New Uranyl Open Framework and Sheet Compounds Formed via In-Situ Protonation of Piperazine by Phosphorous Acid


Abstract
1. Introduction
2. Materials and Methods
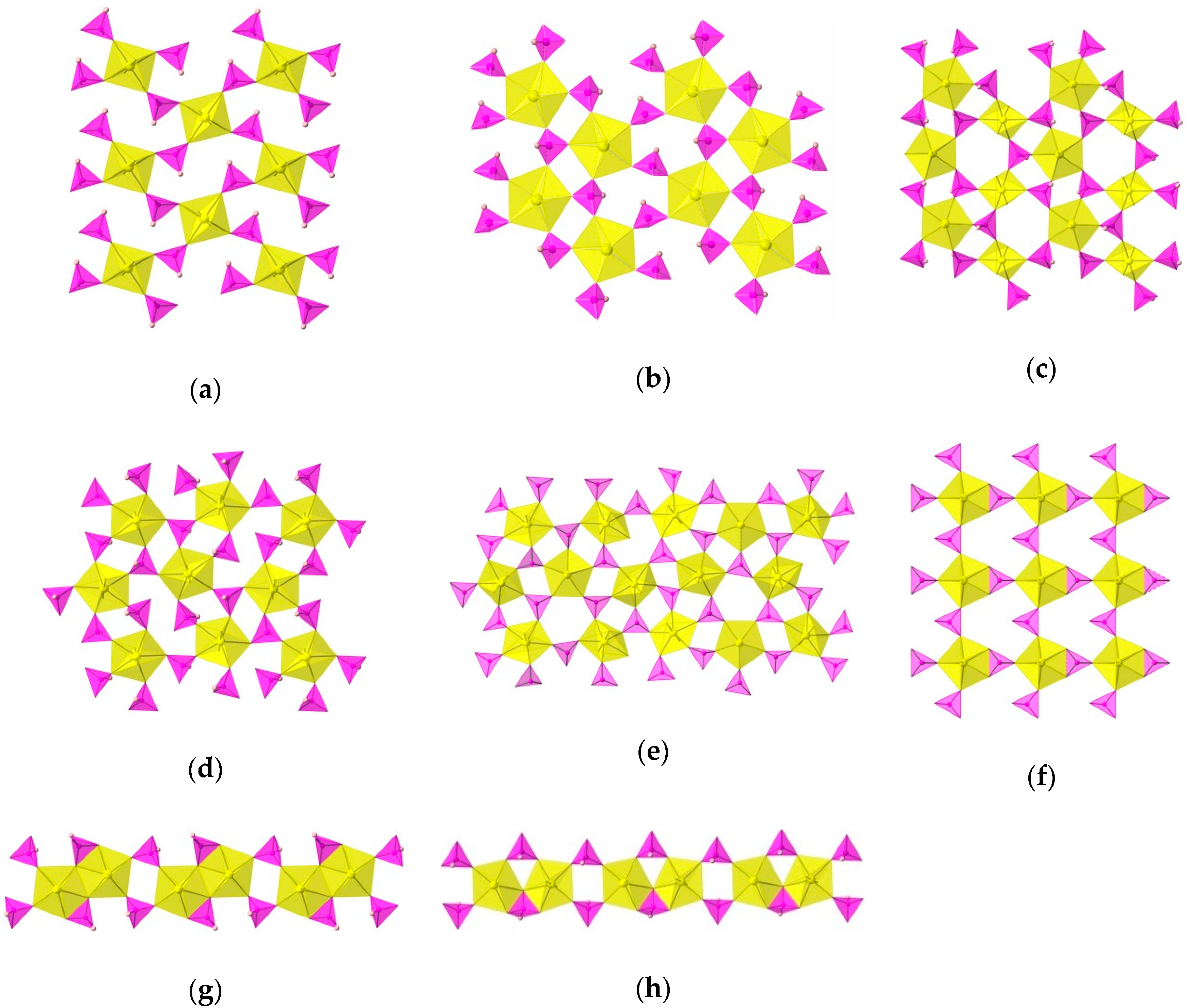
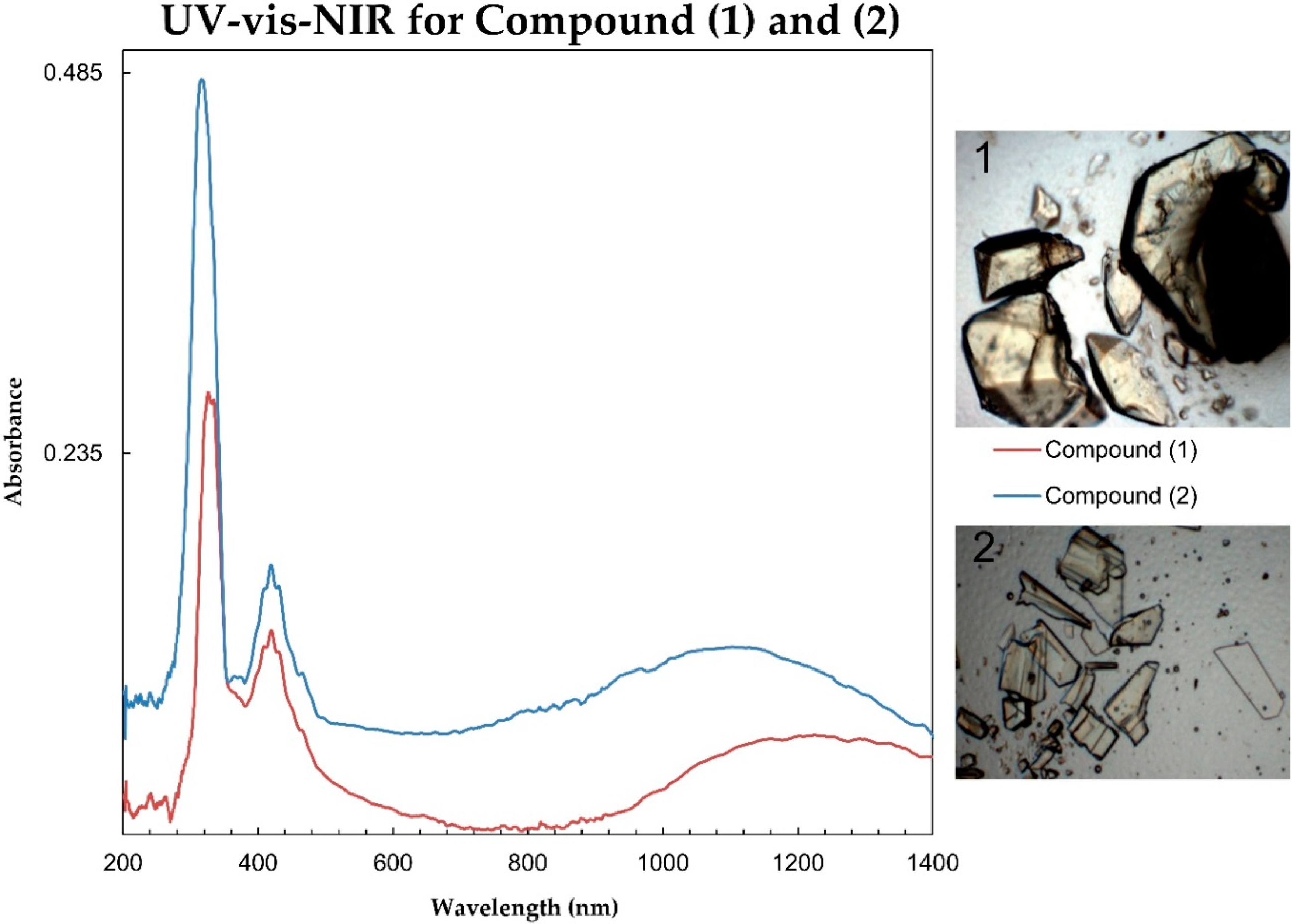
3. Results and Discussion
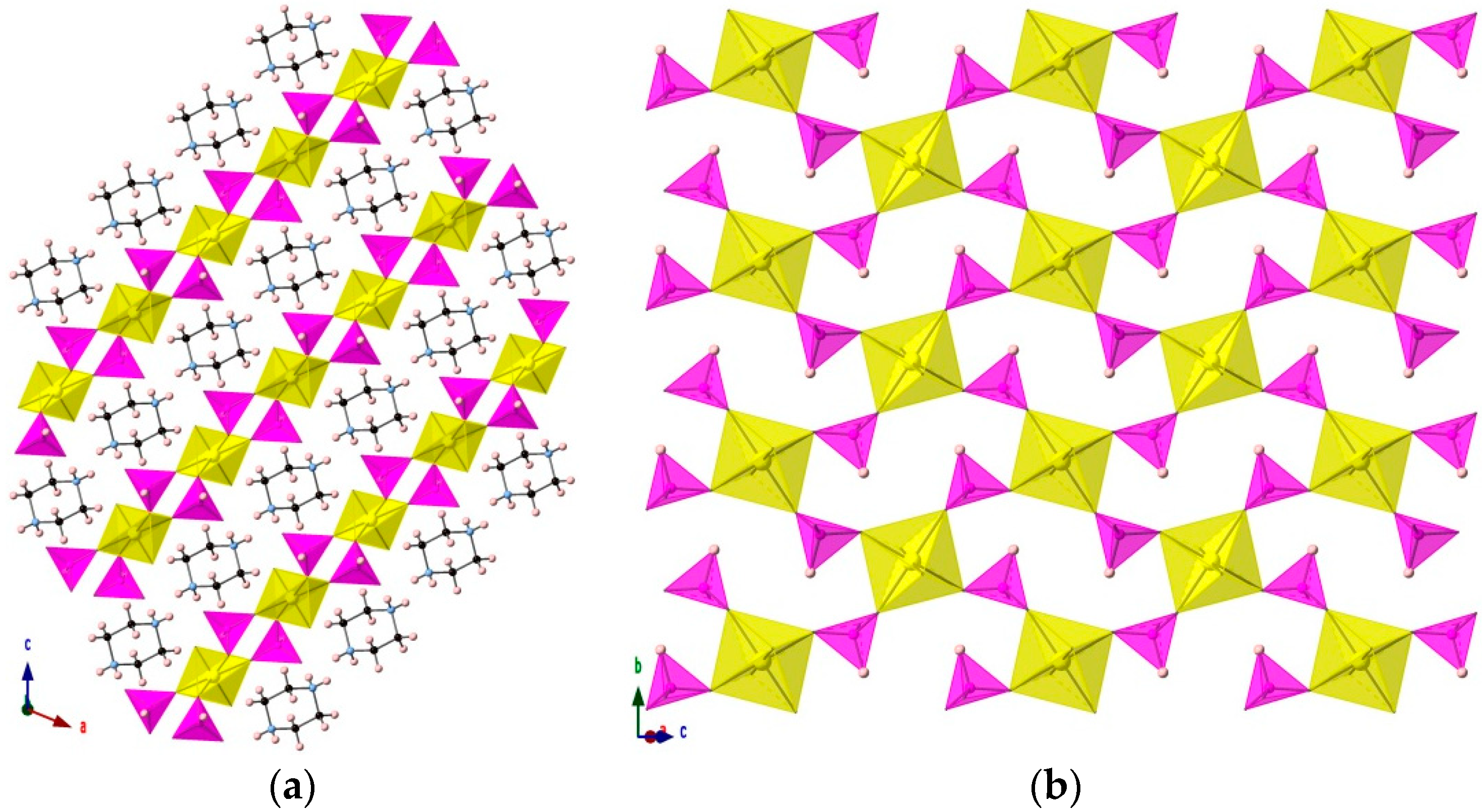
3.1. [H2PZ][UO2(HPO3)2]
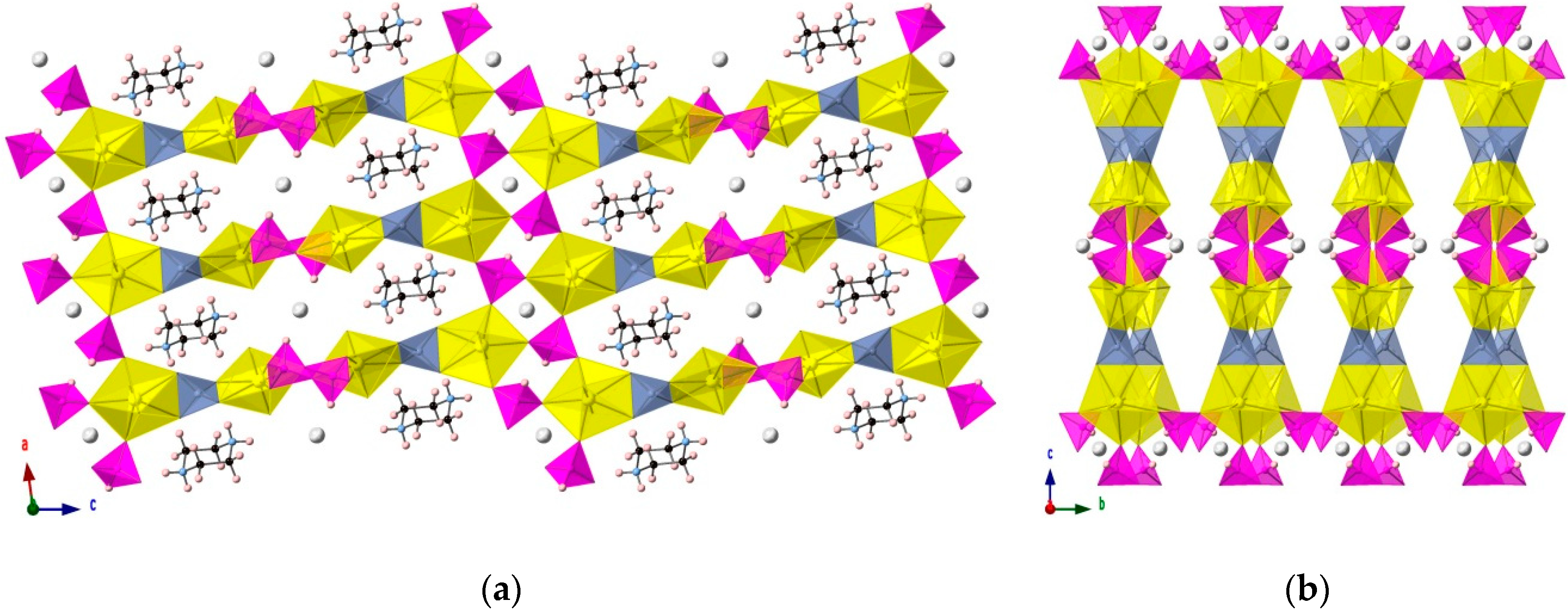
3.2. [(H2PZ)Cs][(UO2)2(HPO3)2(PO4)]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance | vu | Distance | vu | Distance | vu | Distance | vu | Total | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P(1)–O(1) | 1.529 | 1.314 | U(1)–O(1) | 2.265 | 0.662 | 1.976 | ||||||
| P(1)–O(2) | 1.504 | 1.406 | H(1A)–O(2) | 1.887 | 0.275 | H(1B)–O(2) | 1.884 | 0.277 | 1.957 | |||
| P(1)–O(3) | 1.52 | 1.346 | U(1)–O(3) | 2.283 | 0.640 | 1.986 | ||||||
| U(1)–O(4) | 1.785 | 1.669 | 1.669 | |||||||||
| P(1) BVS | 4.066 | U(1) BVS | 5.942 |
| Distance | vu | Distance | vu | Distance | vu | Distance | vu | Total | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P(1)–O(1) | 1.53 | 1.310 | U(1)–O(1) | 2.324 | 0.591 | Cs(1)–O(1) | 3.321 | 0.088 | 1.989 | |||
| P(1)–O(2) | 1.516 | 1.361 | U(1)–O(2) | 2.357 | 0.555 | Cs(1)–O(2) | 3.269 | 0.101 | 2.016 | |||
| P(1)–O(3) | 1.526 | 1.325 | U(1)–O(3) | 2.335 | 0.579 | Cs(1)–O(3) | 3.065 | 0.175 | 2.078 | |||
| P(2)–O(4) | 1.509 | 1.387 | U(2)–O(4) | 2.315 | 0.601 | 1.988 | ||||||
| P(2)–O(5) | 1.522 | 1.339 | U(2)–O(5) | 2.345 | 0.568 | Cs(2)–O(5) | 3.133 | 0.146 | 2.052 | |||
| P(2)–O(6) | 1.527 | 1.321 | U(2)–O(6) | 2.328 | 0.586 | Cs(2)–O(6) | 3.123 | 0.150 | 2.057 | |||
| P(3)–O(7) | 1.529 | 1.225 | U(2)–O(7) | 2.489 | 0.430 | H(1C)–O(7) | 1.795 | 0.352 | 2.007 | |||
| P(3)–O(8) | 1.551 | 1.154 | U(2)–O(8) | 2.473 | 0.443 | H(1C)–O(8) | 1.684 | 0.476 | 2.073 | |||
| P(3)–O(9) | 1.548 | 1.163 | U(1)–O(9) | 2.494 | 0.426 | H(3B)–O(9) | 1.852 | 0.302 | 1.891 | |||
| P(3)–O(10) | 1.543 | 1.179 | U(1)–O(10) | 2.439 | 0.474 | H(3A)–O(10) | 2.151 | 0.135 | 1.787 | |||
| U(1)–O(11) | 1.796 | 1.634 | Cs(1)–O(11) | 3.777 | 0.026 | 1.660 | ||||||
| U(1)–O(12) | 1.788 | 1.660 | Cs(2)–O(12) | 3.426 | 0.066 | 1.726 | ||||||
| U(2)–O(13) | 1.799 | 1.625 | Cs(2)–O(13) | 3.033 | 0.191 | 1.816 | ||||||
| U(2)–O(14) | 1.793 | 1.644 | Cs(2)–O(14) | 3.628 | 0.038 | 1.682 | ||||||
| P(1) BVS | 3.996 | U(1) BVS | 5.918 | |||||||||
| P(2) BVS | 4.047 | U(2) BVS | 5.898 | |||||||||
| P(3) BVS | 4.721 |
References
- Tabuteau, A.; Pages, M.; Livet, J.; Musikas, C. Monazite-like phases containing Tran-Uranium elements (Neptunium and Plutonium). J. Mater. Sci. Lett. 1988, 7, 1315–1317. [Google Scholar] [CrossRef]
- Dacheux, N.; Podor, R.; Chassigneux, B.; Brandel, V.; Genet, M. Actinides immobilization in new matrices based on solid solutions: Th 4−x MxIV(PO4)4P2O7, (MIV = 238U,239Pu). J. Alloys Compd. 1998, 271, 236–239. [Google Scholar] [CrossRef]
- Dacheux, N.; Thomas, A.C.; Chassigneux, B.; PichotI, E.; Brandel, V.; Genet, M. Study of Th4(PO4)4 P2O7 and solid solutions with U(IV), Np(IV) and Pu(IV): Synthesis, characterization, sintering and leaching tests. MRS Proc. 1999, 556, 85. [Google Scholar] [CrossRef]
- Kitaev, D.B.; Volkov, Y.F.; Orlova, A.I. Orthophosphates of tetravalent Ce, Th, U, Np, and Pu with the monazite structure. Radiochemistry 2004, 46, 211–217. [Google Scholar] [CrossRef]
- Wellman, D.M.; Mattigod, S.V.; Parker, K.E.; Heald, S.M.; Wang, C.; Fryxell, G.E. Synthesis of organically templated nanoporous Tin(II/IV) phosphate for radionuclide and metal sequestration. Inorg. Chem. 2006, 45, 2382–2384. [Google Scholar] [CrossRef] [PubMed]
- Bray, T.H.; Nelson, A.D.; Jin, G.B.; Haire, R.G.; Albrecht-Schmitt, T.E. In situ hydrothermal reduction of Neptunium(VI) as a route to Neptunium(IV) phosphonates. Inorg. Chem. 2007, 46, 10959–10961. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.D.; Bray, T.H.; Zhan, W.; Haire, R.G.; Sayler, T.S.; Albrecht-Schmitt, T.E. Further examples of the failure of surrogates to properly model the structural and hydrothermal chemistry of transuranium elements: Insights provided by uranium and neptunium diphosphonates. Inorg. Chem. 2008, 47, 4945–4951. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.D.; Bray, T.H.; Albrecht-Schmitt, T.E. Capitalizing on differing coordination environments and redox potentials to prepare an ordered heterobimetallic U(VI)/Np(IV) diphosphonate. Angew. Chem. Int. Ed. Engl. 2008, 47, 6252–6254. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.D.; Bray, T.H.; Stanley, F.A.; Albrecht-Schmitt, T.E. Periodic trends in actinide phosphonates: Divergence and convergence between thorium, uranium, neptunium, and plutonium systems. Inorg. Chem. 2009, 48, 4530–4535. [Google Scholar] [CrossRef] [PubMed]
- Diwu, J.; Nelson, A.D.; Wang, S.; Campana, C.F.; Albrecht-Schmitt, T.E. Comparisons of Pu(IV) and Ce(IV) diphosphonates. Inorg. Chem. 2010, 49, 3337–3342. [Google Scholar] [CrossRef] [PubMed]
- Diwu, J.; Wang, S.; Liao, Z.; Burns, P.C.; Albrecht-schmitt, T.E. Cerium(IV), Neptunium(IV), and Plutonium(IV) 1,2-phenylenediphosphonates: Correlations and differences between early transuranium elements and their proposed surrogates. Inorg. Chem. 2010, 49, 10074–10080. [Google Scholar] [CrossRef] [PubMed]
- Knope, K.E.; Cahill, C.L. Homometallic UO22+ diphosphonates assembled under ambient and hydrothermal conditions. Dalton Trans. 2010, 39, 8319–8324. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Diwu, J.; Gui, D.; Wang, Y.; Weng, Z.; Chai, Z.; Albrecht-Schmitt, T.E.; Wang, S. Systematic Investigation of the in Situ Reduction Process from U(VI) to U(IV) in a Phosphonate System under Mild Solvothermal Conditions. Inorg. Chem. 2017, 56, 6952–6964. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.M.; Wang, S.; Alekseev, E.V.; Depmeier, W.; Albrecht-Schmitt, T.E. Facile routes to ThIV, UIV, and NpIV phosphites and phosphates. Eur. J. Inorg. Chem. 2011, 3749–3754. [Google Scholar] [CrossRef]
- Cross, J.N.; Villa, E.M.; Wang, S.; Diwu, J.; Polinski, M.J.; Albrecht-Schmitt, T.E. Syntheses, structures, and spectroscopic properties of plutonium and americium phosphites and the redetermination of the ionic radii of Pu(III) and Am(III). Inorg. Chem. 2012, 51, 8419–8424. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.M.; Marr, C.J.; Jouffret, L.J.; Alekseev, E.V.; Depmeier, W.; Albrecht-Schmitt, T.E. Systematic evolution from Uranyl(VI) phosphites to Uranium(IV) phosphates. Inorg. Chem. 2012, 51, 6548–6558. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.M.; Marr, C.J.; Diwu, J.; Alekseev, E.V.; Depmeier, W.; Albrecht-Schmitt, T.E. From order to disorder and back again: In situ hydrothermal redox reactions of uranium phosphites and phosphates. Inorg. Chem. 2013, 52, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.M.; Alekseev, E.V.; Depmeier, W.; Albrecht-Schmitt, T.E. Syntheses, structures, and comparisons of Thallium Uranium phosphites, mixed phosphate-phosphites, and phosphate. Cryst. Growth Des. 2013, 13, 1721–1729. [Google Scholar] [CrossRef]
- Avduevskaya, K.A.; Ragulina, N.B.; Rozanov, I.A. Basic Uranyl Phosphites. Inorg. Mater. 1981, 17, 834–836. [Google Scholar]
- Avduevskaya, K.A.; Rozanov, I.A.; Mironova, V.S. Uranium(IV) Phosphites. Inorg. Mater. 1977, 13, 1515–1517. [Google Scholar]
- Avduevskaya, K.A.; Ragulina, N.B.; Rozanov, I.A. Synthesis and Investigation of Mixed Phosphite Uranylates. Izv. Akad. Nauk SSSR Neorg. Mater. 1978, 14, 2078–2084. [Google Scholar]
- Mistryukov, V.E.E.; Mikhailov, Y.N. Crystal Structure of Rb(hydrophosphito)(phosphito)dioxouranate(VI) trihydrate and K(bis(phosphito)dioxouranate(VI) dihydrate.pdf. Koord. Khimiya 1985, 11, 1393–1398. [Google Scholar]
- Oh, G.N.; Burns, P.C. Solid-state actinide acid phosphites from phosphorous acid melts. J. Solid State Chem. 2014, 215, 50–56. [Google Scholar] [CrossRef]
- Gui, D.; Dai, X.; Zheng, T.; Wang, X.; Silver, M.A.; Chen, L.; Zhang, C.; Diwu, J.; Zhou, R.; Chai, Z.; et al. An ultrastable heterobimetallic Uranium(IV)/Vanadium(III) solid compound protected by a redox-active phosphite ligand: Crystal structure, oxidative dissolution, and first-principles simulation. Inorg. Chem. 2018, 57, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Doran, M.; Walker, S.M.; O’Hare, D. Synthesis and characterisation of (C4N2H12)(UO2)2(PO3H)2{PO2(OH)H}2: A three dimensionally connected actinide framework. Chem. Commun. 2001, 2, 1988–1989. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Cao, Y.; Huang, C.; Zhang, H.; Liu, D.; Yang, Q.; Sun, R. Synthesis and characterization of two new organically templated open-framework Uranium phosphites. Chin. J. Struct. Chem. 2006, 25, 1380–1386. [Google Scholar]
- Mandal, S.; Chandra, M.; Natarajan, S. Synthesis, structure, and upconversion studies on organically templated uranium phosphites. Inorg. Chem. 2007, 46, 7935–7943. [Google Scholar] [CrossRef] [PubMed]
- Felder, J.B.; Smith, M.D.; zur Loye, H.C. Breaking a paradigm: Observation of magnetic order in the purple U(IV) phosphite: U(HPO3)2. Inorg. Chem. 2018, 57, 9851–9858. [Google Scholar] [CrossRef] [PubMed]
- Burns, P.C. The crystal chemistry of hexavalent uranium; polyhedron geometries, bond-valence parameters, and polymerization of polyhedra. Can. Miner. 1997, 35, 1551–1570. [Google Scholar]
- Burns, P.C. The crystal chemistry of uranium. Rev. Miner. Geochem. 1999, 38, 23–90. [Google Scholar]
- Locock, A.J.; Burns, P.C.; Duke, M.J.M.; Flynn, T.M. Monovalent Cations in structures of the meta-autunite group. Can. Mineral. 2004, 42, 973–996. [Google Scholar] [CrossRef]
- Burns, P.C. U6+ Minerals and inorganic compounds: Insights into an expanded structural hierarchy of crystal structures. Can. Miner. 2005, 43, 1839–1894. [Google Scholar] [CrossRef]
- Burns, P.C. Crystal chemistry of uranium oxocompounds: An overview. In Structural Chemistry of Inorganic Actinide Compounds; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–30. [Google Scholar]
- Locock, A.J. Crystal chemistry of actinide phosphates and arsenates. In Structural Chemistry of Inorganic Actinide Compounds; Elsevier: Amsterdam, The Netherlands, 2007; pp. 217–278. [Google Scholar]
- Krivovichev, S.V. Crystal Chemistry of Uranium Oxides and Minerals. In Comprehensive Inorganic Chemistry II; Elsevier: Amsterdam, The Netherlands, 2013; pp. 611–640. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- CrystalMaker, version 9.2.9.48; Software For Technical Computation; CrystalMaker Software Ltd.: Oxfordshire, UK, 2017.
- Bell, J.T.; Biggers, R.E. The absorption spectrum of the uranyl ion in perchlorate media. J. Mol. Spectrosc. 1965, 18, 247–275. [Google Scholar] [CrossRef]
- Burrows, H.D.; Kemp, T.J. The photochemistry of the uranyl ion. Chem. Soc. Rev. 1974, 3, 139. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction CrysAlis PRO 2015; Software For Technical Computation; Rigaku: Woodlands, TX, USA, 2015.
- Linde, S.A.; Gorbunova, Y.E.; Lavrov, A.V. Crystal Structure of K4UO2(PO4)2. Russ. J. Inorg. Chem. 1980, 25, 1992–1994. [Google Scholar]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B Struct. Sci. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Loub, J. Crystal chemistry of inorganic phosphites. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 468–473. [Google Scholar] [CrossRef]
- Vanier, M.; Brisse, F. Structural studies of compounds with aliphatic chains. 7. The structure of piperazinium glutarate and the geometry of the piperazinium cation. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 3060–3063. [Google Scholar] [CrossRef]
- Kang, J.; Ahn, J.; Shin, H.; Pyo, S.; Yun, H.; Do, J. Crystal structure of tris(piperazinium) hexakis[(μ3-oxo)(μ2-oxo)-dioxotungsten]tellurate(VI) hexahydrate, [C4H12N2]3[TeW6O24] 6H2O. Zeitschrift für Krist. New Cryst. Struct. 2011, 226, 129–130. [Google Scholar] [CrossRef]
- Marcotrigiano, G.; Menabue, L.; Pellacani, G.C. Tetrahalo- and (mixed-tetrahalo)cuprates of the piperazinium dication. Coordination geometry changes in some CuX42- anions. Inorg. Chem. 1976, 15, 2333–2336. [Google Scholar] [CrossRef]
- Havlíček, D.; Plocek, J.; Němec, I.; Gyepes, R.; Mička, Z. The Crystal Structure, Vibrational Spectra, and Thermal Behavior of Piperazinium(2+) Selenate Monohydrate and N, N′-Dimethylpiperazinium(2+) Selenate Dihydrate. J. Solid State Chem. 2000, 150, 305–315. [Google Scholar] [CrossRef]
- Havlíček, D.; Chudoba, V.; Němec, I.; Císařová, I.; Mička, Z. Preparation, crystal structure, vibrational spectra and thermal behaviour of piperazinium(2+) selenite monohydrate and piperazinium(2+) diselenite. J. Mol. Struct. 2002, 606, 101–116. [Google Scholar] [CrossRef]
- Moskwa, M.; Bator, G.; Piecha-Bisiorek, A.; Jakubas, R.; Medycki, W.; Ciżman, A.; Baran, J. X-ray structure and investigation of molecular motions by dielectric, vibrational and 1 H NMR methods for two organic-inorganic hybrid piperazinium compounds: (C4H12N2)2[Sb2Cl10]·2H2O and (C4H12N2)2[Sb2Br10]·2H2O. Mater. Res. Bull. 2018, 104, 202–211. [Google Scholar] [CrossRef]
- Berrocal, T.; Mesa, J.L.; Pizarro, J.L.; Lezama, L.; Bazán, B.; Arriortua, M.I.; Rojo, T. Hydrothermal synthesis, crystal structure, thermal behavior and spectroscopic and magnetic properties of two new organically templated fluoro-vanadyl-hydrogenarsenates: (R)0.5[(VO)(HAsO4)F] (R: Ethylenediammonium and piperazinium). J. Solid State Chem. 2008, 181, 884–894. [Google Scholar] [CrossRef]
- Yu, D.; Xue, D.; Ratajczak, H. Microscopic characteristics of hydrogen bonds of hydrated borates. Phys. B Condens. Matter 2006, 371, 170–176. [Google Scholar] [CrossRef]
- Parkin, A.; Oswald, I.D.H.; Parsons, S. Structures of piperazine, piperidine and morpholine. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.M.; Diwu, J.; Alekseev, E.V.; Depmeier, W.; Albrecht-Schmitt, T.E. Structural changes within the alkaline earth uranyl phosphites. Dalton Trans. 2013, 42, 9637–9644. [Google Scholar] [CrossRef] [PubMed]




| Compounds | ||
| Chemical Formula | [H2C4H10N2][UO2(HPO3)2] | [(H2C4H10N2)Cs][(UO2)2(HPO3)2(PO4)] |
| Mr (g/mol) | 518.14 | 1016.05 |
| Crystal System | Monoclinic | Monoclinic |
| Space Group | P21/n | I2/a |
| a, Å | 8.3711(2) | 12.4927(3) |
| b, Å | 7.2898(2) | 6.64030(10) |
| c, Å | 10.8505(3) | 44.7504(9) |
| β, deg. | 112.205(3) | 97.631(2) |
| V, Å3 | 613.03(3) | 3679.40(13) |
| Z | 2 | 8 |
| Density (g/cm3) | 2.807 | 3.668 |
| Absorption Coefficient (mm−1) | 13.531 | 19.864 |
| Crystal Size (mm3) | 0.150 × 0.099 × 0.095 | 0.153 × 0.070 × 0.028 |
| Data Collection | ||
| Temperature, K | 293(2) | 293(2) |
| Radiation type, wavelength | MoKα (λ = 0.71073) | |
| Absorption correction | CrysAlis PRO 1.171.39.44a Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm | |
| 2θ angles range, deg. | 5.288 to 61.008 | 3.674 to 58.358 |
| Reflections collected | 17323 | 20732 |
| Unique reflections | 1870 | 4640 |
| Observed reflections | 1676 | 4304 |
| Structure Refinement | ||
| Rint | 0.0302 | 0.0406 |
| R1 [F2 > 2σ(F2)], wR2 [F2] | R1 = 0.0139, wR2 = 0.0334 | R1 = 0.0473, wR2 = 0.1199 |
| R1 [all data], wR2 [all data] | R1 = 0.0164, wR2 = 0.0347 | R1 = 0.0507, wR2 = 0.1216 |
| Data/Restraints/Parameters | 1870/0/83 | 4640/2/243 |
| S | 1.047 | 1.151 |
| ρmax, ρmin, e·Å−3 | 0.99/−0.78 | 7.63/−4.74 |
| Compound (1) | ||||
| Atom | x | y | z | U(eq) |
| U1 | 5000 | 10000 | 5000 | 14.05(5) |
| P1 | 5888.3(7) | 6339.8(8) | 7398.3(5) | 16.95(10) |
| O1 | 5967(2) | 7415(2) | 6212.5(17) | 27.0(4) |
| O2 | 4379(2) | 6866(2) | 7754.3(17) | 25.2(3) |
| O3 | 7610(2) | 6466(3) | 8567.0(18) | 32.9(4) |
| O4 | 6152(2) | 9354(3) | 3979.2(17) | 26.4(3) |
| N1 | 1300(3) | 5175(2) | 6311(2) | 19.6(4) |
| C1 | 1738(3) | 4512(4) | 5178(2) | 23.2(4) |
| C2 | −208(3) | 6440(3) | 5841(2) | 24.8(5) |
| Compound (2) | ||||
| Atom | x | y | z | U(eq) |
| U1 | 4660.3(3) | 3993.4(6) | 6943.7(2) | 6.84(11) |
| U2 | 3936.9(3) | 4145.2(6) | 5548.8(2) | 9.85(11) |
| Cs1 | 2500 | −2500 | 7500 | 27.0(3) |
| Cs2 | 2500 | 8845.0(19) | 5000 | 33.4(3) |
| P1 | 5148(3) | −1155(4) | 7234.0(7) | 12.0(5) |
| P2 | 6160(2) | 6479(5) | 5252.7(7) | 16.1(6) |
| P3 | 4155(2) | 3975(4) | 6249.3(6) | 8.2(5) |
| O1 | 5331(7) | 789(12) | 7064.4(19) | 16.3(17) |
| O2 | 4370(7) | −2595(12) | 7054.0(18) | 12.4(15) |
| O3 | 4778(7) | −721(12) | 7539.1(18) | 12.5(15) |
| O4 | 5626(7) | 5397(18) | 5490(2) | 27(2) |
| O5 | 6220(7) | 5224(13) | 4970.7(19) | 14.8(16) |
| O6 | 7263(7) | 7299(14) | 5386(2) | 18.0(17) |
| O7 | 4991(7) | 4712(14) | 6053.9(18) | 14.5(16) |
| O8 | 3194(7) | 3271(14) | 6017.5(18) | 16.1(17) |
| O9 | 3819(7) | 5604(13) | 6466.2(19) | 16.9(17) |
| O10 | 4610(7) | 2259(12) | 6461.6(18) | 14.3(16) |
| O11 | 3330(7) | 3277(14) | 7014(2) | 16.6(17) |
| O12 | 6005(6) | 4644(12) | 6884.5(18) | 12.5(15) |
| O13 | 3383(8) | 6631(14) | 5573(2) | 22.5(19) |
| O14 | 4457(7) | 1657(14) | 5508.1(19) | 19.4(18) |
| N1 | 1040(10) | 260(20) | 5970(3) | 28(3) |
| N2 | 2243(10) | −244(18) | 6559(3) | 24(2) |
| C1 | 1330(8) | 2017(17) | 6171(3) | 11(2) |
| C2 | 1876(10) | −1410(18) | 6033(3) | 16(2) |
| C3 | 1962(9) | −2011(17) | 6359(2) | 11(2) |
| C4 | 1452(9) | 1431(19) | 6496(3) | 14(2) |
| Compound (1) | ||||||
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
| U1 | 12.31(6) | 18.32(7) | 9.81(6) | −0.24(3) | 2.26(4) | 1.18(3) |
| P1 | 15.2(2) | 17.2(2) | 15.8(2) | 1.7(2) | 2.79(18) | 1.7(2) |
| O1 | 30.3(9) | 28.8(8) | 24.5(8) | 10.5(7) | 13.5(7) | 10.0(7) |
| O2 | 21.7(8) | 29.4(8) | 26.5(8) | −3.7(7) | 11.3(7) | −1.8(6) |
| O3 | 22.3(8) | 34.3(10) | 28.5(9) | 3.7(8) | −5.9(7) | 4.9(7) |
| O4 | 30.3(9) | 30.7(9) | 22.9(8) | 3.2(7) | 15.4(7) | 7.4(8) |
| N1 | 17.8(9) | 23.4(9) | 15.8(9) | −0.6(6) | 4.4(7) | −0.8(6) |
| C1 | 19.6(11) | 29.3(11) | 20.9(11) | −0.2(9) | 8.1(9) | 4.8(9) |
| C2 | 25.0(11) | 26.5(11) | 21.0(10) | −4.0(9) | 6.5(8) | 4.7(9) |
| Compound (2) | ||||||
| Atom | U11 | U22 | U33 | U23 | U13 | U12 |
| U1 | 9.63(19) | 6.15(18) | 4.72(18) | −0.19(13) | 0.91(13) | −0.39(13) |
| U2 | 9.57(19) | 14.7(2) | 5.37(18) | −0.62(14) | 1.35(13) | −0.71(14) |
| Cs1 | 15.2(5) | 44.4(7) | 20.7(5) | 2.6(5) | −0.3(4) | −1.5(5) |
| Cs2 | 57.2(9) | 16.7(5) | 22.6(6) | 0 | −8.4(6) | 0 |
| P1 | 19.1(14) | 7.5(12) | 9.3(13) | 0.0(10) | 1.6(11) | 1.1(10) |
| P2 | 12.5(14) | 28.0(16) | 7.5(13) | −2.7(12) | 0.2(10) | −0.7(12) |
| P3 | 7.7(12) | 11.4(12) | 5.4(12) | −0.1(9) | 1.0(9) | 0.0(9) |
| O1 | 25(5) | 10(4) | 14(4) | 2(3) | 4(3) | 0(3) |
| O2 | 16(4) | 10(4) | 10(4) | 1(3) | −4(3) | 5(3) |
| O3 | 16(4) | 12(4) | 9(4) | 0(3) | 0(3) | −2(3) |
| O4 | 11(4) | 57(7) | 13(4) | 2(4) | 2(3) | −12(4) |
| O5 | 13(4) | 20(4) | 12(4) | −2(3) | 4(3) | −2(3) |
| O6 | 14(4) | 23(4) | 18(4) | −2(4) | 5(3) | 0(3) |
| O7 | 13(4) | 20(4) | 10(4) | −2(3) | 1(3) | −4(3) |
| O8 | 17(4) | 27(5) | 5(3) | 1(3) | 3(3) | −8(4) |
| O9 | 22(4) | 16(4) | 12(4) | −3(3) | 1(3) | 8(3) |
| O10 | 27(4) | 6(3) | 10(4) | −5(3) | 0(3) | 5(3) |
| O11 | 13(4) | 19(4) | 19(4) | −2(3) | 7(3) | −3(3) |
| O12 | 13(4) | 13(4) | 11(4) | 4(3) | 1(3) | −1(3) |
| O13 | 40(6) | 13(4) | 16(4) | 1(3) | 8(4) | 3(4) |
| O14 | 21(4) | 24(5) | 12(4) | −5(3) | −1(3) | 8(4) |
| N1 | 29(6) | 34(7) | 20(6) | 4(5) | 0(5) | −6(5) |
| N2 | 29(6) | 20(5) | 21(6) | 3(4) | −2(5) | 4(5) |
| C1 | 4(4) | 11(5) | 18(5) | 2(4) | 2(4) | −2(4) |
| C2 | 22(6) | 14(5) | 13(5) | −7(4) | 10(4) | −5(5) |
| C3 | 11(5) | 12(5) | 10(5) | 1(4) | 4(4) | −1(4) |
| C4 | 12(5) | 18(6) | 13(5) | −3(4) | 5(4) | −2(4) |
| Compound (1) | |||||
| Atom | Atom | Length/Å | Atom | Atom | Length/Å |
| U1 | O1 1 | 2.265(2) | P1 | O1 | 1.529(2) |
| U1 | O1 | 2.265(2) | P1 | O2 | 1.504(2) |
| U1 | O3 2 | 2.283(2) | P1 | O3 | 1.520(2) |
| U1 | O3 3 | 2.283(2) | P1 | H1 | 1.44(4) |
| U1 | O4 1 | 1.785(2) | N1 | C1 | 1.490(3) |
| U1 | O4 | 1.785(2) | N1 | C2 | 1.489(3) |
| C1 | C2 4 | 1.508(3) | |||
| Compound (2) | |||||
| Atom | Atom | Length/Å | Atom | Atom | Length/Å |
| U1 | O1 | 2.324(8) | P1 | H1 | 1.48(5) |
| U1 | O2 5 | 2.357(8) | P2 | O4 | 1.509(10) |
| U1 | O3 6 | 2.335(8) | P2 | O5 | 1.522(9) |
| U1 | O9 | 2.494(9) | P2 | O6 | 1.527(9) |
| U1 | O10 | 2.439(8) | P2 | H2 | 1.54(5) |
| U1 | O11 | 1.796(8) | P3 | O7 | 1.529(9) |
| U1 | O12 | 1.788(8) | P3 | O8 | 1.551(9) |
| U2 | O4 | 2.315(9) | P3 | O9 | 1.548(9) |
| U2 | O5 7 | 2.345(8) | P3 | O10 | 1.543(8) |
| U2 | O6 8 | 2.328(9) | N1 | C1 | 1.49(2) |
| U2 | O7 | 2.489(8) | N1 | C2 | 1.52(2) |
| U2 | O8 | 2.473(8) | N2 | C3 | 1.49(2) |
| U2 | O13 | 1.799(9) | N2 | C4 | 1.49(2) |
| U2 | O14 | 1.793(9) | C1 | C4 | 1.49(2) |
| P1 | O1 | 1.530(9) | C2 | C3 | 1.50(2) |
| P1 | O2 | 1.516(9) | |||
| P1 | O3 | 1.526(8) | |||
| Compound (1) | |||||
| D-H | d(D-H) | d(H⋯A) | <DHA | d(D⋯A) | A |
| N1-H1A | 0.890 | 1.887 | 165.67 | 2.758 | O2 |
| N1-H1B | 0.890 | 1.884 | 163.71 | 2.750 | O2 |
| Compound (2) | |||||
| D-H | d(D-H) | d(H⋯A) | <DHA | d(D⋯A) | A |
| C1-H1C | 0.970 | 1.795 | 167.22 | 2.749 | O7 |
| C1-H1D | 0.970 | 1.684 | 171.37 | 2.647 | O8 |
| C3-H3B | 0.970 | 1.852 | 163.72 | 2.796 | O9 |
| C3-H3A | 0.970 | 2.151 | 151.44 | 3.039 | O10 |
| Compound | KUPO3 | RbUPO3 | CsUPO3 | TlUPO3 | RbUH2PO3 | CsUH2PO3 | TlUH2PO3 | TlUPO3-2 | CaUPO3 | SrUPO3 | BaUPO3 | (1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure Type | (b) | (c) | (c) | (c) | (c) | (d) | (d) | (e) | (f) | (g) | (h) | (a) |
| a | 7.598(3) | 11.1654(2) | 10.7049(2) | 11.1774(2) | 8.589(3) | 7.975(1) | 6.8795(3) | 14.705(2) | 22.8646(15) | 7.746(2) | 10.5731(5) | 8.3711(2) |
| b | 6.938(1) | 11.3309(2) | 11.8511(2) | 11.4215(2) | 10.275(1) | 10.564(2) | 9.7980(4) | 10.448(1) | 6.6182(3) | 8.651(2) | 16.6827(8) | 7.2898 (2) |
| c | 10.617(4) | 12.1573(2) | 12.6431(2) | 12.1390(2) | 13.230(4) | 10.677(2) | 13.3598(6) | 26.641(3) | 7.0309(4) | 8.723(2) | 13.3432(7) | 10.8505 (3) |
| α | 90 | 90 | 90 | 90 | 90 | 94.147(3) | 83.848(3) | 90 | 90 | 98.81(2) | 90 | 90 |
| β | 110.84(2) | 109.376(1) | 101.585(1) | 108.907(1) | 106.83(3) | 95.880(3) | 79.249(3) | 91.203(8) | 94.762(4) | 108.52(2) | 113.273(2) | 112.205(3) |
| γ | 90 | 90 | 90 | 90 | 90 | 90.411(4) | 74.623(3) | 90 | 90 | 110.15(2) | 90 | 90 |
| Crystal System | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space Group | P21 | P21/n | P21/c | P21/n | P21/n | P-1 | P-1 | P21/n | C2 | P-1 | P21/c | P21/n |
| Network | Sheet | Sheet | Sheet | Sheet | Sheet | Sheet | Sheet | Sheet | Sheet | Chain | Chain | Sheet |
| Uranyl Polyhedra | PBP | SBP PBP | SBP PBP | SB PBP | PBP | PBP | PBP | PBP | PBP | PBP | PBP | SBP |
| U–U Polyhedra Interactions | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Isolated Polyhedra | Edge-Sharing Dimers | Corner-Sharing Dimers | Isolated Polyhedra |
| U=O Avg. Bond Length (Å) | 1.79(1) | 1.788(8) 1.76(1) | 1.785(4) 1.786(5) | 1.772(7) 1.770(6) | 1.78(2) | 1.786(4) | 1.77(1) | 1.75(2) | 1.767(9) | 1.795(7) | 1.780(6) | 1.785(2) |
| U–O Avg. Bond Length (Å) | 2.37(2) | 2.286(4) 2.363(8) | 2.280(4) 2.363(5) | 2.277(7) 2.391(8) | 2.51(1) | 2.363(4) | 2.37(1) | 2.36(2) | 2.393(9) | 2.377(6) | 2.394(6) | 2.274(2) |
| Phosphite Oxygen | Terminal & Bridging | All Bridging | All Bridging | All Bridging | Terminal & Bridging | Terminal & Bridging | Terminal & Bridging | All Bridging | Terminal & Bridging | Terminal & Bridging | Terminal & Bridging | Terminal & Bridging |
| P–O Avg. Bond Length (Å) | η-1.46(2) μ2-1.53(2) | μ2-1.519(8) | μ2-1.522(5) | μ2-1.499(7) | η-1.57(1) μ2-1.48(1) | η-1.578(5) μ2-1.516(5) | η-1.56(1) μ2-1.50(1) | μ2-1.51(2) | η-1.53(1) μ2-1.526(9) | η-1.490(7) μ2-1.540(6) | η-1.511(5) μ2-1.534(6) μ3-1.559(5) | η-1.504(2) μ2-1.525(2) |
| P–H Avg. Bond Length (Å) | 1.13 | 1.33 | 1.46 | 1.37 | 1.23 | 1.31 | 1.41 | N/A | N/A | 1.49 | 1.24 | 1.44 |
| Reference | [22] | [16] | [16] | [18] | [22] | [16] | [18] | [18] | [56] | [56] | [56] | This Work |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, E.M.; Cross, J.N.; Albrecht-Schmitt, T.E. New Uranyl Open Framework and Sheet Compounds Formed via In-Situ Protonation of Piperazine by Phosphorous Acid. Minerals 2018, 8, 497. https://doi.org/10.3390/min8110497
Villa EM, Cross JN, Albrecht-Schmitt TE. New Uranyl Open Framework and Sheet Compounds Formed via In-Situ Protonation of Piperazine by Phosphorous Acid. Minerals. 2018; 8(11):497. https://doi.org/10.3390/min8110497
Chicago/Turabian StyleVilla, Eric M., Justin N. Cross, and Thomas E. Albrecht-Schmitt. 2018. "New Uranyl Open Framework and Sheet Compounds Formed via In-Situ Protonation of Piperazine by Phosphorous Acid" Minerals 8, no. 11: 497. https://doi.org/10.3390/min8110497
APA StyleVilla, E. M., Cross, J. N., & Albrecht-Schmitt, T. E. (2018). New Uranyl Open Framework and Sheet Compounds Formed via In-Situ Protonation of Piperazine by Phosphorous Acid. Minerals, 8(11), 497. https://doi.org/10.3390/min8110497