1. Introduction
The Fe–Ni laterite deposits in Greece are a major source of nickel, a metal of strategic significance. Greece’s Fe–Ni ore reserves are currently estimated to be in the range of 200 Mt at 0.9%–1.4% Ni ([
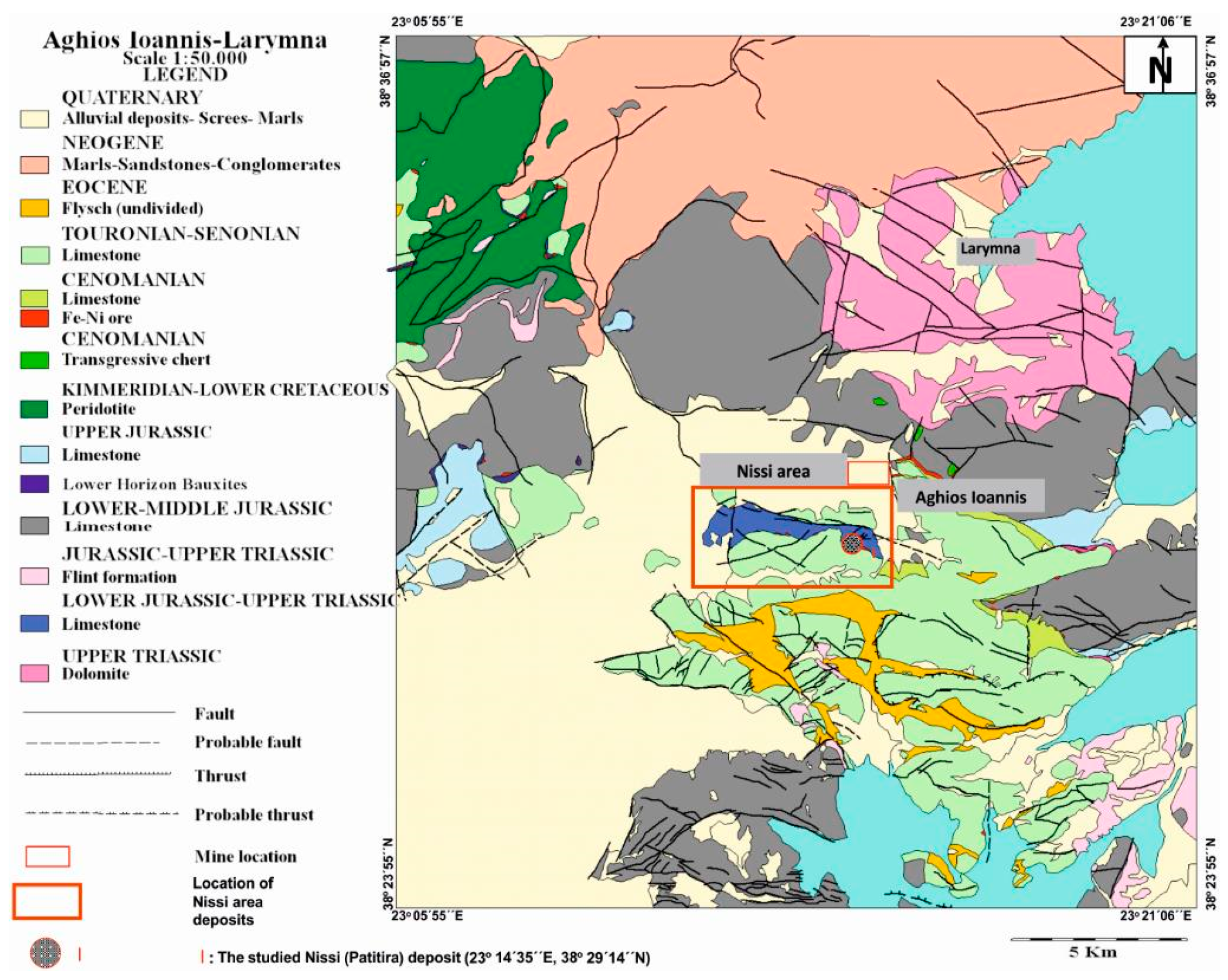
1] and references therein). Aghios Ioannis mining area includes Nissi area deposits (located SSW of the Aghios Ioannis deposit) (
Figure 1; [
2]). The laterite ores at Aghios Ioannis and Nissi area deposits have been eroded, transported, and deposited onto karstified Triassic–Jurassic limestones and are conformably overlain by Upper Cretaceous limestones (
Figure 1). The Nissi area deposits are comprised of a series of lenses, that may occur either as isolated typical Fe–Ni–laterite or bauxite laterite ores or as an association of Fe–Ni ore at the lowest part of the deposit, followed by bauxite laterite towards its upper part ([
3] and references therein)—such as the Patitira deposit (located at the eastern part of the Nissi area) (
Figure 1).
Geochemical characteristics of the Ni-laterite deposits in Lokris have been studied for major and trace elements, including rare earth elements and chemistry of minerals in those deposits [
1,
3,
4,
5,
6,
7,
8,
9]. At the Nissi deposits, a strong enrichment of light rare-earth elements (LREE) relative to heavy rare-earth elements (HREE), due to the presence of the authigenic hydroxylbastnaesite-(Nd) minerals, has already been described [
8,
10]. Nickel-bearing minerals, such as takovite, Mn–Co–Ni asbolane, hydrated halloysite and an unknown Al–Ni–silicate from the Aghios Ioannis deposit have been described [
1,
11,
12,
13,
14].
Present study focuses on two vertical profiles from the Nissi (Patitira) bauxite laterite deposit, in Lokris area, characterized by the presence of goethite microtextures resembling a bacteriomorphic mineral (Profile I) and significant REE enrichment (Profile II). Geochemical variations and a detailed description of the REE-minerals and associated minerals are given in an attempt to define the factors controlling the compositional variation in REE minerals and the REE concentration process.
3. Methods of Investigation
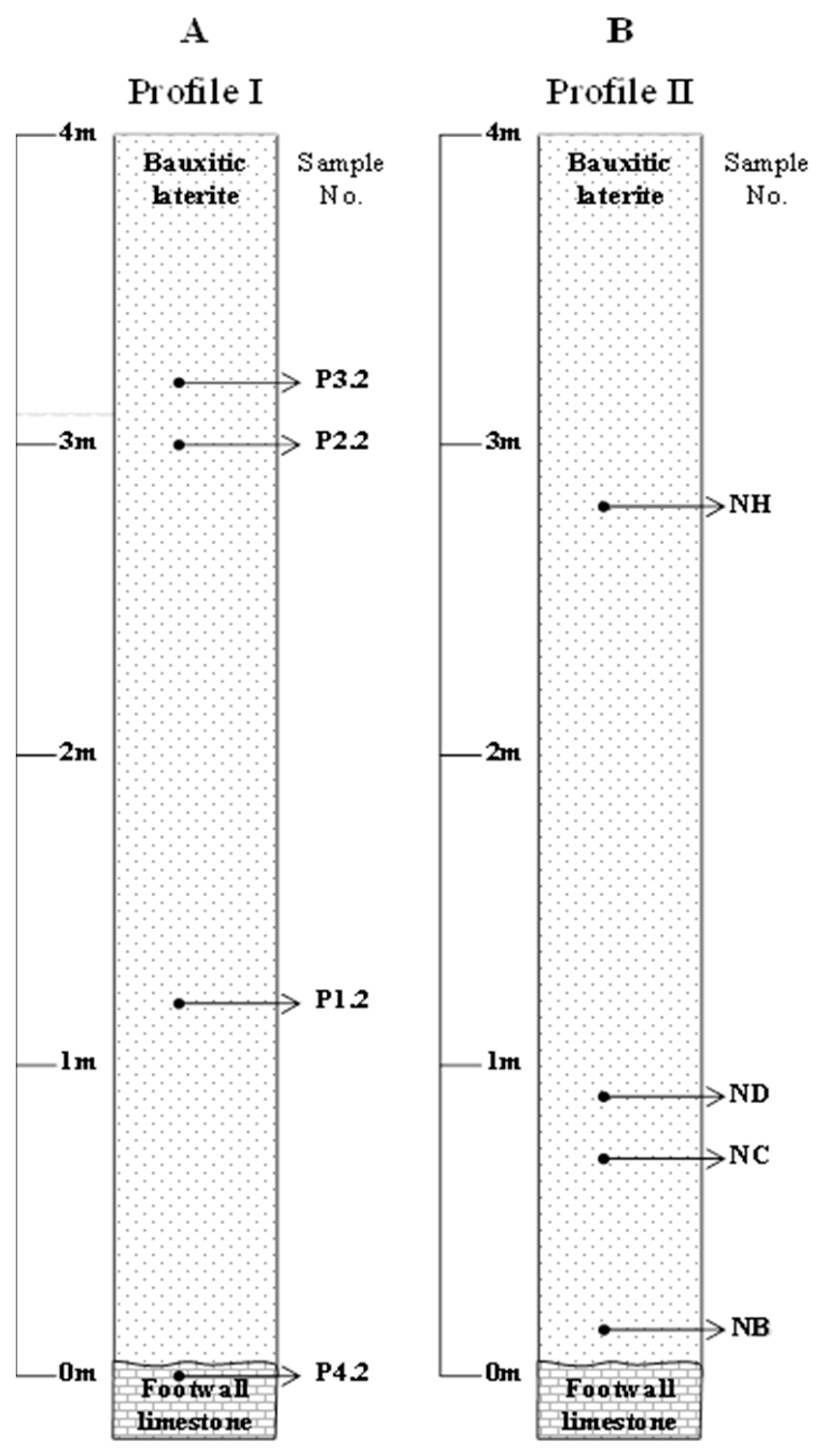
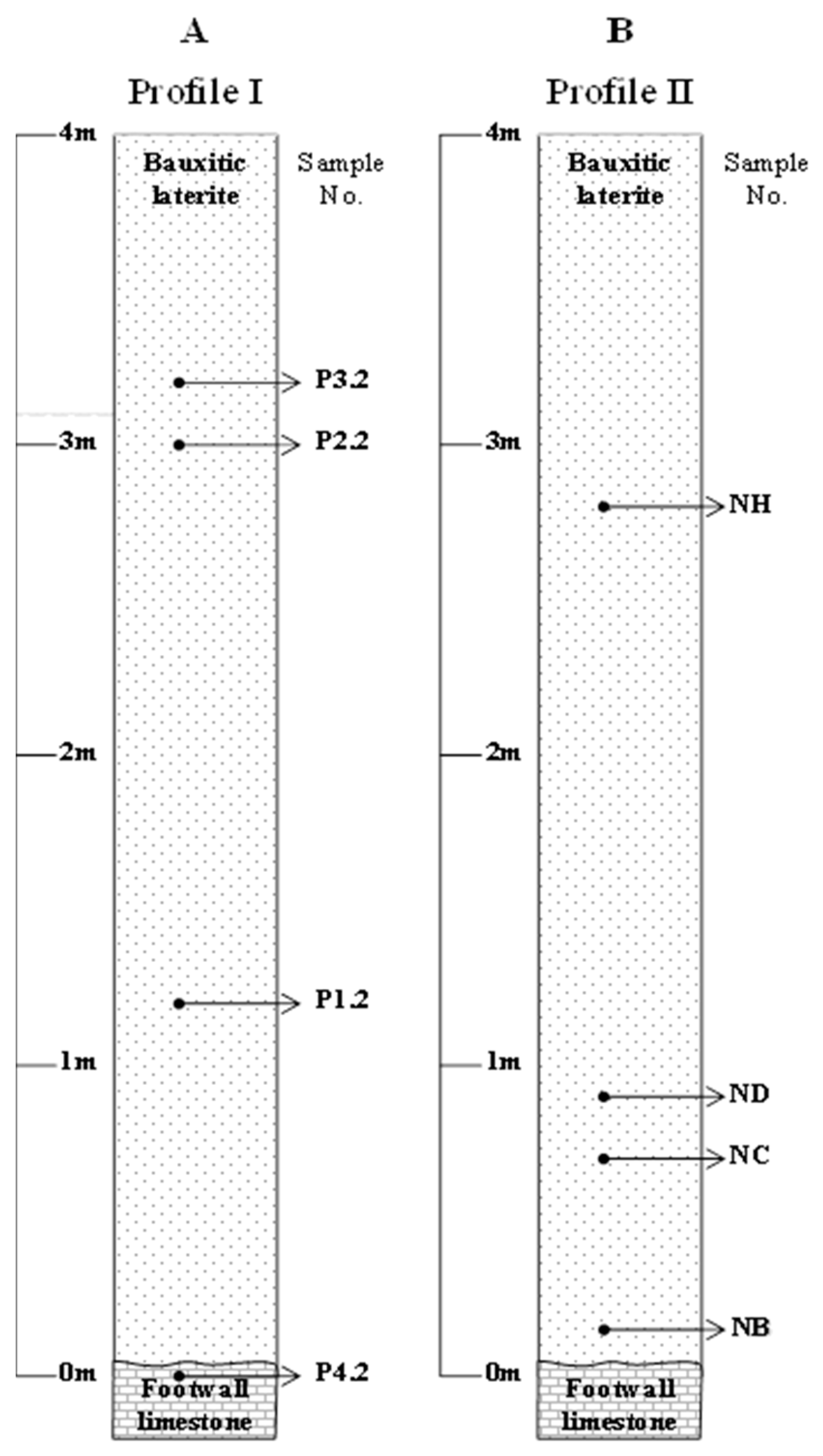
In order to identify geochemical trends from the overlying limestone to the footwall limestone, sampling along two relatively small vertical laterite profiles (Profile I and Profile II), covering the exposed area of the Patitira deposit (
Figure 1 and
Figure 2), was carried out. Eight samples weighing about 3 kg each were selected (out of 20 collected) as representative ores. Their stratigraphic position and description are given in
Table 1. Polished sections of all samples were examined using reflected light microscope, scanning electron microscope (SEM), equipped with energy dispersive spectroscopy (EDS). The SEM-EDS analyses were carried out at the Faculty of Geology and Geoenvironment, National and Kapodistrian University of Athens (NKUA), using a JEOL JSM 5600 scanning electron microscope (Tokyo, Japan), equipped with ISIS 300 OXFORD automated energy dispersive analysis system (Oxford shire, UK). Analytical conditions were 20 kV accelerating voltage, 0.5 nA beam current, <2 μm beam diameter and 50 s count times. The following X-ray lines were used: AsLa, FeKa, NiKa, CoKa, CuKa, CrKa, AlKa, TiKa, CaKa, SiKa, MnKa, MgKa, ClKa, LaLa, CeLa, PrLa, NdLa, GdLa, and YLa. Pure metals standards were used for Cu, Ni, Co and Cr, and pyrite for S and Fe. Standard indium arsenide and 300 s counting time were used for As and the presence of arsenic was confirmed by the X-ray spectrum. Careful calibration for REE was carried out using LaB
6 for La, CeO
2 for Ce, PrF
3 for Pr, NdF
3 for Nd, GdF
3 for Gd and Y. In addition to the above carbon coated samples, a few polished sections showing unpolished parts, revealing the presence of goethite micro-textures resembling bacterial cell were coated by gold and analyzed, in order to identify traces of carbon and other components of organic material.
Major and trace elements in laterite samples were determined by ICP–MS analysis, at the XRAL Laboratories Ltd., Ancaster, ON, Canada. Detection limits are 0.1 ppm for Ni and Co and all analyzed rare earth elements, 0.5 ppm for As, 1 ppm for Cr and Mn, 0.01 wt % for Fe, Ca, Mg, and Al, 0.05 wt % for S, and 0.001 wt % for Ti. According to the results for the quality control samples, the precision of the analyses of the minor and trace elements is in good agreement with international standards.
The samples were dissolved using a strong multi-acid (HNO
3–HClO
4–HF) digestion and the residues dissolved in concentrated HCl. The moisture content was determined by drying the samples at 105 °C and the organic matter content was determined by igniting the oven-dried samples (from moisture content determination) in a muffle furnace at 440 °C for 3 h [
19] and calculating the weight difference as a percentage (NKUA).
X-ray powder diffraction (XRD) data were obtained using a Siemens Model 5005 X-ray diffractometer (Bruker AXS GmbH., Karlsruhe, Germany), Cu Ka radiation at 40 kV, 40 nA, 0.020° step size and 1.0 s. step time. The XRD patterns were evaluated using the EVA 10.0 program of the Siemens DIFFRAC (Bruker AXS GmbH.) and the D5005 software package (NKUA).
Raman spectra were recorded with a Renishaw RM1000® Raman micro-spectrometer (Renishaw, UK) at the School of Mining and Metallurgical Engineering of the Technical University of Athens, Greece. Spectra were excited at room temperature with the 632.8 nm line of a red 19 mW He–Ne laser through an OLYMPUS® ×100 objective (Olympus Co., Tokyo, Japan). Each unpolarized spectrum represents the accumulation of three acquisitions of 20 s each. The spectra were collected at a constant laboratory temperature (20 °C) with a Peltier-cooled charge coupled device (CCD) detector, and the positions of the Raman bands were controlled and eventually corrected by regularly measuring the position of the Rayleigh line. The spectral resolution was about 0.4 cm−1. The numerical aperture of the objective was 0.9. The laser spot on the surface had a diameter of approximately 1 μm and a power of 4 mW. The entrance slit into the spectrometer was set to 40 µm. Light was dispersed by a holographic grating with 1800 grooves/mm. No baseline correction was performed to the spectra. Spectra were treated with the GRAMS32 software (Version 7.0, Galactic Industries Corp., Salem, NH, USA).
5. Mineralogical Characteristics
Samples collected from the Patitira Profile I (
Figure 2A;
Table 1) are mostly composed of goethite, limonite, hematite, chromite, boehmite and gibbsite, calcite, Fe-chlorite (chamosite), Ni-bearing chlorite, Ti-oxides and quartz. Angular and/or rounded fragments of goethite often martitized, contain detrital grains of quartz, chromite and chlorite (
Figure 5A,B;
Table 4). Gibbsite, illite, kaolinite, montmorillonite and Mn–Co–Ni asbolane are abundant towards fractures and the lowest parts of the deposit. The chromite grains or fragments are residual components inherited from the ophiolitic parent rocks of a wide compositional variation, including high-Cr and high-Al types. The most salient feature is the presence of goethite in small sizes resembling bacterial cell coated by goethite (
Figure 5C–F;
Table 4) ([
16] and references therein). In addition, the SEM-EDS study of gold-coated samples (
Figure 5F) showed the presence of C, N, Fe, Al, K, Si, Ca, Mn and Ni. Although quantitative analyses were not obtained, due to unpolished surface and the small size of the analyzed material, the presence of the above components may reflect the presence of microorganisms.
Samples labeled as NC and ND, upward in the profile II (
Figure 2B;
Table 1) consist mainly of goethite, hematite, chlorite, illite, quartz, calcite, chromite, kaolinite, boehmite, and Ti-oxides, whereas the sample labeled as NH consisting mainly of boehmite, gibbsite, goethite, chlorite, quartz, kaolinite, chromite, hematite and Ti-oxides. There is an abundance of gibbsite, illite, kaolinite, montmorillonite, smectite, takovite, Mn–Co–Ni asbolane, halloysite and Al–Ni–Co–Mn silicates towards fractures and the lowest parts of the deposit (
Figure 2B).
The XRD pattern (
Figure 6) shows the presence of gibbsite, halloysite, lizardite, bastnaesite, goethite, takovite and six reflections recorded at 11.8 Å, 5.90 Å, 3.94 Å, 2.575 Å, 2.443 Å, and 1.503 Å, matching the reflections of an “unknown Al–Ni–silicate mineral” already described by Maksimovic [
11,
12]. It is notable that this “unknown Al–Ni–silicate mineral” has been described in the localities of Ba and Takovo in Serbia in association with takovite, gibbsite and halloysite and in the karstic nickel deposit of Aghios Ioannis in Lokris area with hydrated halloysite [
11,
12].
Besides, the XRD pattern shows reflections assigned to any of the following minerals: bastnaesite–La [La(CO
3)F], bastnaesite–Ce [CeCO
3F] and bastnaesite–(Ce,La) [(Ce,La)CO
3F)], in consistency with the relevant data base of the used evaluation software program (EVA 10.0 diffrac plus). Combined XRD and SEM/EDS data (
Figure 6 and
Figure 7;
Table 5 and
Table 6) indicated the dominance of a (La,Nd,Y)(CO
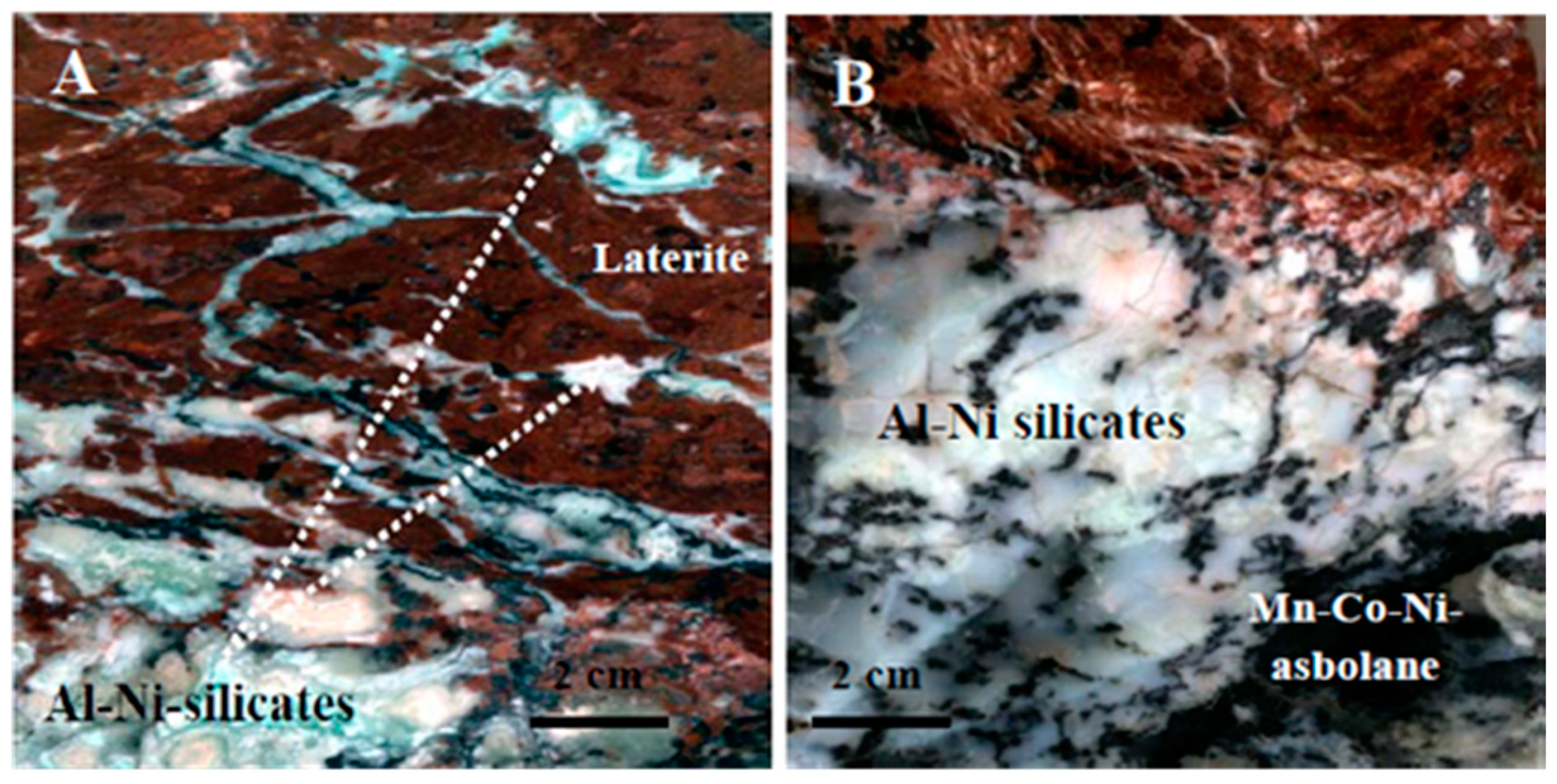
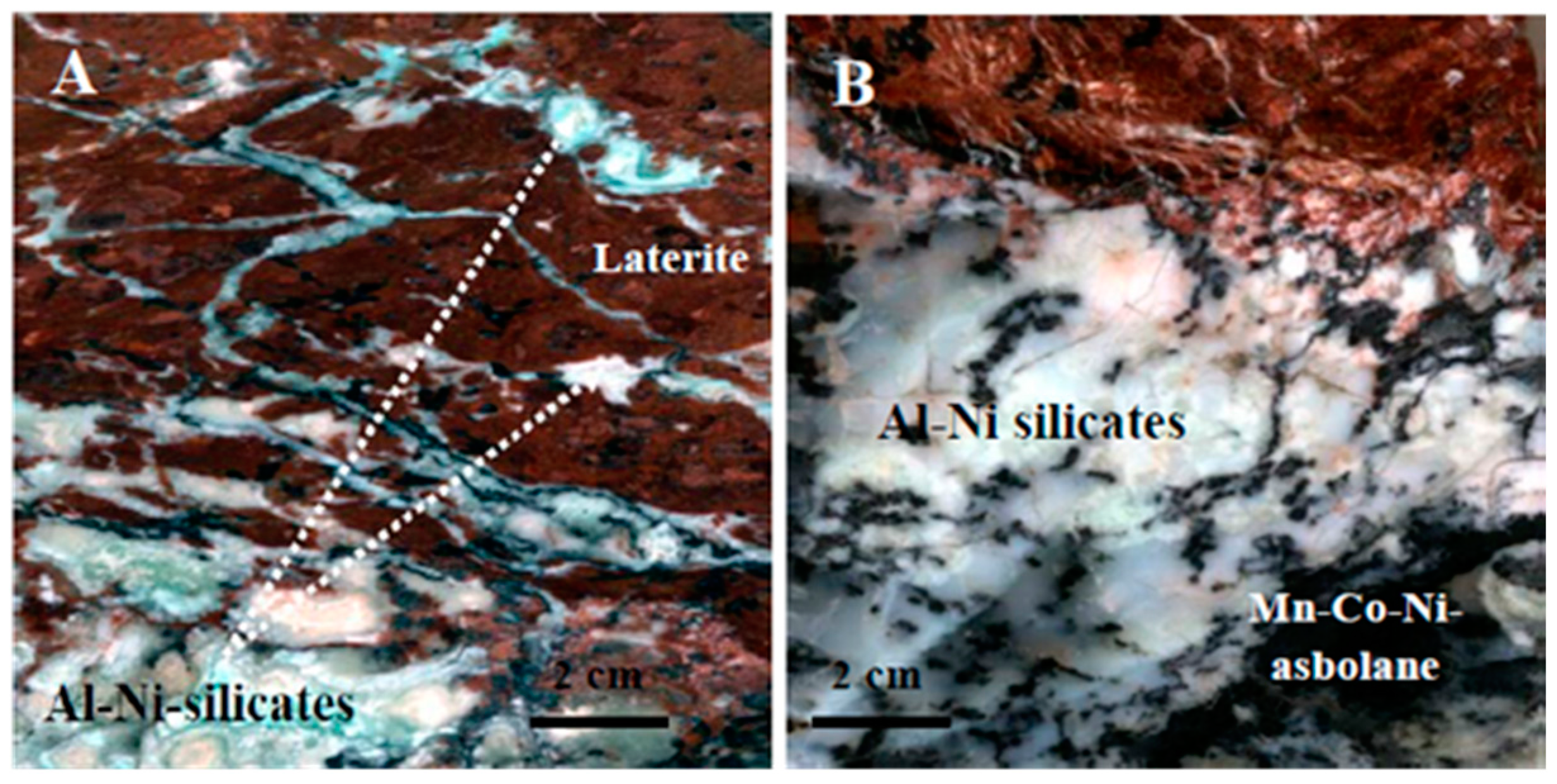
3)F member of the bastnaesite group. More specifically, bastnaesite occurs mostly in a closed association with intergrowths between takovite and an unknown Al–Ni–silicate mineral (
Figure 6 and
Figure 7A–E,
Table 5 and
Table 7) or as rounded crystals at the margins of calcite (
Figure 7A–C,F). The Al–Ni–silicates display commonly acicular or needle-shaped morphology, ranging in length from a few tens of nm to several microns. Intergrowths of takovite–Al–Ni–silicate are commonly found as successive thin (a few to tens of μm) layers, composed of fine fibrous crystals of nm to a few μm (
Figure 7D,E).
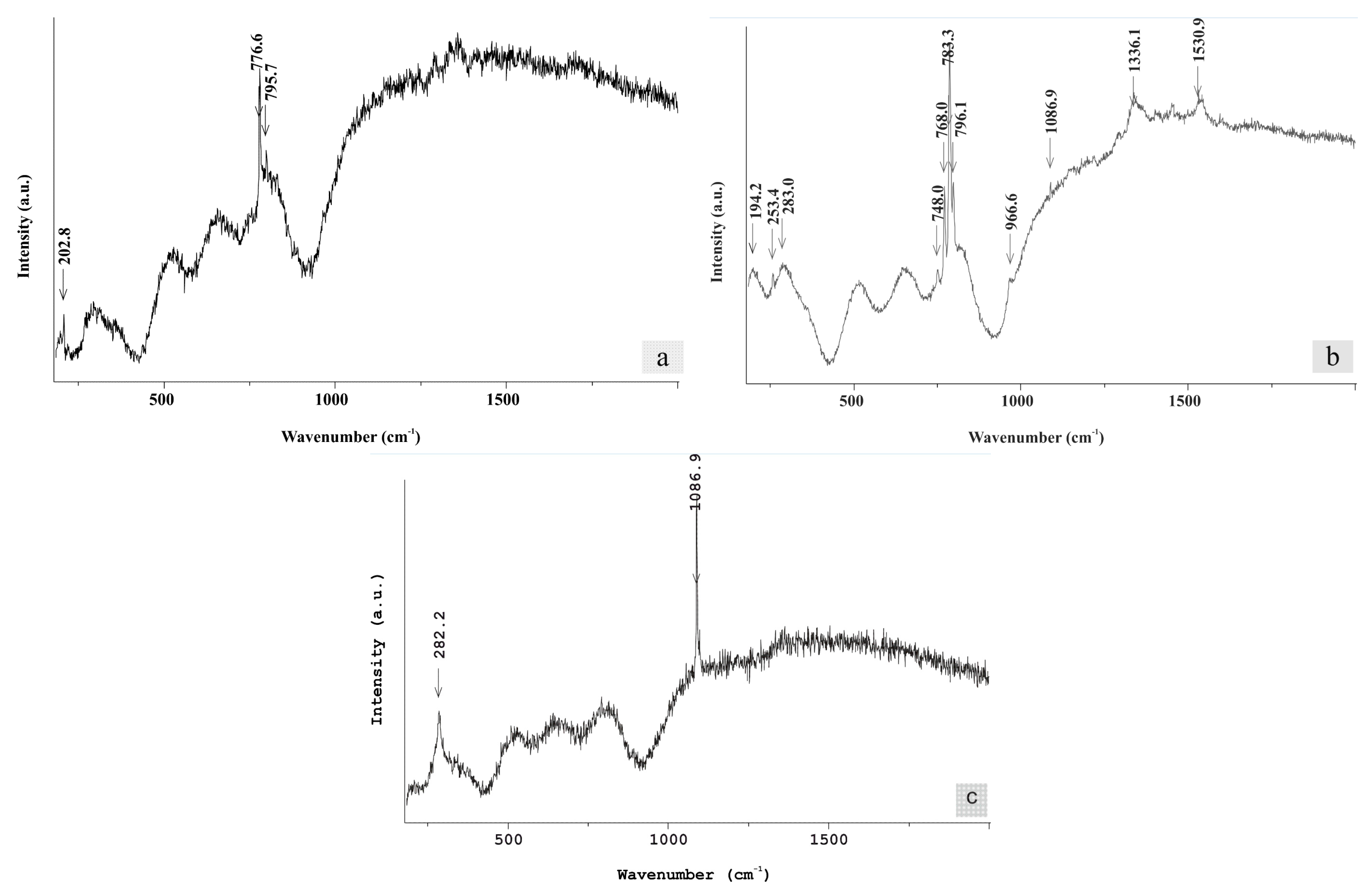
Raman spectroscopy was additionally employed to investigate the REE-minerals from the studied profile. Representative Raman spectra of the REE-carbonates, studied herein, are given in
Figure 8. They refer to the bastnaesite group minerals and exhibit three distinguished Raman features: (
Figure 8a) Raman spectra with dominant bands at around 795, 776 and 202 cm
−1 and minor ones at around 1350 and 1290 cm
−1 (e.g., spectrum nissi 1_8); (
Figure 8b) Raman spectra characterized by intense Raman bands at around 1086, 796, 783 and 768 cm
−1 and minor ones at around 1530, 1336, 965, 748, 283, 253 and 195 cm
−1 (e.g., spectrum nissi 1_1); and (
Figure 8c) Raman spectra with main Raman bands arising at around 1086, 712 and 281 cm
−1 (e.g., spectrum nissi 1_12).
The Raman spectrum of bastnaesite/hydroxylbastnaesite has been reported to exhibit significant variations based on its chemical composition. A single intense band due to the symmetric stretching vibration (ν1) of the carbonate groups appears at 1085 cm
−1 for Canada bastnaesite-(Ce,La), at 1095 cm
−1 for Pakistan bastnaesite-(Ce,La), at 1096 cm
−1 for Norway bastnaesite-(Y,La) and at 965 cm
−1 for Malawi hydroxylbastnaesite-(Ce,La) [
21]; the latter remains controversial as it has been attributed either to carbonate bending mode [
22] or to the OH deformation mode of hydroxylbastnaesite [
21] and it is absent in the Raman spectra of any other halogenated carbonate. The variation of the ν1 band position is a function of the chemical composition of the mineral. Fundamentally, the higher the ionic radius the lower the wavenumber of the symmetric stretching mode. Raman modes around the 700 cm
−1 region are due to the in-plane bending mode (ν4). As the carbonate groups become distorted from regular planar symmetry, this mode splits into two components. In bastnaesite, the variation in the position of the ν4 band, usually observed at 729 cm
−1, reflects its composition. Two bands are observed for hydroxylbastnaesite at 602 and 569 cm
−1 probably ascribed to the ν4 mode [
23].
Taking into consideration the afore-mentioned studies, we could come to the following band assignments:
The shifts of the Raman band observed in this study compared to the published ones are considered to relate to their chemical composition, i.e., to the presence of Nd instead of Ce in the chemical composition of the carbonates studied herein, as well as to the presence of a considerable amount of Ca.
6. Discussion
In a global scale, the large REE deposits are associated with carbonatites and alkaline-peralkaline igneous rocks and hydrothermally altered silicate rocks, including pegmatites; on the contrary, REE concentrations by metamorphic or diagenetic processes are low ([
6,
14,
24,
25,
26] and references therein). Fluorocarbonates, phosphates, silicates and oxides occur as disseminated and very fine-grained REE-minerals hosted by peralkaline felsic volcanic/volcaniclastic rocks. The bastnaesite-group minerals are among the most economically important ones, whereas their origin is related to late stages of magma evolution [
26].
Fluorocarbonates in the Nissi (Patitira) bauxite laterite deposit, Lokris, are related with the lateritic weathering, which is an important process in the REE-enrichment in laterites [
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
24]. The mineralogy of the weathered rocks and the physico/chemical conditions (temperature, acidity, redox potential) are among major controlling factors of the composition of the aqueous phase, which in turn can control the degree to which REEs are released to the downward moving fluids in contrast to iron that is quite reactive [
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
24,
25,
26]. Multistage mineral phase deposition and transformations in the Lokris karst-type laterites is apparent (
Figure 4; [
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16]). The epigenetic mobilization of Fe, Mn, Ni, Co and rare earth elements (REE) under reducing (due to the decomposition of organic material) and acidic (due to the decomposition of pyrite) conditions and subsequently re-deposition under alkaline conditions, during a diagenetic-post diagenetic stage at the Nissi (Lokris) laterite deposits, has been well established [
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
27].
Experimental data on the Fe-(oxyhydr)oxides hematite, goethite, lepidocrocite and ferrihydrite suggest that Al-substitution in these materials may influence particle stability and phase transformation behavior, typically stabilizing the oxyhydroxide phases relative to the oxide one (hematite). Lower Al-substitution energies for the oxyhydroxides goethite, lepidocrocite and ferrihydrite indicate that Al-substitution in these minerals might be more readily accomplished in a mixed-Al/Fe precipitation environment than for the oxide, hematite [
28]. The presence of significant organic matter and Fe–Al–(oxyhydr)oxides may suggest that subsequent to the deposition of varying forms of pyrite [
16], the deposition of goethite and Al-hydroxides, co-existing and/or cross-cutting earlier goethite and matrix, took place. Besides, goethite replacing pyrite (
Figure 5B) and exhibiting elevated contents of aluminum and arsenic (
Table 4), may be related with sorption and replacement processes, due to the metastable oxyhydroxide ferrihydrite with respect to goethite, since nanoparticulate under typical surface conditions, has high specific surface area and significant reactivity toward the sorption of aqueous contaminants (AsO
43−, SO
42−, among others [
29]).In addition, the negative Gibbs free energy (ΔG°) values for arsenite and arsenate sorption on Fe-rich soil concretions are consistent with spontaneous reaction between the species and the medium [
29].
There is a debate on whether the mineral is of direct organic or inorganic origin [
30,
31,
32,
33,
34,
35,
36,
37,
38,
39]. The relatively high content of organic matter (
Table 2 and
Table 3), the SEM images, resembling bacteriomorphic goethite (
Figure 5) coupled with the negative δ
34S values for sulfide-bearing samples from the Nissi area [
16] may suggest the existence of the appropriate conditions for element bio-leaching and bio-mineralization. However, the detection of molecular and elemental bio-signatures regarding bacteriomorphic goethite requires furthermore research. Employing additional techniques, such as a mass spectrometry (MS, based on laser or ion-beams), in microscale and/or in nanoscale, is amongst our interests for future research.
Bastnaesite series can be classified into four types based on the predominant rare earth element [
40,
41]: bastnaesite-(Ce), [(Ce,La)CO
3,F], bastnaesite-(La), [(La,Ce)CO
3,F], bastnaesite-(Y), [(Y,Ce)CO
3,F] and bastnaesite-(Nd). The most frequent REE minerals in karstic deposits are members of the bastnaesite group, including: Synchysite–(Nd), bastnaesite–(Ce) and bastnaesite–(Nd), hydroxylbastnaesite–(Nd,La) and hydroxylcarbonate–(Nd,La). The occurrence of REE-phosphates and alkaline earth phosphates, such as monazite–(Nd), monazite–(La) and neodymian goyasite is relatively rare [
14,
27]. The REE minerals as identified by previous authors at the Nissi laterite deposits have been described within black Mn-oxide assemblages and are dominated by bastnaesite, hydroxylcarbonate–(Nd,La) and hydroxylbastnaesite–(Nd,La) [
7,
10].
In addition to REE-minerals described by above authors, a predominance of La (average 23 wt % La
2O
3) over Ce (0–2 wt % CeO
2) and the presence of Pr (average 5 wt % Pr
2O
3), Nd (average 23 wt% Nd
2O
3), Y (3–6 wt % Y
2O
3) and F (4–8 wt % F) in REE-minerals presented (
Figure 7;
Table 6) suggests that the investigated bastnaesites differ compared to those described by previous authors in terms of:
- (a)
Their chemical composition compared to the hydroxylbastnaesite described at the Nissi deposits. Ideal bastnaesite simplified and calculated as LaCO
3F [
42] contains ~8% F. Since
Table 6 presents two data with >5% F these are not hydroxyl bastnaesite, but normal (fluorine) bastnaesite, although analysis in column 2 could be the OH variant.
- (b)
Their association with margins of calcite grains (
Figure 7F) and its intergrowths with Al–Ni–silicates (
Figure 7B–E).
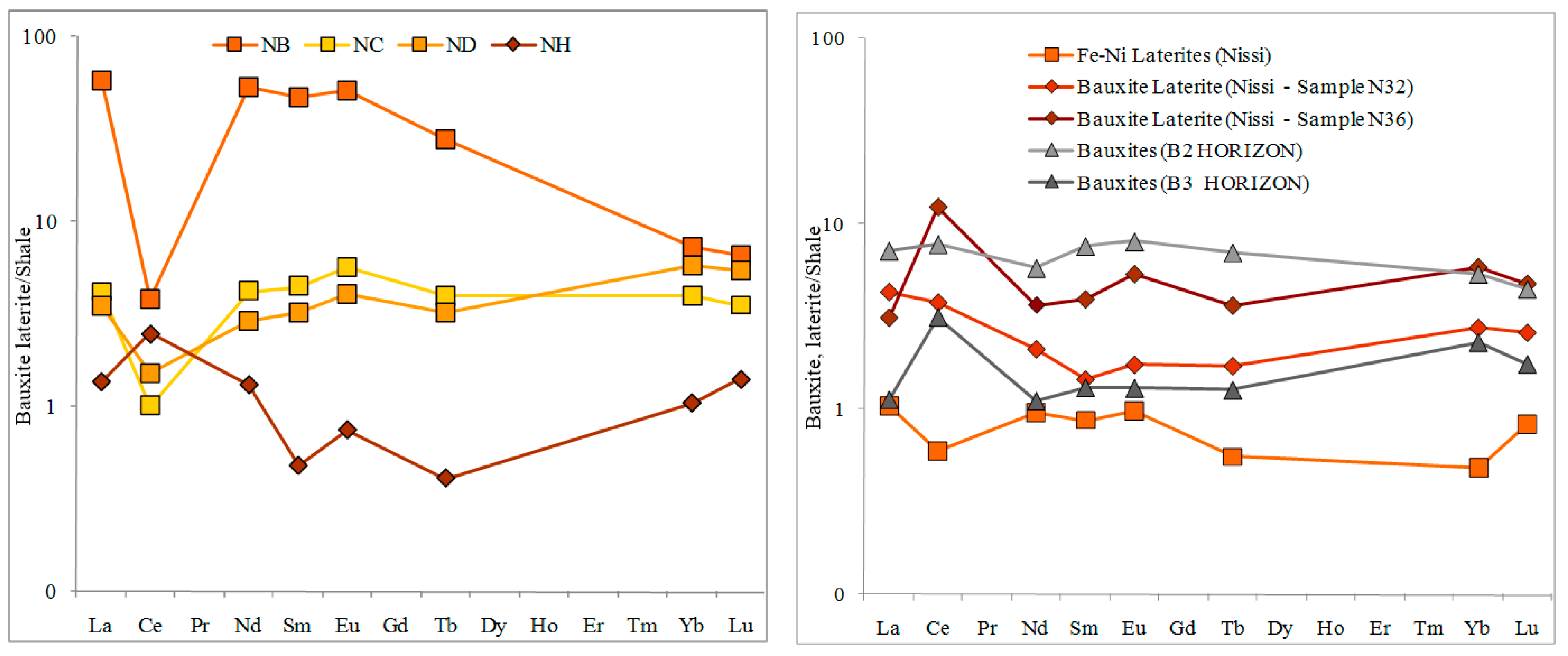
The latter texture relationships, combined with the major negative Ce-anomaly in the corresponding REE-patterns (
Figure 4a), may indicate a late stage deposition of the investigated bastnaesites under reducing alkaline conditions. In contrast the Ce-positive anomaly in the uppermost part of the profile II (
Figure 2 and
Figure 4a) may reflect the oxidation of Ce
3+ to Ce
4+ and its incorporation in other Ce-rich minerals.
According to the literature, bastnaesite (CeFCO
3), synchysite (CeFCO
3·CaCO
3), parisite (2CeFCO
3·CaCO
3), and roentgenite (3CeFCO
3·2CaCO
3) belong to the REE fluorocarbonate group known as the bastnaesite series ([
43] and references therein). The bastnaesite mineral group presents two common features: (i) the structure can be broadly described as the stacking of three types of layers (CeF, CO
3 and Ca) along the
c-axis; and (ii) the Ce/F ratio in all the phases is 1, indicating that this feature is common in all members of the group. Thus, these minerals differ in the orientation of carbonate ion within the carbonate layers, the order of staking of the different type of layers and the length of the crystallographic
c-axis. Commonly, fluorocarbonates are polycrystals with syntaxial intergrowth of two species in contact along an irregular surface or along repeated parallel planes (0001). The
a-axes of the two species may have the same or opposite directions. In the latter case, the two species are related to each other as two individual twins with a rotation of 60° around the
c ternary axis. All pairs have been observed, except the bastnaesite-synchysite pair [
43].
The syntactic intergrowths can be described as mixtures of bastnaesite and synchysite. Manfredi et al. [
43] considered bastnaesite-(Ce) and synchysite-(Ce) as two end-members and that parisite and roentgenite are ordered mixtures of bastnaesite (B) and synchysite (S) in single layers stacked along the
c crystallographic axis direction. Parisite can be considered a BS stacking and roentgenite a BS2 stacking [
43] and references therein.
Minerals of this series are often affected by stacking faults, disorder of chemical composition, superstructures, twinning, polytypism and syntactic intergrowths during the course of crystallization and crystal growth along with other mineral phases, making it difficult to obtain crystallographic data on the one structures of the minerals [
44].
Additionally, crystal structures of these minerals tend to have a layered topology. The planar carbonate groups are inclined (standing on-edge) with respect to the overall structural layering. The addition of another anion, F
−, can influence this topology. The development of a structural hierarchy of fluoride carbonates requires consideration of ratios of these two anions and their attached cationic polyhedral groups [
45].
According to Grice et al. [
45], as a consequence of the presence of similar rare earth fluoride carbonate layers, twinning is frequently observed, together with syntactic intergrowth. All associations of the above minerals exist and were early recognized from morphological measurements. These features also explain the very long resistance of these minerals toward accurate structure determinations [
45].
In present study, the XRD pattern shows reflections assigned to bastnaesite. The observed high contents of Ca (CaO from 6.37 wt % to 8.04 wt %,
Table 6) in REE-minerals presented indicate that they are not pure bastnaesites and that probably is a case of syntaxic intergrowth of parisite with bastnaesite, synchysite or roentgenite, which is typical for these minerals.
The poor quality exhibiting by the powder X-ray diffraction pattern concerning the fluorocarbonate minerals (
Figure 6), is probably due to:
- (a)
lattice deformation or disordering as a result of substitutions of Ce or La by Nd and of Ce by Y;
- (b)
poor crystallization of the mineral;
- (c)
very small size of the mineral crystallites; and
- (d)
syntaxic intergrowth that may occur.
For the clarification of the minerals and structural features of the studied phases, additional techniques (such as high-resolution transmission electron microscopy (HRTEM)), should be conducted and this subject is amongst our interests for further research.
On the Origin of REE Fluorocarbonate Minerals
Parent rocks of the Ni–Fe deposits in the Lokris area, are primarily harzburgites, which represent the erosional outliers of a probable “complete” ophiolitic nappe that were transformed to serpentinites. Within the serpentine bodies of the basal tectonic mélange, xenoliths of basic and sedimentary rocks are included, while a continuous transition from karstic Ni-Fe deposits towards bauxite material in a southern direction has been reported [
4,
6,
7].
Although the REE content in ultramafic rocks is usually very low, there is a significant REE content along with other metals such as Al, Fe, Mn, Ni contents in mafic rocks and serpentinites parent rocks [
6,
7] suggesting that the ophiolitic rocks cannot be precluded as a REE source. Recently, a review of the regularities of REE distribution in minerals contained in mafic and ultramafic rocks as accessory phases was performed [
46]. These minerals include garnets, zircons, apatites and perovskites, which can accumulate high concentrations of REE in their structure, as well as chrome spinels, ilmenites, and micas which can accumulate, only limited contents of these trace elements [
46]. In addition, the Europium in other samples (
Table 6) is not depleted relative to neighboring REE (Sm and Gd) in the bulk ores, suggesting an original mantle source for the REE, since crustal rocks are mostly Eu-negative. In addition, the apparent Yttrium enrichment (
Table 6) is consistent with the USGS model of origin from ultramafic rocks [
47] including along contacts between ultramafic rocks and carbonate rocks.
However, mineralogical characteristics, in particular the occurrence of different morphological forms of goethite (
Figure 5) of varying composition (
Table 4), with inclusions of chromite (
Figure 5A) inhered from different sources [
1] point to a dynamic cycling of the parent material. The occurrence of the above rare earth minerals described herein and by previous authors, all in the lowermost part of laterite ores at their contact with the carbonate basement may suggest small differences in the conditions during their precipitation and/or variations in the REE source. In addition to the weathering process the studied profiles as allochthonous laterites lying on a carbonate basement, with well-established characteristics of a multistage re-mobilization and re-deposition [
3,
4,
5,
6,
13,
48] may have been affected by other processes.
The extensive presence of total organic matter (O.M.) at the contact of the Nissi (Patitira) bauxite laterites and the carbonate basement (
Table 2; [
16]) appears to create the appropriate conditions for iron and REE leaching and re-deposition [
20,
33,
49,
50,
51,
52,
53,
54,
55]. The preferential occurrence of REE-minerals in the lower transitional zone and within the underlain limestone (
Figure 7) itself suggests that their formation was controlled by physico-chemical conditions, such as an alkaline barrier, redox potential, solubility, supersaturation [
29,
56] rather than by the precursor rocks. The presence of rare earth minerals between calcite crystals (
Figure 7) probably indicates late stage deposition under reducing alkaline conditions.
The enrichment of the REE at the Nissi (Patitira) concentrated in bastnaesite-group minerals is thought to have been caused by long-term leaching and re-deposition on karstified limestone, during a subsequent stage under alkaline conditions [
10,
24,
29]. Although the dominant oxidation state of the REE in aqueous solution at 25 °C is the 3+ state, Ce behaves differently from the rest of the REE, due to the possibility of being oxidized from Ce
3+ to Ce
4+ in a relatively strong oxidizing environment [
8,
40,
57]. Therefore the positive cerium anomalies occurring in the upper parts of the Nissi profile, and the negative one at the lowest part of the deposit, in contrast to the rest of the REE transported downward, are probably due to the deference in the breakdown of REE-bearing minerals under similar physico/chemical conditions. With respect to the fluorine source, the highest fluorine contents (up to 1200 ppm) that have been recorded in mafic rocks [
58] and the presence of fluor-apatite in ophiolitic mafic rock [
59,
60], suggest a contribution of F-bearing minerals in laterites associated the weathering of such country rocks. In addition, the presence of fluor-hydroxyl-apatite in Fe-Ni-laterites of northern Greece, either as a clastic and/or authigenic mineral, coupled with its texture feature, showing gradual dissolution of apatite crystals [
60] may indicate that apatite dispersed throughout the upper parts of the Nissi (Patitira) laterite deposit was unstable during an epigenetic stage. Fluorine is slightly mobile, under natural soil conditions, but in acid soils its solubility increases [
58], following probably the mobilization of REE.
Since precipitation of REE-hydroxides markedly decreases from La to Lu, from pH 8.4 to 6.0, the REE plots showing a negative slope for samples at the footwall limestone (
Figure 4a) confirm the importance of the pH for the fractionation of REE in the sedimentary cycle. More specifically, in a karstic environment, where the water is weakly alkaline to alkaline, the argillaceous material collected in depressions will be enriched in light REE (LREE) whereas heavy REE (HREE) will mostly be removed by solutions, through different channels in karst [
61,
62]. The compositional variability of all authigenic REE minerals in karstic deposits has been well established [
14] and it is caused basically by two types of substitution: Nd and La, but more frequently Nd, can substitute for Ce, forming Nd- or La-dominant minerals, and OH ions can substitute for F in the structure of bastnaesite, forming hydroxylbastnaesites ([
14] and references therein).
Although the factors controlling REE-stability on molecular scale are still unclear, the stability of the described (La,Nd,Y)(CO
3)F member of the bastnaesite-group and the associated minerals in the Nissi (Patitira) deposit may point to the significant role of the redox and pH conditions during mobilization and re-precipitation processes caused Fe-leaching and residual Al-enrichment at the top of the deposit (
Figure 2B).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}