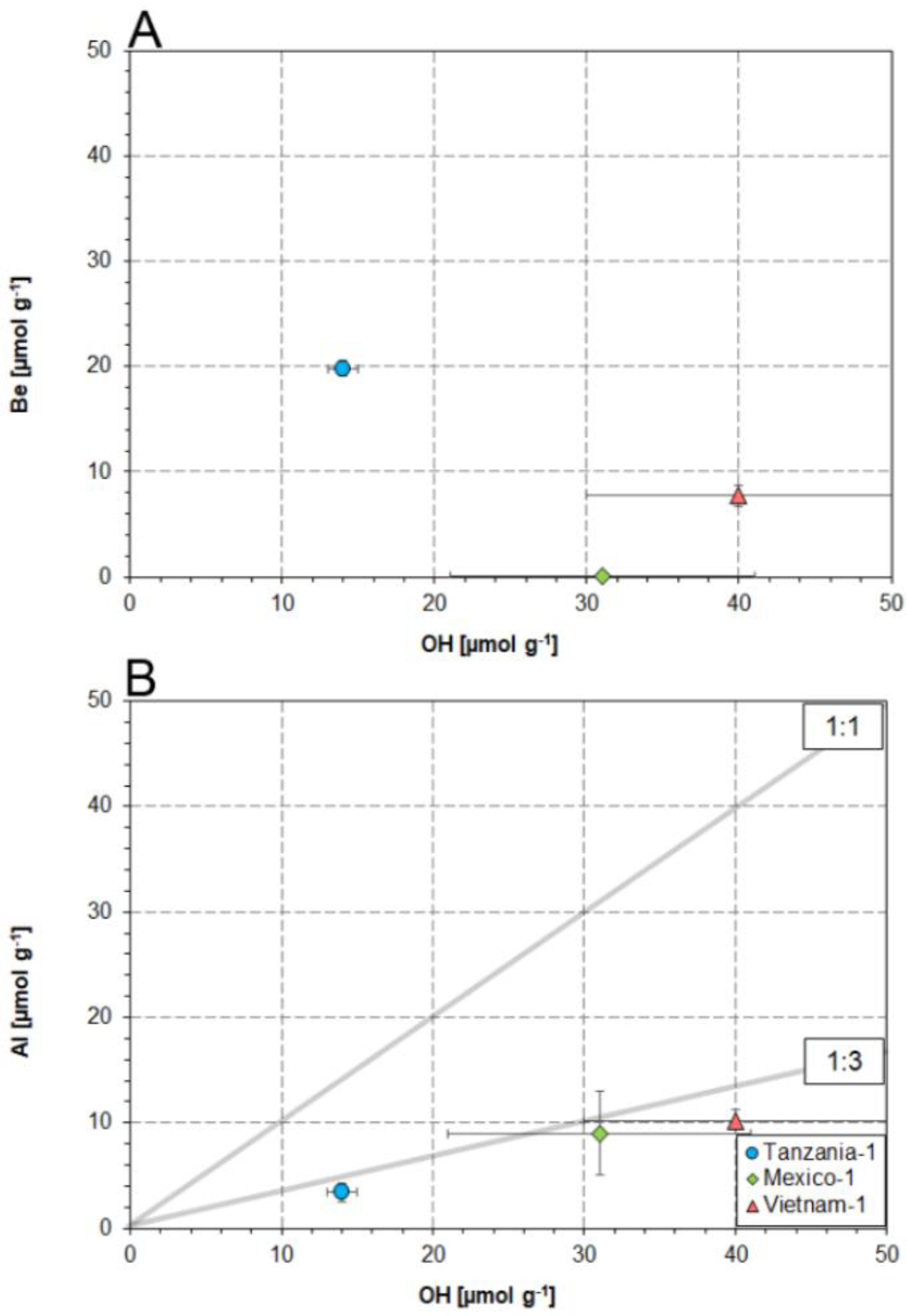
4.2. Substitution Mechanisms Involving OH and Al in the Danburite Lattice
4.3. Constraints on the Geochemical Formation Environment of Danburite
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindbloom, J.T.; Gibbs, G.V.; Ribbe, P.H. Crystal Structure of Hurlbutite—Comparison with Danburite and Anorthite. Am. Mineral. 1974, 59, 1267–1271. [Google Scholar]
- Best, S.P.; Clark, R.J.H.; Hayward, C.L.; Withnall, R. Polarized single-crystal Raman spectroscopy of danburite, CaB2Si2O8. J. Raman Spectrosc. 1994, 25, 557–563. [Google Scholar] [CrossRef]
- Sugiyama, K.; Takéuchi, Y. Unusual thermal expansion of a B–O bond in the structure of danburite CaB2Si2O8. Z. Kristallogr. -Cryst. Mater. 1985, 173, 293–304. [Google Scholar] [CrossRef]
- Beran, A. OH groups in nominally anhydrous framework structures: An infrared spectroscopic investigation of danburite and labradorite. Phys. Chem. Miner. 1987, 14, 441–445. [Google Scholar] [CrossRef]
- Hurwit, K.N. Gem Trade Lab Notes: Golden yellow danburite from Sri Lanka. Gems Gemol. 1986, 22, 47. [Google Scholar]
- Chadwick, K.M.; Laurs, B.M. Gem News International: Yellow danburite from 346 Tanzania. Gems Gemol. 2008, 44, 169–171. [Google Scholar]
- Huong, L.T.-T.; Otter, L.M.; Häger, T.; Ullmann, T.; Hofmeister, W.; Weis, U.; Jochum, K.P. A New Find of Danburite in the Luc Yen Mining Area, Vietnam. Gems Gemol. 2016, 52. [Google Scholar] [CrossRef]
- De Vito, C.; Pezzotta, F.; Ferrini, V.; Aurisicchio, C. Nb–Ti–Ta oxides in the gem-mineralized and “hybrid” Anjanabonoina granitic pegmatite, central Madagascar: A record of magmatic and postmagmatic events. Can. Mineral. 2006, 44, 87–103. [Google Scholar] [CrossRef]
- Cook, R.B. Connoisseur’s: Danburite, Charcas, San Luis Potosí, Mexico. Rocks Miner. 2003, 78, 400–403. [Google Scholar] [CrossRef]
- Hintze, J. Safari njema—AFRIKANISCHES TAGEBUCH (I): Reise zu den gelben Danburiten von Morogoro, Tansania. Lapis Die Aktuelle Monatsschrift Fuer Liebhaber Und Sammler Von Mineralien Und 2010, 35, 25. [Google Scholar]
- Dyar, M.D.; Wiedenbeck, M.; Robertson, D.; Cross, L.R.; Delaney, J.S.; Ferguson, K.; Francis, C.A.; Grew, E.S.; Guidotti, C.V.; Hervig, R.L. Reference minerals for the microanalysis of light elements. Geostand. Geoanal. Res. 2001, 25, 441–463. [Google Scholar] [CrossRef]
- Ottolini, L.; Cámara, F.; Hawthorne, F.C.; Stirling, J. SIMS matrix effects in the analysis of light elements in silicate minerals: Comparison with SREF and EMPA data. Am. Mineral. 2002, 87, 1477–1485. [Google Scholar] [CrossRef]
- Balmer, W.A.; Hauzenberger, C.A.; Fritz, H.; Sutthirat, C. Marble-hosted ruby deposits of the Morogoro Region, Tanzania. J. Afr. Earth Sci. 2017, 134, 626–643. [Google Scholar] [CrossRef]
- Möller, A.; Mezger, K.; Schenk, V. U–Pb dating of metamorphic minerals: Pan-African metamorphism and prolonged slow cooling of high pressure granulites in Tanzania, East Africa. Precambrian Res. 2000, 104, 123–146. [Google Scholar] [CrossRef]
- Huong, L.T.-T.; Krenn, K.; Hauzenberger, C. Sassolite- and CO2-H2O-bearing Fluid Inclusions in Yellow Danburite from Luc Yen, Vietnam. J. Gemmol. 2017, 35, 544–550. [Google Scholar] [CrossRef]
- Garnier, V.; Ohnenstetter, D.; Giuliani, G.; Maluski, H.; Deloule, E.; Trong, T.P.; Van, L.P.; Quang, V.H. Age and significance of ruby-bearing marble from the Red River Shear Zone, northern Vietnam. Can. Mineral. 2005, 43, 1315–1329. [Google Scholar] [CrossRef]
- Jochum, K.P.; Stoll, B.; Weis, U.; Jacob, D.E.; Mertz-Kraus, R.; Andreae, M.O. Non-Matrix-Matched Calibration for the Multi-Element Analysis of Geological and Environmental Samples Using 200 nm Femtosecond LA-ICP-MS: A Comparison with Nanosecond Lasers. Geostand. Geoanal. Res. 2014, 38, 265–292. [Google Scholar] [CrossRef]
- Jochum, K.P.; Stoll, B.; Herwig, K.; Willbold, M. Validation of LA-ICP-MS trace element analysis of geological glasses using a new solid-state 193 nm Nd: YAG laser and matrix-matched calibration. J. Anal. At. Spectrome. 2007, 22, 112–121. [Google Scholar] [CrossRef]
- Jochum, K.P.; Nohl, U.; Herwig, K.; Lammel, E.; Stoll, B.; Hofmann, A.W. GeoReM: A new geochemical database for reference materials and isotopic standards. Geostand. Geoanal. Res. 2005, 29, 333–338. [Google Scholar] [CrossRef]
- Palme, H.; Jones, A. Solar system abundances of the elements. Treat. Geochem. 2003, 1, 711. [Google Scholar]
- Plessen, B.; Harlov, D.E.; Henry, D.; Guidotti, C.V. Ammonium loss and nitrogen isotopic fractionation in biotite as a function of metamorphic grade in metapelites from western Maine, USA. Geochim. Cosmochim. Acta 2010, 74, 4759–4771. [Google Scholar] [CrossRef]
- Fowler, A.D.; Doig, R. The significance of europium anomalies in the REE spectra of granites and pegmatites, Mont Laurier, Quebec. Geochim. Cosmochim. Acta 1983, 47, 1131–1137. [Google Scholar] [CrossRef]
- Braun, J.-J.; Pagel, M.; Muller, J.-P.; Bilong, P.; Michard, A.; Guillet, B. Cerium anomalies in lateritic profiles. Geochim. Cosmochim. Acta 1990, 54, 781–795. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shimizu, H.; Kagi, H.; Yoshida, H.; Usui, A.; Nomura, M. A new method for the determination of CeIII/CeIV ratios in geological materials; application for weathering, sedimentary and diagenetic processes. Earth Planet. Sci. Lett. 2000, 182, 201–207. [Google Scholar] [CrossRef]
- Taunton, A.E.; Welch, S.A.; Banfield, J.F. Microbial controls on phosphate and lanthanide distributions during granite weathering and soil formation. Chem. Geol. 2000, 169, 371–382. [Google Scholar] [CrossRef]
- Bao, Z.; Zhao, Z. Geochemistry of mineralization with exchangeable REY in the weathering crusts of granitic rocks in South China. Ore Geol. Rev. 2008, 33, 519–535. [Google Scholar] [CrossRef]
- Wunder, B.; Stefanski, J.; Wirth, R.; Gottschalk, M. Al-B substitution in the system albite (NaAlSi3O8)-reedmergnerite (NaBSi3O8). Eur. J. Mineral. 2013, 25, 499–508. [Google Scholar] [CrossRef]
- Stubican, V.; Roy, R. Boron substitution in synthetic micas and clays. Am. Mineral. 1962, 47, 1166. [Google Scholar]
- Chen, C.; Liu, Y.; Foley, S.F.; Ducea, M.N.; He, D.; Hu, Z.; Chen, W.; Zong, K. Paleo-Asian oceanic slab under the North China craton revealed by carbonatites derived from subducted limestones. Geology 2016, 44, 1039–1042. [Google Scholar] [CrossRef]
- Gozzi, F.; Gaeta, M.; Freda, C.; Mollo, S.; Di Rocco, T.; Marra, F.; Dallai, L.; Pack, A. Primary magmatic calcite reveals origin from crustal carbonate. Lithos 2014, 190, 191–203. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Operating Conditions of NWRFemto200 Laser System | |
| Wavelength λ (nm) | 200 |
| Fluence (J·cm−2) | 0.51 |
| Pulse length (fs) | 150 |
| Pulse repetition rate (Hz) | 50 |
| Laser energy output (%) | 100 |
| Spot size (μm) | 55 |
| Line length (µm) | 300 |
| Scan speed (μm∙s−1) | 5 |
| Warm-up time (s) | 28 |
| Dwell time (s) | 60 |
| Washout time (s) | 30 |
| Operating Conditions of the Element2 Mass Spectrometer | |
| RF power (W) | 1055 |
| Cooling gas (Ar) flow rate (L·min−1) | 16 |
| Auxiliary gas (Ar) flow rate (L∙min−1) | 1.19 |
| Additional gas (He) flow rate (L∙min−1) | 0.7 |
| Sample gas (Ar) flow rate (L∙min−1) | 0.7 |
| Sample time (s) | 0.002 |
| Samples per peak | 100 |
| Mass window (%) | 10 |
| Time per pass (s) | 2 |
| Scan mode (Escan/Bscan) | both |
| Mass resolution | 300 |
| BE-N Altered Basalts (SARM) | H TCD | C TCD | N TCD | S TCD | S IR |
| n | 14 | 20 | 17 | 21 | 8 |
| Average (µg·g−1) | 2771 ± 534 | 2301 ± 147 | 197 ± 42 | 301 ± 37 | 298 ± 23 |
| RSD % | 19 | 6 | 21 | 12 | 8 |
| BAM-U110 | |||||
| n | 13 | 18 | 18 | 17 | – |
| Average (µg·g−1) | 12,258 ± 1758 | 72,340 ± 2640 | 4237 ± 165 | 9114 ± 1082 | – |
| RSD % | 14 | 4 | 4 | 12 | – |
| JP-1 Peridotite massif (JGS) | |||||
| n | 4 | 12 | 14 | 14 | 14 |
| Average (µg·g−1) | 3195 ± 170 | 763 ± 82 | 91 ± 23 | 27 ± 14 | 26 ± 7 |
| RSD % | 5 | 11 | 26 | 51 | 27 |
| Element | Isotope Used | L.O.D. | Charcas, San Luis Potosi, Mexico | Morogoro, Tanzania | Luc Yen, Vietnam | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mex-1 | Mex-2 | Tanz-1 | Tanz-2 | Viet-1 | Viet-2 | |||||||||||||||
| Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | Ø (µg∙g−1) | Ø (µmol∙g−1) | RSD (%) | |||
| CaO* | – | – | 22.47 | 0.04 | 0.49 | 22.43 | – | 0.71 | 22.63 | 0.04 | 0.60 | 22.48 | 0.04 | 0.57 | 22.32 | 0.03 | 0.51 | 22.40 | 0.03 | 0.37 |
| B | 11 | 10 | 86218 | 7975 | 6.67 | 90471 | 8368 | 4.05 | 86280 | 7981 | 1.56 | 87270 | 8072 | 2.69 | 85698 | 7927 | 3.13 | 89138 | 8245 | 3.19 |
| SiO2 * | – | – | 48.31 | 0.8 | 0.40 | 48.81 | 0.8 | 0.33 | 48.80 | 0.8 | 0.36 | 48.88 | 0.8 | 0.38 | 48.81 | 0.8 | 0.43 | 48.43 | 0.8 | 0.37 |
| La | 139 | 0.01 | 0.15 | 0.0011 | 63.5 | 0.01 | 0.0001 | 124.0 | 675 | 4.9 | 35.3 | 807 | 5.8 | 4.7 | 368 | 2.7 | 42.3 | 400 | 2.9 | 22.0 |
| Ce | 140 | 0.01 | 0.20 | 0.0014 | 48.1 | 0.05 | 0.0004 | 10.00 | 827 | 5.9 | 34.7 | 964 | 6.9 | 3.4 | 508 | 3.6 | 36.9 | 525 | 3.8 | 19.6 |
| Pr | 141 | 0.03 | <0.03 | – | – | <0.03 | – | – | 51.4 | 0.37 | 35.1 | 59.5 | 0.42 | 4.0 | 37.4 | 0.27 | 35.3 | 36.5 | 0.26 | 17.5 |
| Nd | 143 | 0.08 | <0.08 | – | – | <0.08 | – | – | 91.1 | 0.63 | 34.5 | 106 | 0.74 | 4.9 | 80.9 | 0.56 | 32.2 | 72.8 | 0.51 | 16.2 |
| Sm | 147 | 0.05 | <0.05 | – | – | <0.05 | – | – | 4.65 | 0.03 | 28.7 | 5.68 | 0.04 | 9.6 | 5.94 | 0.04 | 27.1 | 5.23 | 0.03 | 18.7 |
| Eu | 151 | 0.05 | <0.05 | – | – | <0.05 | – | – | 0.28 | 0.002 | 37.7 | 0.31 | 0.002 | 14.1 | 0.34 | 0.002 | 28.5 | 0.41 | 0.003 | 38.2 |
| Gd | 157 | 0.04 | <0.04 | – | – | <0.04 | – | – | 2.05 | 0.013 | 28.1 | 2.38 | 0.015 | 10.9 | 2.46 | 0.016 | 37.1 | 2.13 | 0.014 | 28.3 |
| Tb | 159 | 0.01 | <0.01 | – | – | <0.01 | – | – | 0.15 | 0.001 | 22.4 | 0.16 | 0.001 | 15.5 | 0.18 | 0.001 | 29.5 | 0.20 | 0.001 | 26.1 |
| Dy | 163 | 0.05 | <0.05 | – | – | <0.05 | – | – | 0.58 | 0.004 | 35.7 | 0.68 | 0.004 | 7.80 | 0.85 | 0.005 | 30.5 | 0.92 | 0.006 | 33.7 |
| Ho | 165 | 0.01 | <0.01 | – | – | <0.01 | – | – | 0.10 | 0.001 | 35.9 | 0.11 | 0.001 | 11.3 | 0.13 | 0.001 | 40.3 | 0.20 | 0.001 | 31.1 |
| Er | 167 | 0.04 | <0.04 | – | – | <0.04 | – | – | 0.25 | 0.001 | 30.0 | 0.28 | 0.002 | 38.6 | 0.46 | 0.003 | 41.0 | 0.61 | 0.004 | 33.3 |
| Tm | 169 | 0.02 | <0.02 | – | – | <0.02 | – | – | 0.03 | 0.0002 | 40.5 | 0.04 | 0.0002 | 49.3 | 0.09 | 0.0005 | 31.4 | 0.12 | 0.0007 | 40.1 |
| Yb | 173 | 0.04 | <0.04 | – | – | <0.04 | – | – | 0.23 | 0.0013 | 44.4 | 0.26 | 0.0015 | 32.5 | 0.64 | 0.0037 | 42.1 | 0.90 | 0.0052 | 22.4 |
| Lu | 175 | 0.02 | <0.02 | – | – | <0.02 | – | – | 0.02 | 0.0001 | 50.3 | 0.03 | 0.0002 | 43.5 | 0.12 | 0.0007 | 23.7 | 0.13 | 0.0007 | 32.2 |
| Al | 27 | 10 | 325 | 12.0 | 14.9 | 171 | 6.4 | 40.5 | 84.3 | 3.1 | 36.2 | 101 | 3.7 | 7.58 | 288 | 10.7 | 12.8 | 257 | 9.5 | 6.41 |
| As | 69 | 1 | 46.6 | 0.6 | 8.74 | 7.05 | 0.1 | 19.0 | <1 | 0.01 | 56.5 | <1 | 0.01 | 67.8 | 1.13 | 0.02 | 103 | 1.10 | 0.01 | 46.0 |
| Ba | 135, 137 | 0.1 | 0.50 | 0.004 | 54.1 | 0.19 | 0.001 | 36.60 | 0.29 | 0.002 | 25.06 | 0.22 | 0.002 | 14.77 | 0.48 | 0.003 | 89.66 | 0.63 | 0.005 | 27.03 |
| Be | 9 | 3 | 3.06 | 0.3 | 12.5 | 4.94 | 0.5 | 26.0 | 156 | 17.4 | 32.8 | 178 | 19.8 | 2.42 | 71.7 | 8.0 | 14.09 | 67.0 | 7.4 | 12.2 |
| Cr | 53 | 5 | <5 | – | – | <5 | – | – | <5 | – | – | <5 | – | – | 6.44 | 0.12 | 43.8 | <5 | – | – |
| Cu | 65 | 1 | 1.56 | 0.02 | 65.3 | <1 | – | – | <1 | – | – | <1 | – | – | 1.10 | 0.02 | 78.2 | 2.00 | 0.03 | 56.4 |
| Fe | 57 | 20 | <20 | – | – | <20 | – | – | <20 | – | – | <20 | – | – | <20 | – | – | <20 | – | – |
| K | 39 | 7 | <7 | – | – | <7 | – | – | <7 | – | – | <7 | – | – | <7 | – | – | <7 | – | – |
| Mg | 25 | 9 | <9 | – | – | <9 | – | – | <9 | – | – | <9 | – | – | <9 | – | – | <9 | – | – |
| Mn | 55 | 1 | <1 | – | – | <1 | – | – | <1 | – | – | <1 | – | – | 18.4 | 0.34 | 8.93 | 14.57 | 0.27 | 14.3 |
| Na | 23 | 50 | <50 | – | – | <50 | – | – | <50 | – | – | <50 | – | – | <50 | – | – | <50 | – | – |
| Ni | 62 | 18 | <18 | – | – | <18 | – | – | <18 | – | – | <18 | – | – | <18 | – | – | <18 | – | – |
| Pb | 207, 208, 209 | 0.1 | 0.27 | 0.0013 | 49.2 | 0.23 | 0.0011 | 40.0 | 7.46 | 0.0360 | 33.8 | 8.05 | 0.0389 | 8.94 | 11.1 | 0.0534 | 5.05 | 10.6 | 0.0514 | 5.60 |
| Sb | 121, 123 | 1 | 13.0 | 0.1 | 81.9 | 35.8 | 0.3 | 75.6 | <1 | – | – | <1 | – | – | <1 | – | – | <1 | – | – |
| Sr | 88 | 0.1 | 767 | 8.8 | 12.09 | 1611 | 14.8 | 40.6 | 387 | 4.4 | 1.44 | 381 | 4.4 | 1.02 | 66.5 | 0.8 | 2.81 | 66.1 | 0.8 | 5.08 |
| Th | 232 | 0.01 | <0.01 | – | – | <0.01 | – | – | 0.47 | 0.0020 | 41.3 | 0.66 | 0.0028 | 8.45 | 0.11 | 0.0005 | 51.7 | 0.11 | 0.0005 | 44.6 |
| Ti | 49 | 3 | <3 | – | – | <3 | – | – | <3 | – | – | <3 | – | – | <3 | – | – | <3 | – | – |
| V | 51 | 0.5 | 0.71 | 0.01 | 36.54 | 0.98 | 0.02 | 19.1 | 0.84 | 0.02 | 10.8 | 0.83 | 0.02 | 11.3 | 0.73 | 0.01 | 33.8 | 0.95 | 0.02 | 22.5 |
| Y | 89 | 0.1 | <0.1 | – | – | <0.1 | – | – | 4.21 | 0.05 | 35.1 | 5.23 | 0.1 | 6.19 | 10.6 | 0.1 | 21.2 | 14.1 | 0.2 | 27.9 |
| Zn | 67 | 10 | 59.6 | 0.9 | 34.6 | 27.5 | 0.4 | 53.1 | 15.8 | 0.2 | 32.5 | 15.5 | 0.2 | 41.7 | 63.2 | 1.0 | 39.3 | 89.9 | 1.4 | 49.0 |
| Sample Location | Unpowdered Samples | Powdered Samples | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H | eq. H2O+ (wt %) | C | N | S | H | eq. H2O+ (wt %) | C | N | S | |
| Mexico | <10 | <0.01 | 200 ± 5 | 50 ± 10 | 13 ± 1 | 31 ± 10 | 0.028 | 130 ± 50 | 230 ± 130 | 25 ± 7 |
| Tanzania | <10 | <0.01 | 120 ± 40 | 100 ± 50 | 8 ± 1 | 14 ± 1 | 0.012 | 70 ± 40 | 100 ± 50 | 22 ± 3 |
| Vietnam | 300 ± 50 | 0.244 | 5600 ± 20 | 1000 ± 10 | 15 ± 3 | 40 ± 10 | 0.036 | 3000 ± 40 | 520 ± 20 | 19 ± 11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huong, L.T.-T.; Otter, L.M.; Förster, M.W.; Hauzenberger, C.A.; Krenn, K.; Alard, O.; Macholdt, D.S.; Weis, U.; Stoll, B.; Jochum, K.P. Femtosecond Laser Ablation-ICP-Mass Spectrometry and CHNS Elemental Analyzer Reveal Trace Element Characteristics of Danburite from Mexico, Tanzania, and Vietnam. Minerals 2018, 8, 234. https://doi.org/10.3390/min8060234
Huong LT-T, Otter LM, Förster MW, Hauzenberger CA, Krenn K, Alard O, Macholdt DS, Weis U, Stoll B, Jochum KP. Femtosecond Laser Ablation-ICP-Mass Spectrometry and CHNS Elemental Analyzer Reveal Trace Element Characteristics of Danburite from Mexico, Tanzania, and Vietnam. Minerals. 2018; 8(6):234. https://doi.org/10.3390/min8060234
Chicago/Turabian StyleHuong, Le Thi-Thu, Laura M. Otter, Michael W. Förster, Christoph A. Hauzenberger, Kurt Krenn, Olivier Alard, Dorothea S. Macholdt, Ulrike Weis, Brigitte Stoll, and Klaus Peter Jochum. 2018. "Femtosecond Laser Ablation-ICP-Mass Spectrometry and CHNS Elemental Analyzer Reveal Trace Element Characteristics of Danburite from Mexico, Tanzania, and Vietnam" Minerals 8, no. 6: 234. https://doi.org/10.3390/min8060234
APA StyleHuong, L. T.-T., Otter, L. M., Förster, M. W., Hauzenberger, C. A., Krenn, K., Alard, O., Macholdt, D. S., Weis, U., Stoll, B., & Jochum, K. P. (2018). Femtosecond Laser Ablation-ICP-Mass Spectrometry and CHNS Elemental Analyzer Reveal Trace Element Characteristics of Danburite from Mexico, Tanzania, and Vietnam. Minerals, 8(6), 234. https://doi.org/10.3390/min8060234