5.1. Comparison of Analytical Results
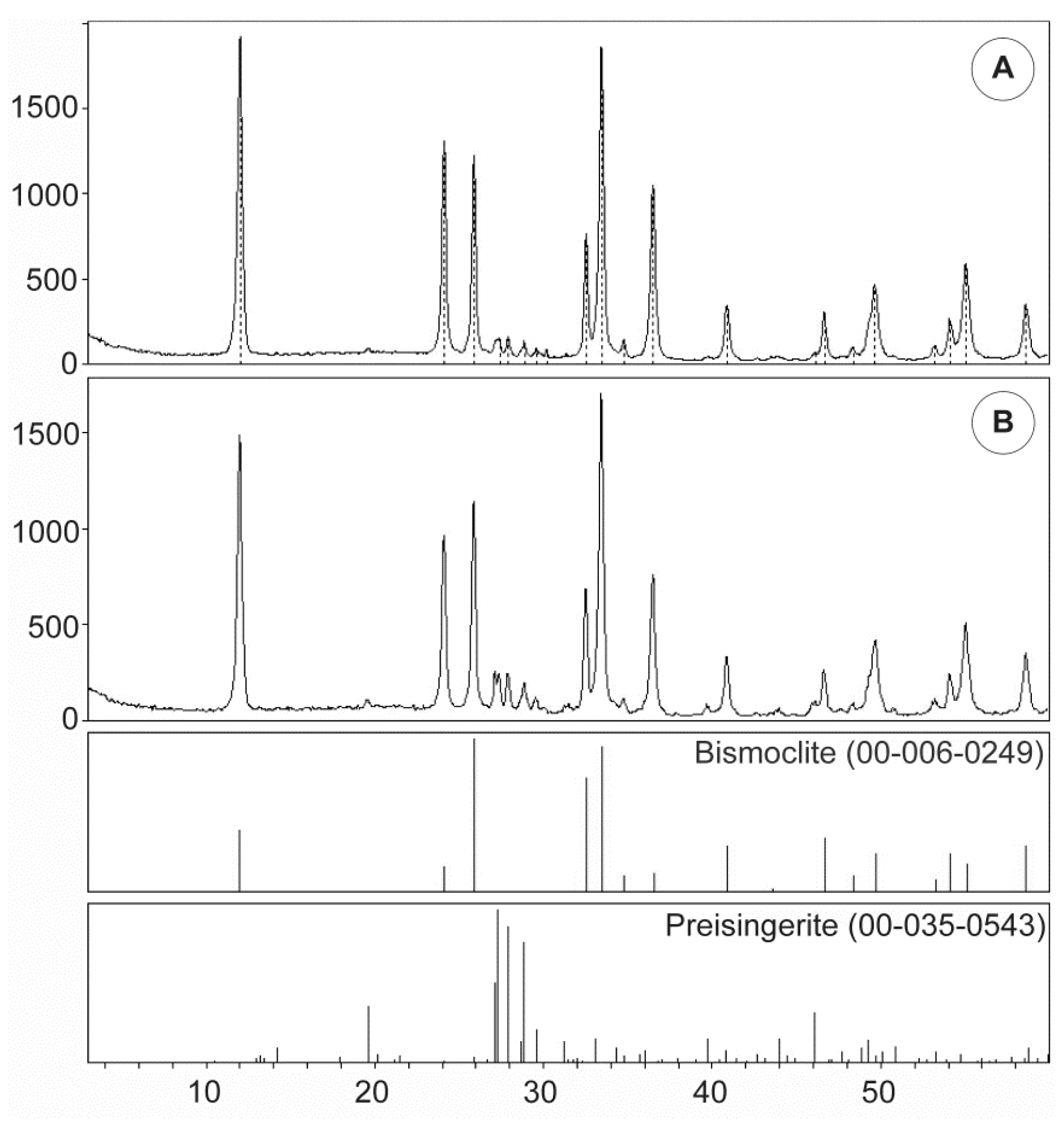
The crystal structure of bismoclite and other bismuth oxyhalides has only been studied by a few authors. Major contributions were made by Bannister and Hey (1934, 1935) [
50,
51] who carried out structural research by photographic methods. The bismuth oxychloride structure was described in detail by Keramidas et al. (1993) [
52], who synthesized bismoclite plates, selected a small single crystal, centered it on a single crystal diffractometer and then refined the cell constants by a least–squares procedure.
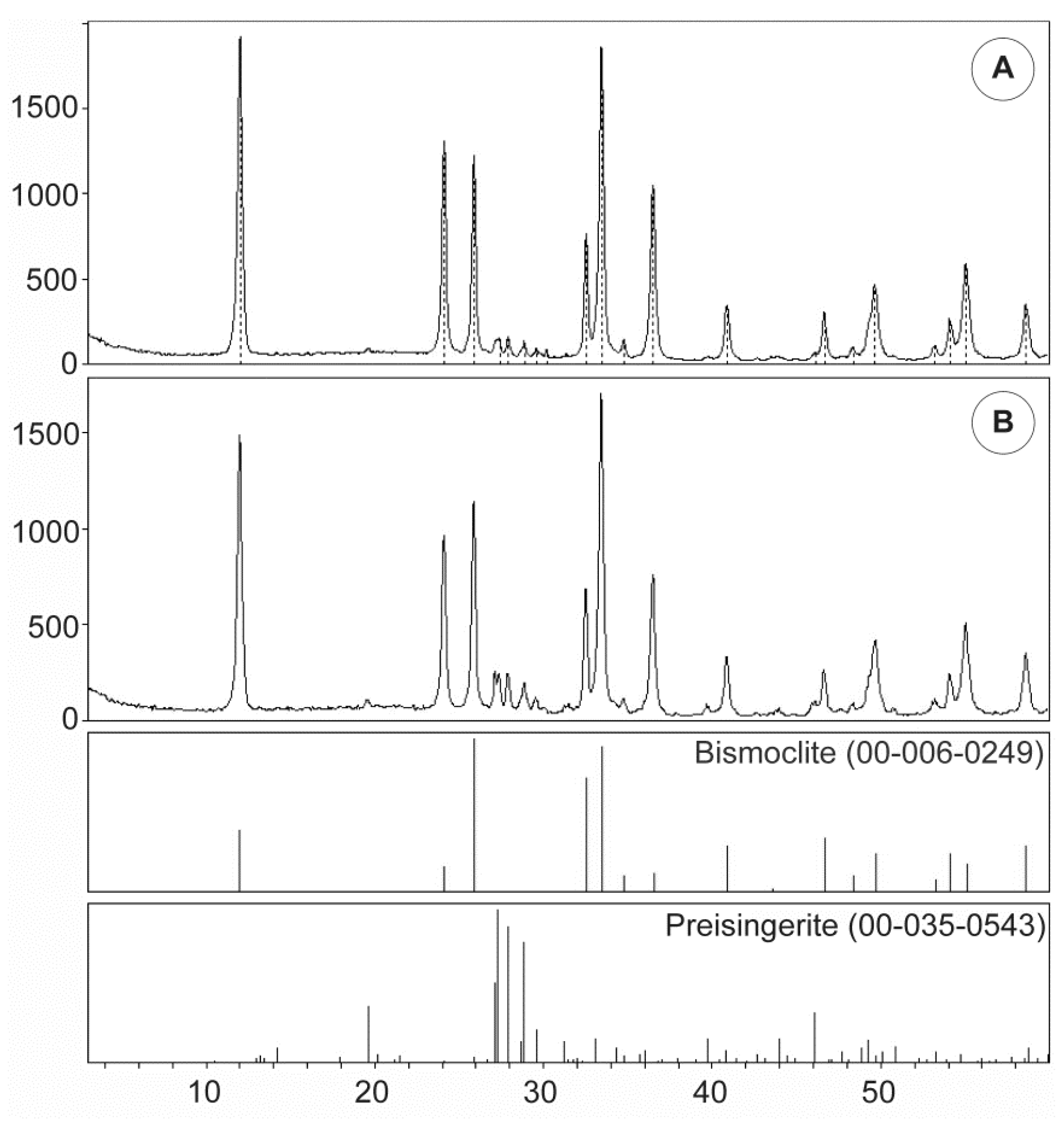
Table 6 summarizes the unit cell dimensions and crystal structure parameters for synthetic and naturally occurring bismoclite, and highlights the consistency between previous studies and our XRD results. Ketterer and Krämer (1986) [
53] documented and refined the structure of bismuth oxybromide, a compound analogous to bismuth oxychloride that is a member of the bismuth oxyhalide family.
Table 6 also lists the calculated crystallite size yielded for bismoclite from San Francisco de los Andes. As the 2θ angle increases, the instrumental error decreases, thus the crystallite size obtained for the (003) reflection is more reliable than for (001) and even (002). In addition to crystallite size, a diversity of factors can contribute to the diffraction peak widening. The most significant factors include inhomogeneous strain and instrumental effects [
54]. The Scherrer equation only provides a lower limit on the particle size, therefore a larger crystallite size than its actual dimensions may be obtain as a result of a non-zero contribution.
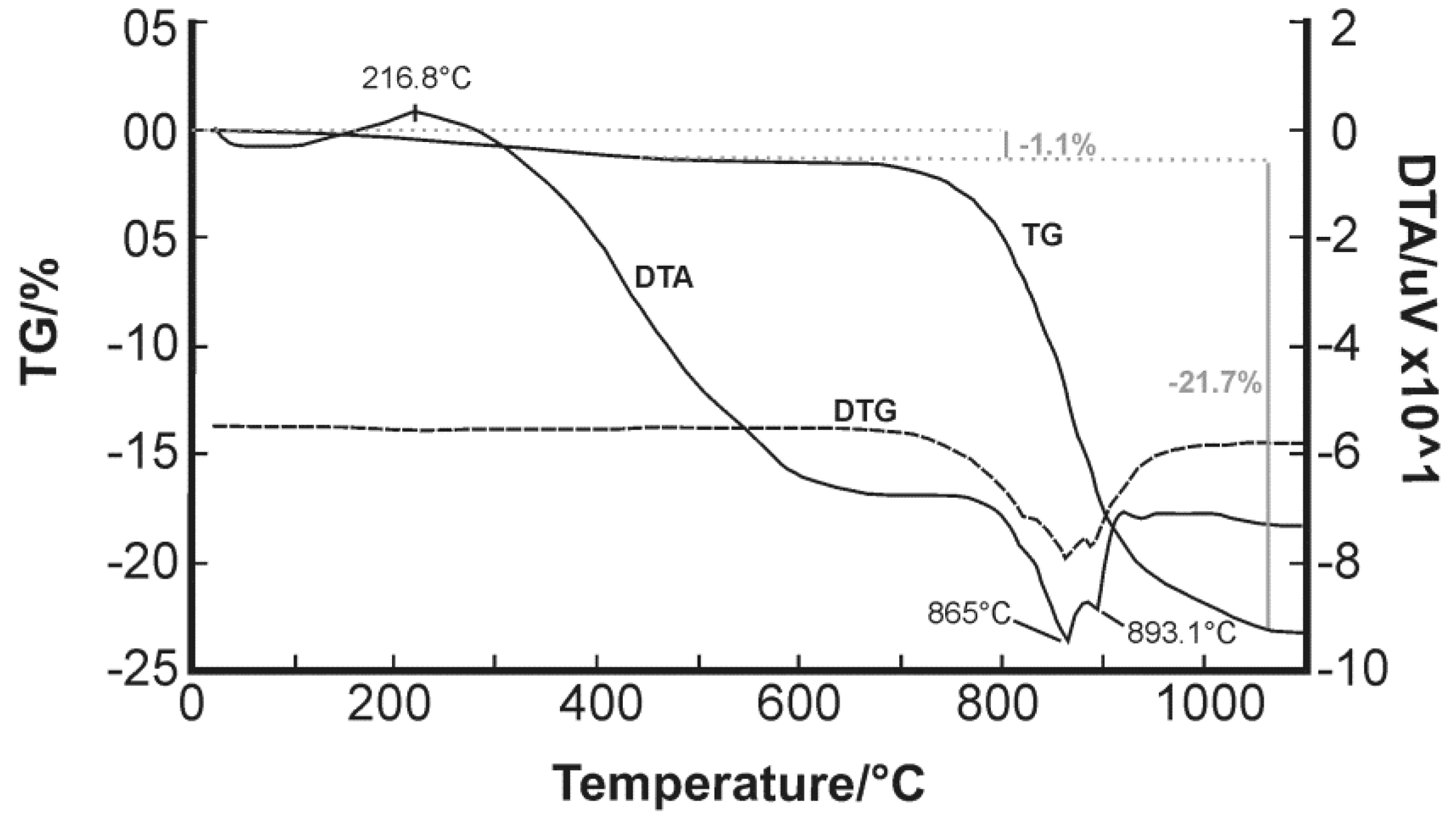
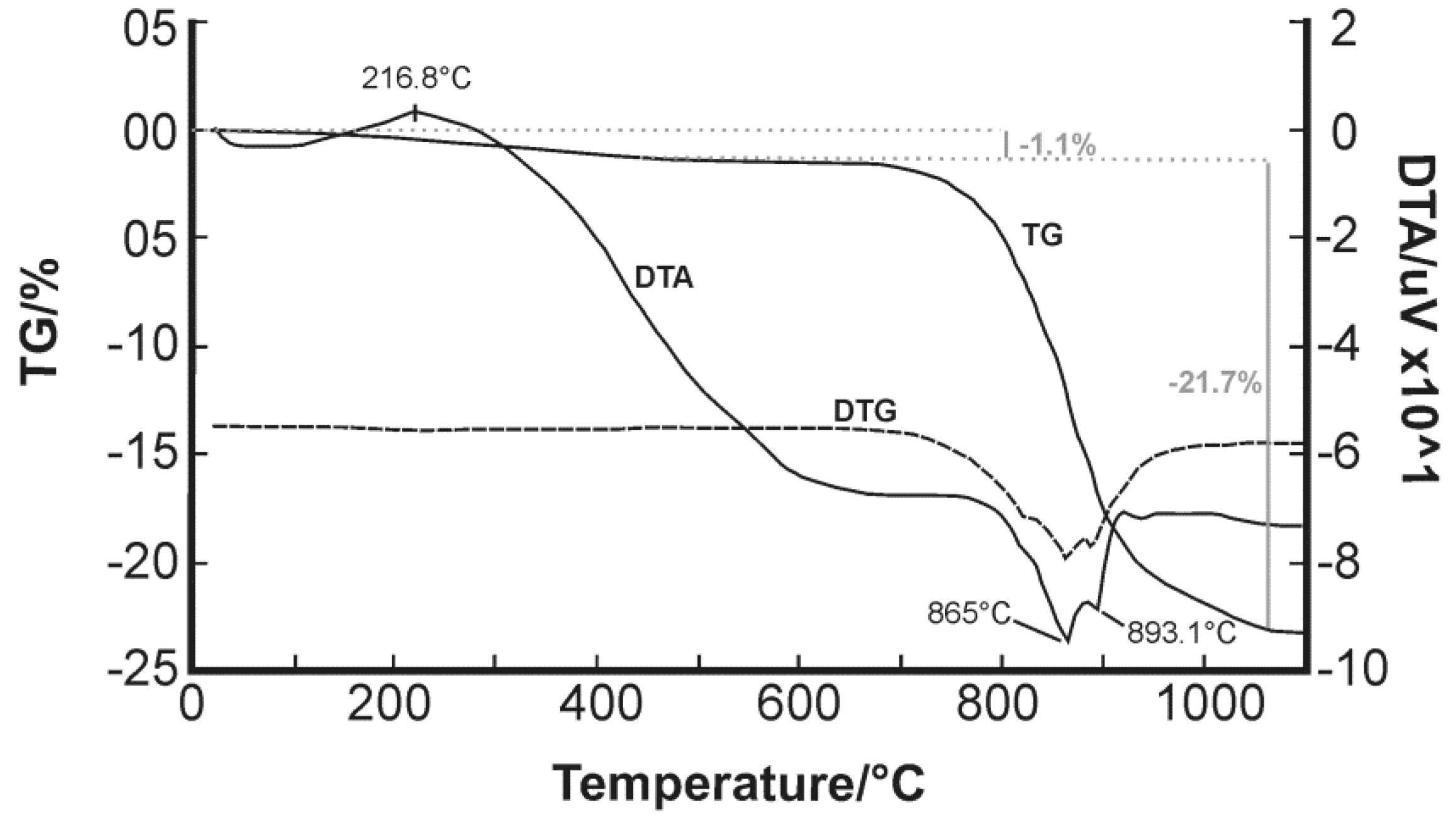
Only scarce information is available regarding thermal experiments on bismuth oxychlorides. Chukhrov et al. (1960) [
55] reported a curve for bismoclite which shows characteristic peaks at 625–675 °C and 825–875 °C [
4], whereas Rao and Adusumilli (1965) [
4] studied a bismoclite sample from Brazil and observed only one endothermic curve between 920 and 930 °C prior to material fusion at 945 °C (
Table 7). Despite Rao and Adusumilli (1965) [
4] finding no correlation between the data they obtained and those of Chukhrov et al. (1960) [
55], the current study has found a clear overlap between 865 and 875 °C (
Figure 6). The double endothermic spike is shifted to slightly higher temperatures compared to Chukhrov et al. (1960) [
55], possibly due to the effect of trace amounts of preisingerite.
The preparation and photocatalytic activity of BiOCl catalyst was documented recently by Shi et al. (2011) [
56]. Their experiment revealed that the TG curve displayed no significant mass loss between 40 and 600 °C, but there was a sharp mass loss after 600 °C with an exothermic peak on the DTA curve, which indicates the point of BiOCl decomposition (
Table 7). The DTA curve they obtained had a large endothermic peak below 150 °C, which indicates the evaporation of water molecules and hydrochloric acid. They observed a significant exothermic phenomenon between 600 and 900 °C that might be triggered by a phase change (
Table 7). Previous studies have documented the products of thermal decomposition of BiOCl (e.g., 3BiOCl = Bi
2O
3 + BiCl
3, a reaction which occurs between 575 and 600 °C; or Bi
24O
31Cl
10 (‘Arppe compound’), a complex layer structure formed when BiOCl is heated above 600 °C; [
57,
58]). The thermal experiment carried out in this study identified a constant mass below 600 °C and a sharp mass decrease at higher temperatures. Furthermore, on the DTA curve, spikes below 150 °C and between 600 and 900 °C were recorded. The main difference between this study and that of Shi et al. (2011) [
56] is that the exothermic and endothermic spikes are interchanged.
After the evaporation of HCl and H
2O, the ideal remaining substance is Bi
2O
3. Schröder et al. (2010) [
59] verified the temperature range of α- to δ-Bi
2O
3 transitions with in-situ Raman spectroscopy, XRD, DTA and impedance spectroscopy (IS). During the DTA heating process, they only detected one transition from the monoclinic α- to δ-Bi
2O
3 at 727 ± 2 °C before reaching its melting point at 822 ± 3 °C (
Table 7). During the cooling process, the δ-phase transformed into tetragonal β-Bi
2O
3 at 639 ± 2 °C and finally back into α-Bi
2O
3 at 563 ± 2 °C. Although not detected in their DTA, the δ-phase sometimes transformed into the cubic γ-phases between 630 and 650 °C, depending on the experimental conditions, and at about 550 °C back into the α-phase. Bi
2O
3 is the ideal heating product after pure bismoclite; none of the DTA spikes and melting point of Bi
2O
3 are analogous to the DTA results of bismoclite from San Francisco de los Andes. This fact is consistent with the presence of trace amounts of preisingerite as indicated by our XRD and chemical analyses. Those features below 250 °C are interpreted as the evaporation of water molecules and hydrochloric acid. The resulting substance is thought to be BiAsO
4 and the peaks between 865 and 893 °C are presumably due to phase changes between the monoclinic α-BiAsO
4 and the tetragonal β-BiAsO
4.
Lee (2008) [
60] studied synthetic monoclinic α-BiAsO
4 (rooseveltite) and recorded a phase transformation at 871 °C (
Table 7). Her results are consistent with the maximum endothermic peak at 865 °C recorded in our study.
Monoclinic rooseveltite (α-BiAsO
4) has a monazite-like structure. It is dimorphous with tetragonal tetrarooseveltite (β-BiAsO
4), which has a scheelite-like structure [
61,
62,
63,
64,
65]. Monazite and scheelite structures are adopted by numerous minerals and synthetic compounds including BiAsO
4 and NdAsO
4. Mooney (1948) [
62] synthesized bismuth arsenate crystals and noted that the monoclinic form precipitated quickly, whereas the tetragonal form tended to precipitate after a longer period of time. Isostructural compounds such as synthetic NdAsO
4 exhibit a monazite-like structure at room temperature and pressure but have a scheelite-like structure at higher temperature and pressure [
66,
67]. Finch and Hanchar (2003) [
65] published a schematic P–T phase diagram for this group of minerals where monazite-like structure is stable at lower temperatures, making the scheelite-like structure the higher temperature polymorph.
Based on the above discussion, and the consistency with Lee’s (2008) [
60] DTA results, we consider the endothermic peak at 865 °C (
Figure 6) to represent the transition from the monoclinic α-BiAsO
4 (rooseveltite) monzonite-like structure to the tetragonal β-BiAsO
4 (tetrarooseveltite) scheelite-like structure.
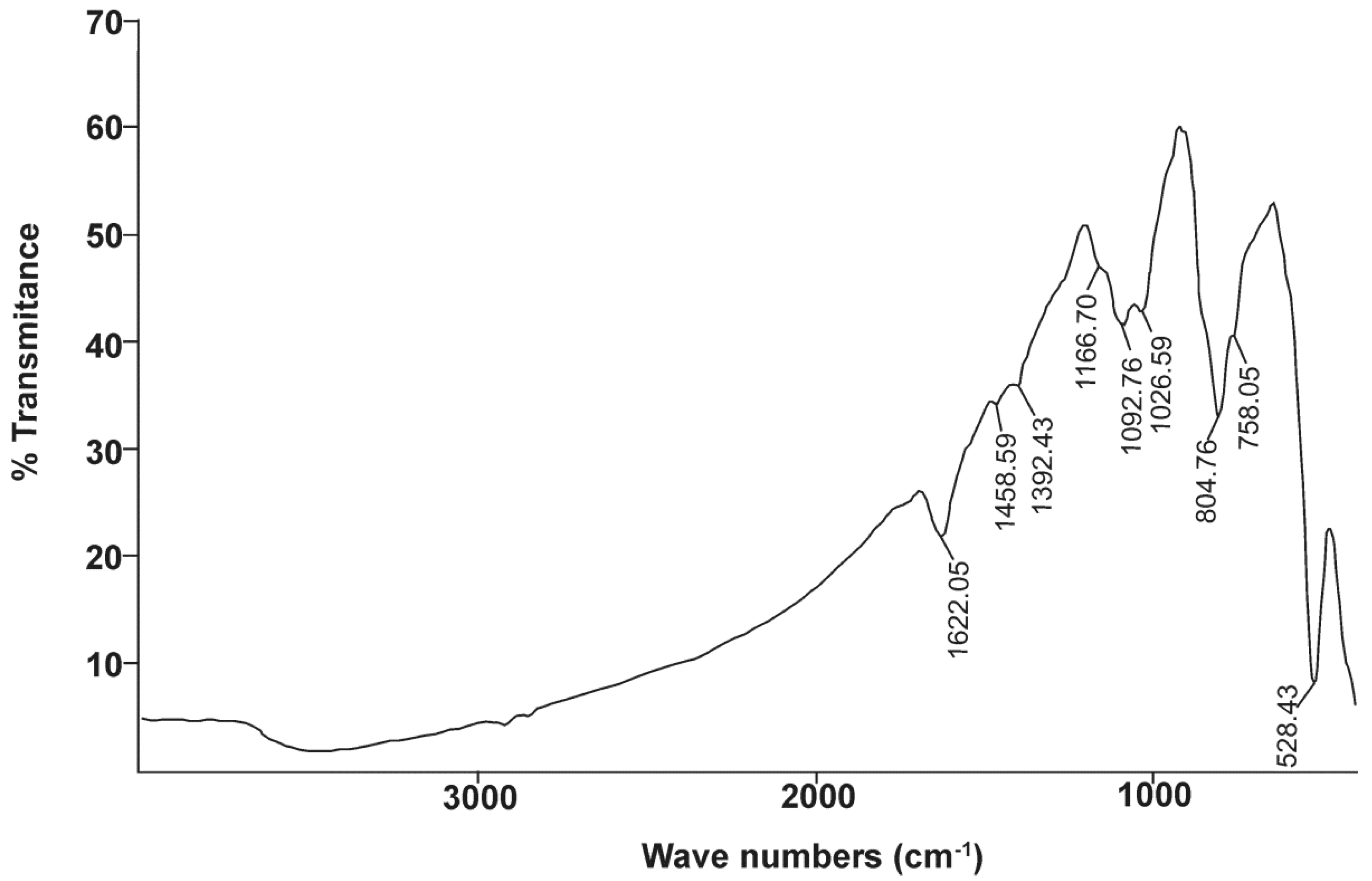
Bismoclite is an ionic salt comprising Bi
+3 cations and O
−2 and Cl
− anions. Due to the lack of covalent bonds, most of the absorption features are located in the far infrared region [
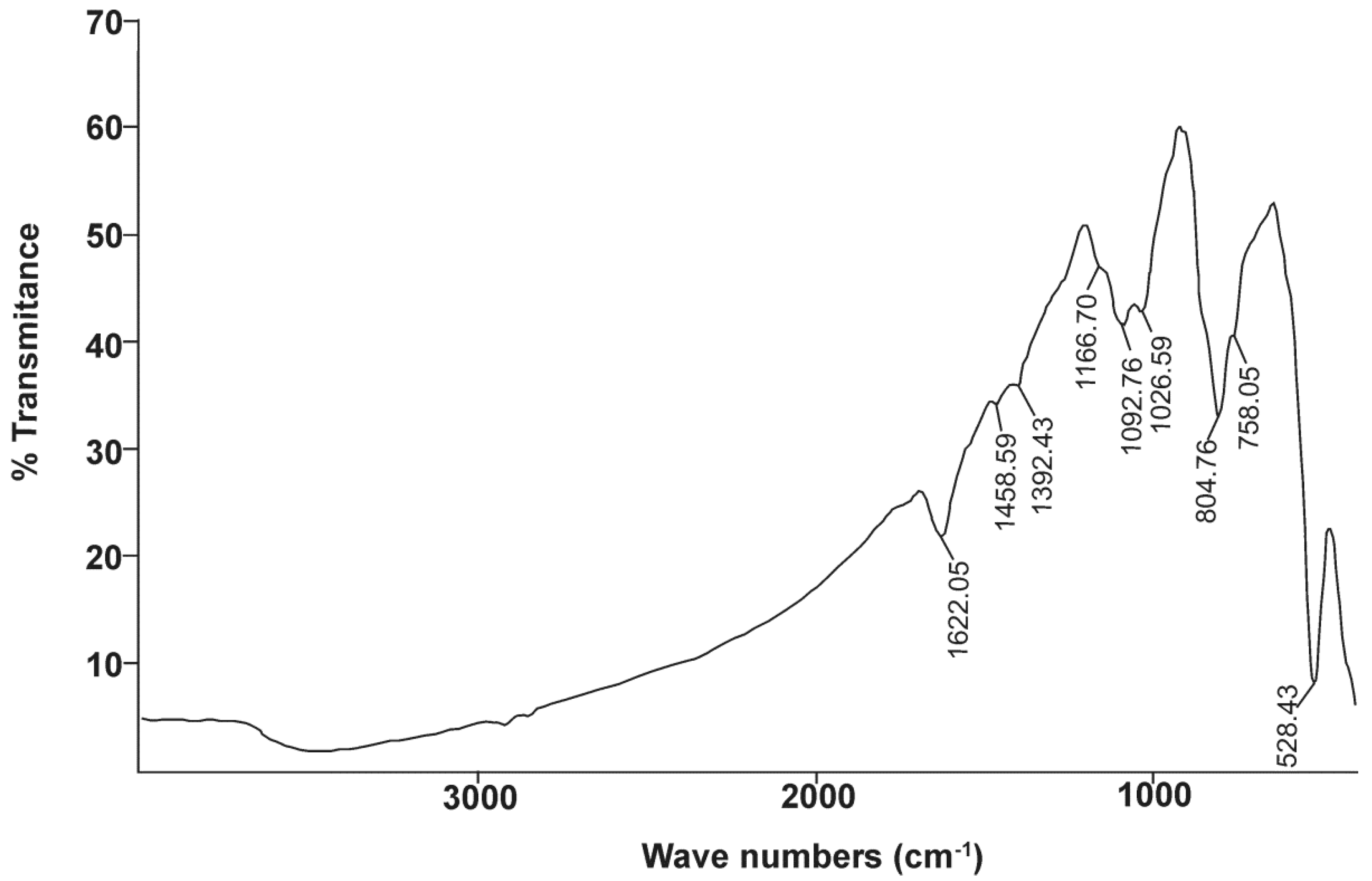
34]. Bismoclite has two sharp absorptions at approximately 528 and 180 cm
−1, as well as two broad absorptions across the ranges 470 to 250 cm
−1 and 170 to 85 cm
−1 (
Table 8) [
68]. In the current study, the first absorption peak is consistent with Nyquist et al. (1997) [
68]. The second absorption peak could not be fully recorded due to equipment limitations, although
Figure 7 shows the beginning of a clear infrared absorption band at wavelength numbers lower than 470 cm
−1.
The photocatalytic activity of BiOCl was recently documented by Ye et al. (2012), Bai et al. (2014), Chang et al. (2014), and Xie et al. (2014) [
69,
70,
71,
72]. By means of FTIR spectroscopy, they recorded the sharpest absorption bands of pure BiOCl at 529, 524, 535 and 523 cm
−1, respectively, and a weak peak at approximately 1650 cm
−1 (
Table 8). Chang et al. (2014) and Xie et al. (2014) [
71,
72] reported a broad, weak absorption at 3450 cm
−1 (
Table 8). Ye et al. (2012) [
69] and Bai et al. (2014) [
70] published IR spectrums up to 2000 cm
−1 and documented a minor peak at 1395 cm
−1 (
Table 8). The main absorption at 528 cm
−1, along with the three minor absorption features, were detected in our FTIR study of naturally occurring BiOCl (
Figure 7).
The presence of preisingerite at San Francisco de los Andes has been confirmed by means of infrared spectrometry. These results are in agreement with the presence of trace amounts of preisingerite as detected by XRD, geochemistry and DTA. The strong and wide absorption band of OH
− group is located in the portion of the electromagnetic spectrum with wavelengths between 4000 and 1700 cm
−1. Sejkora et al. (2004) [
73] noted that the OH
− stretching vibrations of H
2O are located between 2862–3520 cm
−1, whereas the δH
2O bending vibrations occur between 1604 and 1634 cm
−1 (
Table 8). The last absorption is clearly recorded at 1622 cm
−1 (
Figure 7).
Sumin de Portilla (1976) [
74] studied the nature and role of the hydrogen bonds in legrandite and two other Zn bearing hydroxyl-arsenate minerals. She noted that the (AsO
4)
3− ν
3(T
2), ν
1(A
1) and ν
4(T
2) vibration bands are located at around 800, 750 and 400 cm
−1, respectively (
Table 8). These data are consistent with the sharp spike at 804.76 cm
−1, the shoulder detected at 758.05 cm
−1, and the beginning of a strong absorption band to the right side of
Figure 7. Povarennykh (1978) [
75] grouped the characteristic ν
3 and ν
4 vibrations of arsenates between 900–760 cm
−1 and 420–310 cm
−1 (
Table 8). More recent studies documented that the tetrahedral (AsO
4)
3− exhibits the ν
1 symmetric stretching vibration (A
1) at 818 cm
−1, the ν
3 antisymmetric stretching vibration (F
2) at 786 cm
−1, the ν4 bending vibration (F
2) at 405 cm
−1, and the ν
2 symmetric bending vibration (E) at 350 cm
−1 (
Table 8) [
76,
77,
78,
79]. The position of IR absorption bands for (AsO
4)
3− molecules of preisingerite from San Francisco de los Andes are in agreement with the studies cited above; the main difference between them is the interpretation of absorption peaks.
The stretching vibrations of Bi–O and Bi–O–Bi polyhedral occur between 370 and 620 cm
−1 (
Table 8) [
73,
80,
81,
82]. These vibrations partially overlap with the main absorptions of bismoclite at 528 cm
−1 and the beginning of a broad absorption band at 470 cm
−1, as well as with the ν
4 bending vibration of (AsO
4)
3− at 405 cm
−1.
Fu et al. (2003) [
83] noted that the Cl–O stretching vibrations can also be observed at wavelengths above 800 cm
−1 (
Table 8). The absorption at 804 cm
−1 in
Figure 7, can be considered as an overlap of (AsO
4)
3− and Cl–O vibrations of different bonds. The double shoulders at 1392 and 1458 cm
−1, as well as the minor inflexion close to 3000 cm
−1, are the three major peaks of Nujol (
Table 8) [
74,
84]. Due to a scarcity of references to compare with both Bi-bearing minerals, the double peak at 1092 and 1026 cm
−1 (
Figure 7) could not be clearly attributed to any specific bond, and the origins of these peaks remain unclear.
5.2. Bismoclite from San Francisco de los Andes: Its Geneisis and Related Supergene Processes
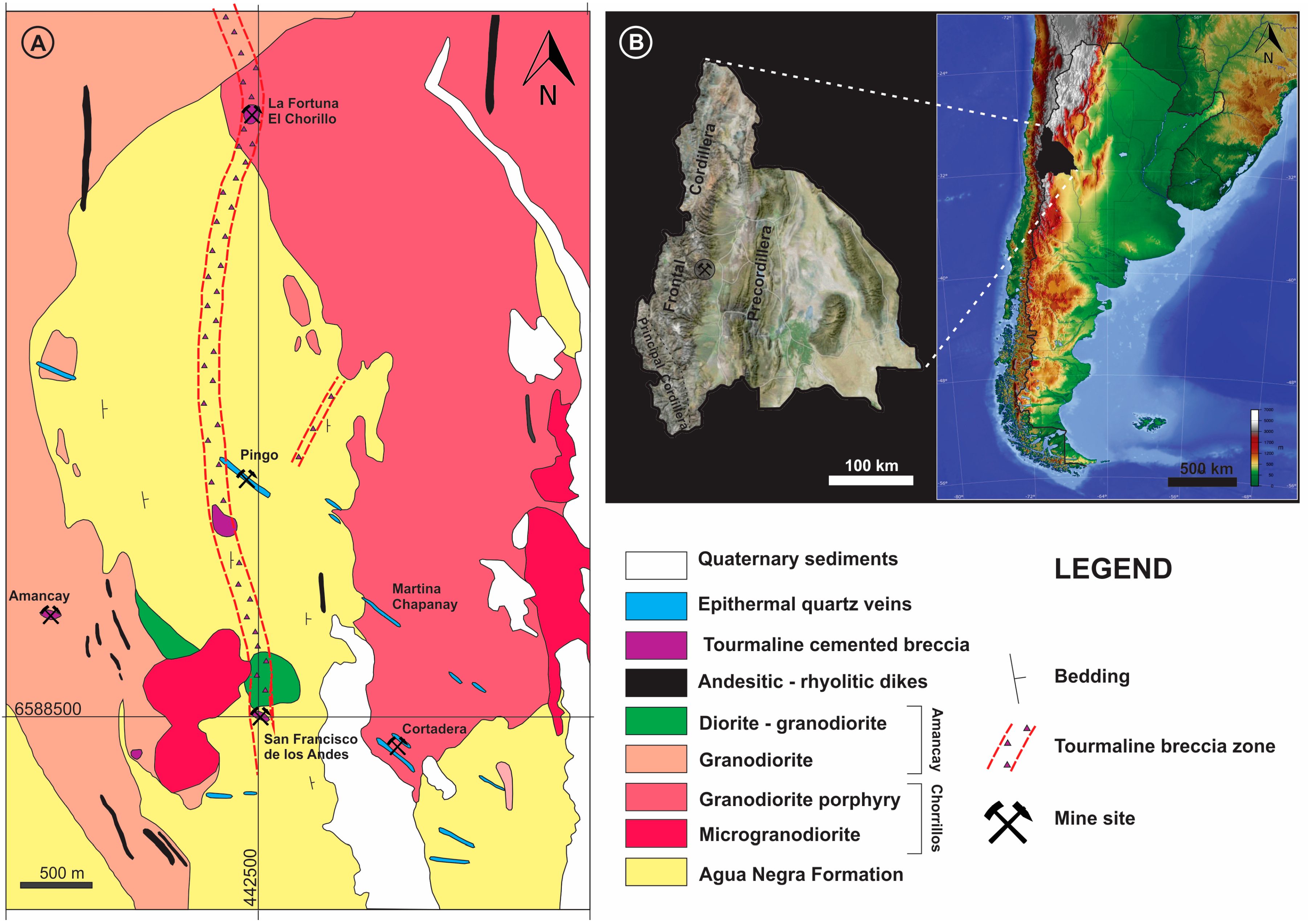
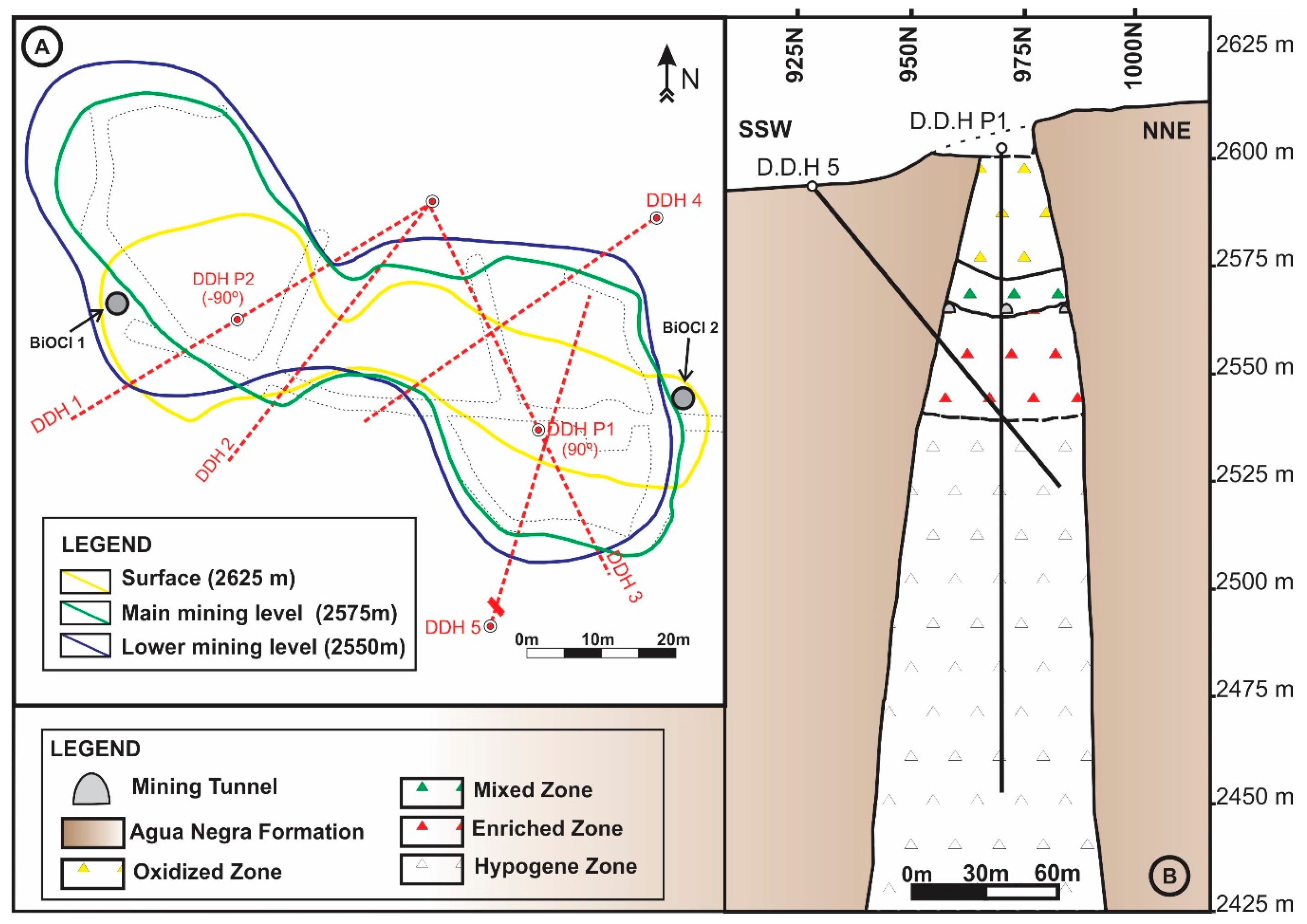
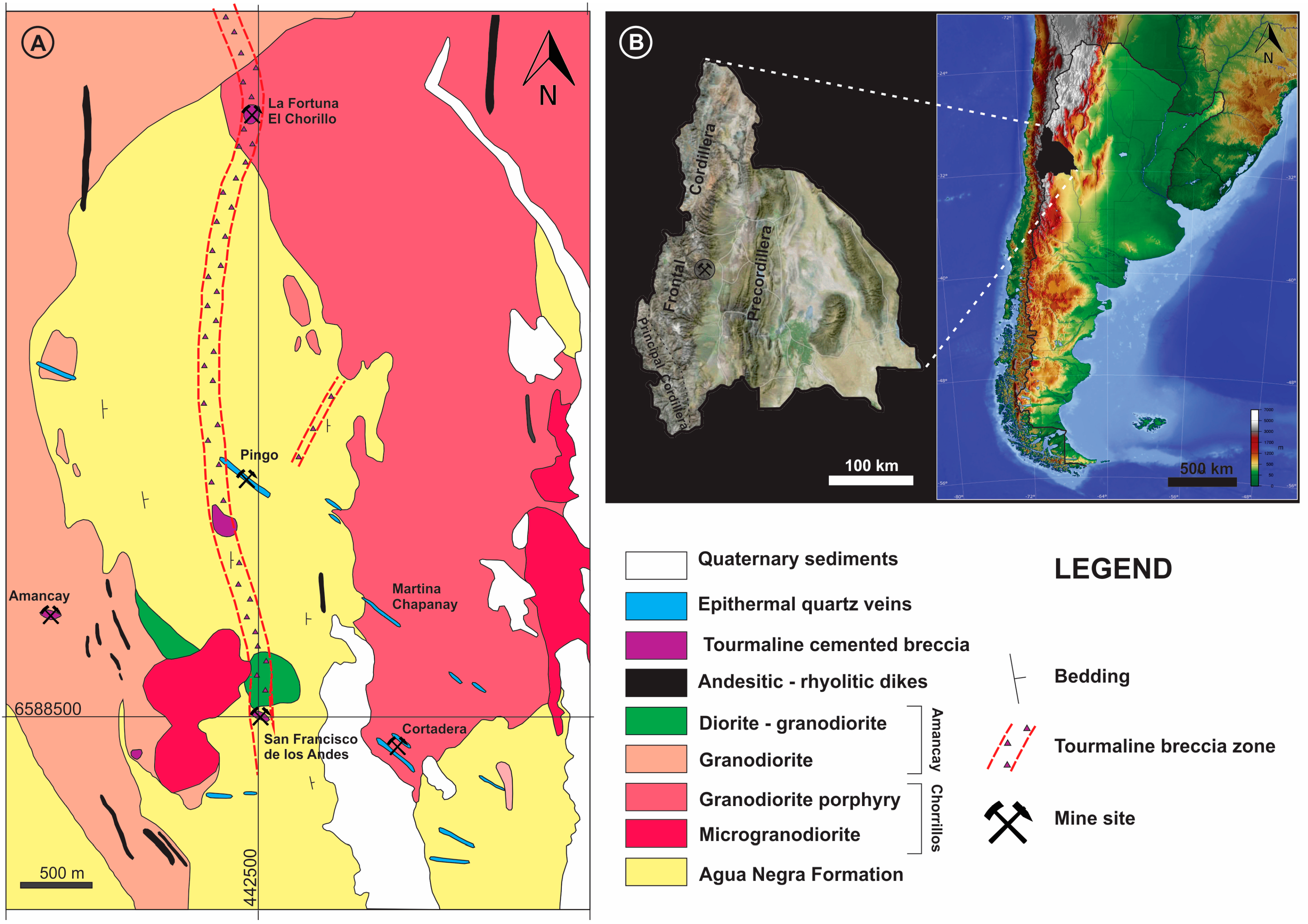
Bismoclite is insoluble and thus may occur as a dominant phase in the oxidized zones of Bi-rich ore deposits. It has proven to be a key mineral, intimately associated with ores in certain deposits, which includes the Diablillos high-sulfidation epithermal Ag–Au deposit in Argentina. Here, bismoclite is commonly associated with the highest silver grades in the oxide zone [
15]. The semi-arid climate favored weathering and supergene enrichment processes, which concentrated native gold and silver minerals. The later occur as chlorargyrite (AgCl), and locally iodargyrite (AgI) [
15]. The climate at San Francisco de los Andes is mostly desert with little precipitation, which enabled the occurrence of water-soluble species at surface (e.g., chalcanthite) together with bismoclite. The presence of bismoclite (±preisingerite) at surface is, therefore, an indicator for areas where supergene and hypogene ores may be found at depth. At San Francisco de los Andes, Cu–sulfates locally can be distinguished at surface due to their brighter colors. However, in areas with higher average rainfall, bismoclite may be the only oxidized mineral present to suggest the presence of hypogene mineralization at depth.
Natural bismoclite typically occurs as small aggregates. Bismoclite from the Diablillos deposit occurs as small masses and fracture coatings, with some aggregates visible in hand specimens [
15]. It is commonly intergrown with chlorargyrite in the high-grade silver zones. It is inferred that bismuth at the Diablillos deposit was probably abundant in the hypogene hydrothermal fluids although bismuth minerals (i.e., bismuthinite, matildite) are rare in the preserved hypogene zone [
15]. At San Francisco de los Andes, bismoclite forms larger aggregates, some of them up to 3 cm size. In the study area, the bismuth-bearing hypogene mineralogy is evident with particular abundance of bismuthinite and cosalite.
As summarized in
Table 1, bismoclite has been documented in the oxidized zones of pegmatites, greisens, W–Mo–(Bi) quartz veins, high-, intermediate-, and low-sulfidation epithermal systems, polymetallic, orogenic, sediment–hosted, volcanogenic massive sulfides, banded iron formations and orthomagmatic Ni–Cu–Fe deposits, but not in porphyries or related magmatic–hydrothermal breccias. When considering element mobility and zoning patterns in mineralized porphyry systems, elements such as Bi, Sb, As, Te, Ag, Pb and Zn are typically anomalously high lateral to the ore zones and/or above them (e.g., Govett, 1983 [
85]). They are particularly characteristic in the outer propylitic alteration zones of porphyry deposits and are concentrated at shallow levels in high-, and low-sulfidation epithermal deposits. All the aforementioned elements are characteristic in most of the hypogene S-rich mineral phases that occur as cement in the San Francisco de los Andes breccia pipe complex (
Table 2).
Magmatic–hydrothermal breccia systems commonly occur at the apices of porphyry deposits, and often host large amounts of economic mineralization; these ore phases are consistent with those of porphyry-style mineralization (e.g., chalcopyrite, bornite and molybdenite at the Rio Blanco–Los Bronces district, [
86]). At the San Francisco de los Andes breccia complex, the characteristic breccia facies and their morphology resemble those of magmatic–hydrothermal breccias in porphyry systems, but are cemented by minerals species more common to distal porphyry and/or shallow epithermal levels.
The rarity of abundant Bi-bearing mineral phases in porphyry and magmatic–hydrothermal breccia deposits is the main reason why bismoclite (and associated preisingerite) have not previously been reported from these types of deposits. For bismoclite to form, it would require available hypogene bismuth minerals; followed by suitable supergene conditions to allow its formation.
In the oxidized zone of San Francisco de los Andes, bismuthinite can be oxidized to bismoclite due to an increase in both Eh and HCl fugacity; e.g.:
| Bi2S3(s) + 2Cl− + 2H2O + 6O2(g) | = | 2BiOCl(s) + 3SO42− + 4H+ | | (1) |
| Bismuthinite | | Bismoclite | | |
Although it is possible that bismoclite from San Francisco de los Andes may have also formed from Pb–Bi sulfosalt cosalite (Pb2Bi2S5) or Cu–Bi sulfosalts such as emplectite (CuBiS2) and cuprobismutite (Cu10Bi12S23), several lines of evidence support bismuthinite as the precursor for bismoclite:
Cosalite is probably the second most abundant hypogene Bi mineral after bismuthinite. Lead is an immobile element in an oxidized and acidic environment such as the San Francisco de los Andes’ sulfate-rich oxidized and mixed zones [
87,
88]. If cosalite was responsible for bismoclite formation, we would expect to find anglesite (PbSO
4) intergrown with the oxychloride as a product; e.g.:
| Pb2Bi2S5(s) + 2Cl− + 2H2O + 10O2(g) | = | 2BiOCl(s) + 2PbSO4(s) + 3SO42− + 4H+ | | (2) |
| Cosalite | | Bismoclite Anglesite | | |
At San Francisco de los Andes, cosalite is unlikely to have been responsible for bismoclite formation because anglesite was only rarely found associated with galena and never with bismoclite.
The only two Cu–Bi sulfosalts found at San Francisco de los Andes are emplectite and cuprobismutite. Either of these hypogene minerals could be responsible for the production of bismoclite, together with the very common chalcanthite (CuSO
4·5H
2O) or less common brochantite (Cu
4(SO
4)(OH)
6); e.g.:
| CuBiS2(s) + Cl− + 5H2O + 4.5O2(g) | = | BiOCl(s) + CuSO4·5H2O(s) + SO42− | | (3) |
| Emplectite | | Bismoclite Chalcanthite | | |
| Cu10Bi12S23(s) + 12Cl− + 50H2O + 52O2(g) | = | 12BiOCl(s) + 10CuSO4·5H2O(s) + 13SO42− | | (4) |
| Cuprobismutite | | Bismoclite Chalcanthite | | |
Emplectite and cuprobismutite are unlikely to have been the hypogene reactants responsible for the formation of bismoclite, as these are minor to rare minerals at San Francisco de los Andes. Furthermore, Cu sulfates were never found in association with bismoclite.
For all of the above, Equation (1) is considered to be the reaction that best describes the formation of bismoclite at San Francisco de los Andes. The sulfate anions liberated from this reaction mostly precipitated as sulfate minerals (
Table 2). These, together with arsenates, are the most common supergene mineral groups in the San Francisco de los Andes’ oxidized (and to a lesser extent mixed) zones. In a similar manner to bismuthinite, the sulfides and sulfosalts listed in
Table 2 most likely reacted with acidic and oxidized meteoric waters to produce additional sulfate anions. Due to the abundance of arsenopyrite at San Francisco de los Andes, a key supergene reaction was the decomposition of arsenopyrite; e.g.:
| FeAsS(s) + 7/2O2 +H2O = Fe3+(aq) + SO42− + H2AsO4−(aq) | | (5) |
| Arsenopyrite | | |
Arsenic speciation after arsenopyrite decomposition depends on many factors, including pH, oxygen availability, presence of bacteria, and/or Fe
3+ in solution. In strongly oxidized and moderately acidic conditions, such as the San Francisco de los Andes oxidized zone, the As species produced is predicted to be dihydrogen arsenate [
87,
89,
90]. This anion is most likely responsible for the formation of supergene preisingerite and the abundant Bi, Cu, Pb and Fe arsenates listed in
Table 2.
In summary, hypogene S-rich minerals from San Francisco de los Andes were weathered under desert conditions by O2-rich waters, to produce solutions that dissolved further reduced mineral phases Hypogene sulfides and sulfosalts were oxidized at the surface and produced sulfuric acid. These reactions led to acidified meteoric waters, which leached metals as they percolated through the oxidized and mixed zones. Above the water table, bismoclite, oxides, arsenates, sulfates and trace carbonates formed; whereas below it, reduced conditions prevailed and hypogene sulfides and sulfosalts remained unaltered. As the supergene solutions penetrated the water table, their metallic content precipitated as secondary reduced species to form the supergene enriched ores. Hypogene iron sulfides reacted with the copper sulfate solution, prompting supergene Cu sulfides and sulfosalt precipitation (i.e., covellite, chalcocite and luzonite) and releasing iron into solution.
Several lines of evidence suggest that bismoclite from San Francisco de los Andes formed due to weathering of bismuthinite by oxidized and moderately acidic fluids. Bismoclite is predicted to form in the supergene environment wherever bismuthinite is exposed to oxygenated saline meteoric waters. Evaporative conditions led to the formation of Cl-bearing minerals, as wet climates preclude their formation.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}