3.1. Relaxations of the (110) Surface
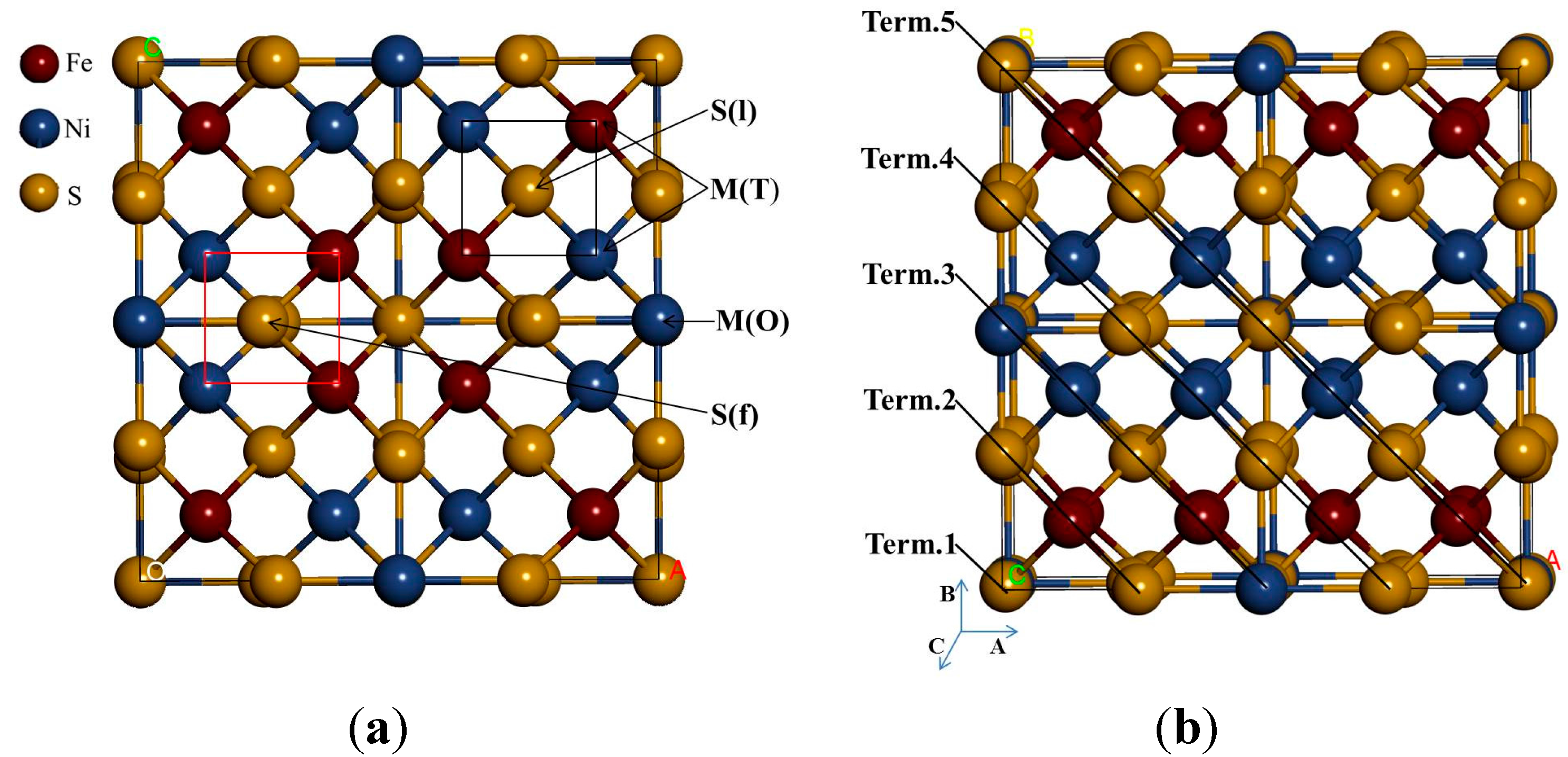
Based on the stable surface geometry (
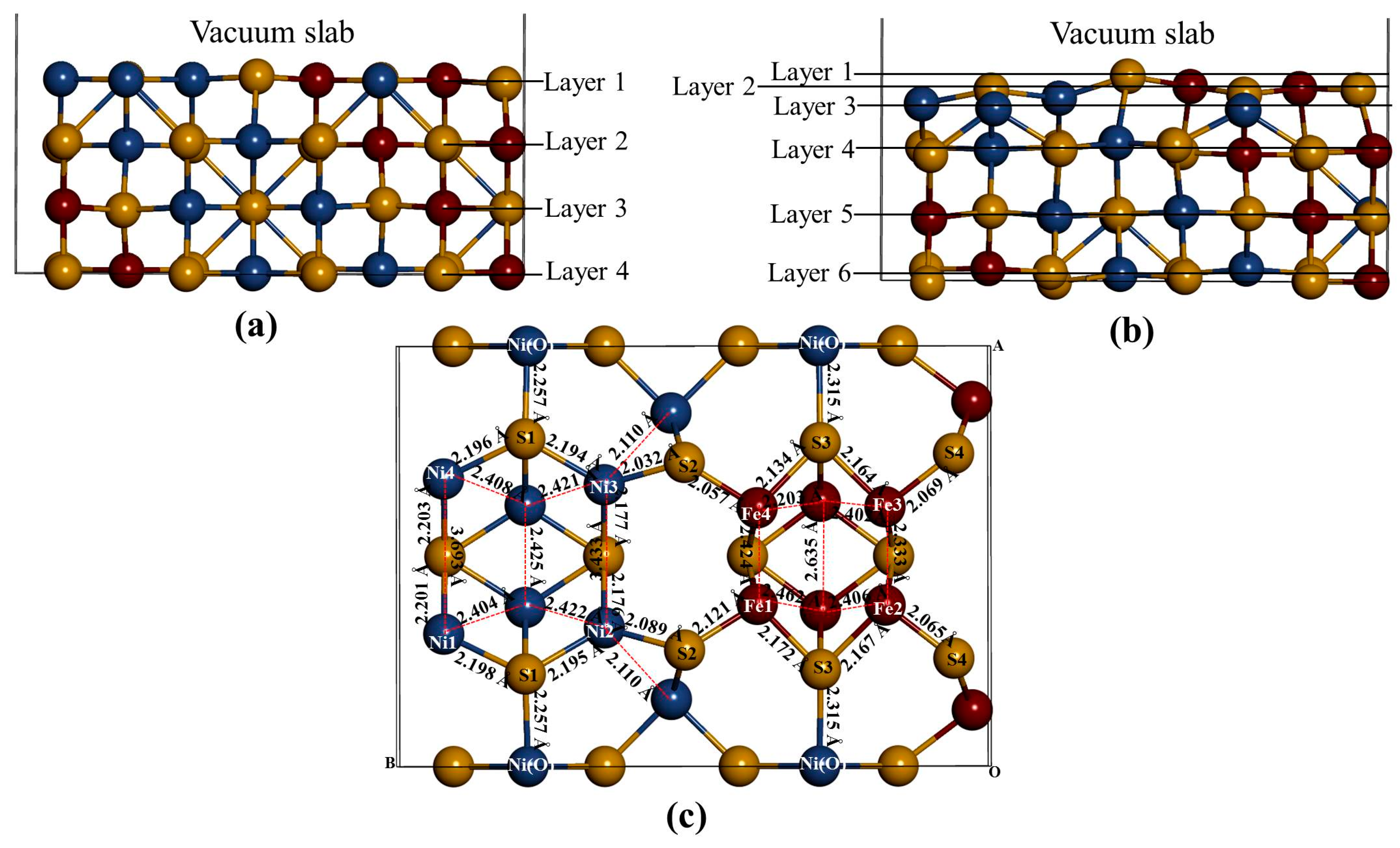
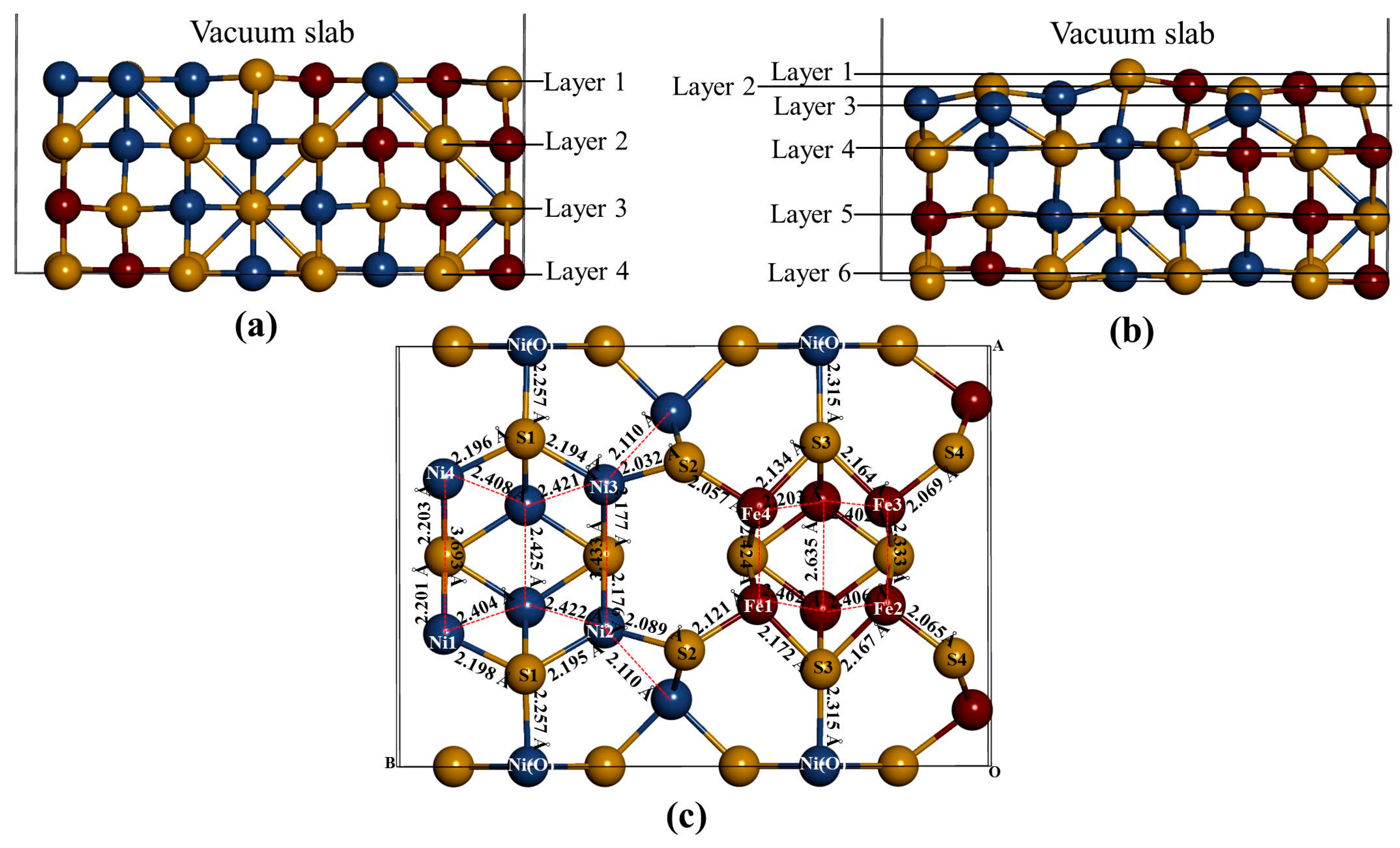
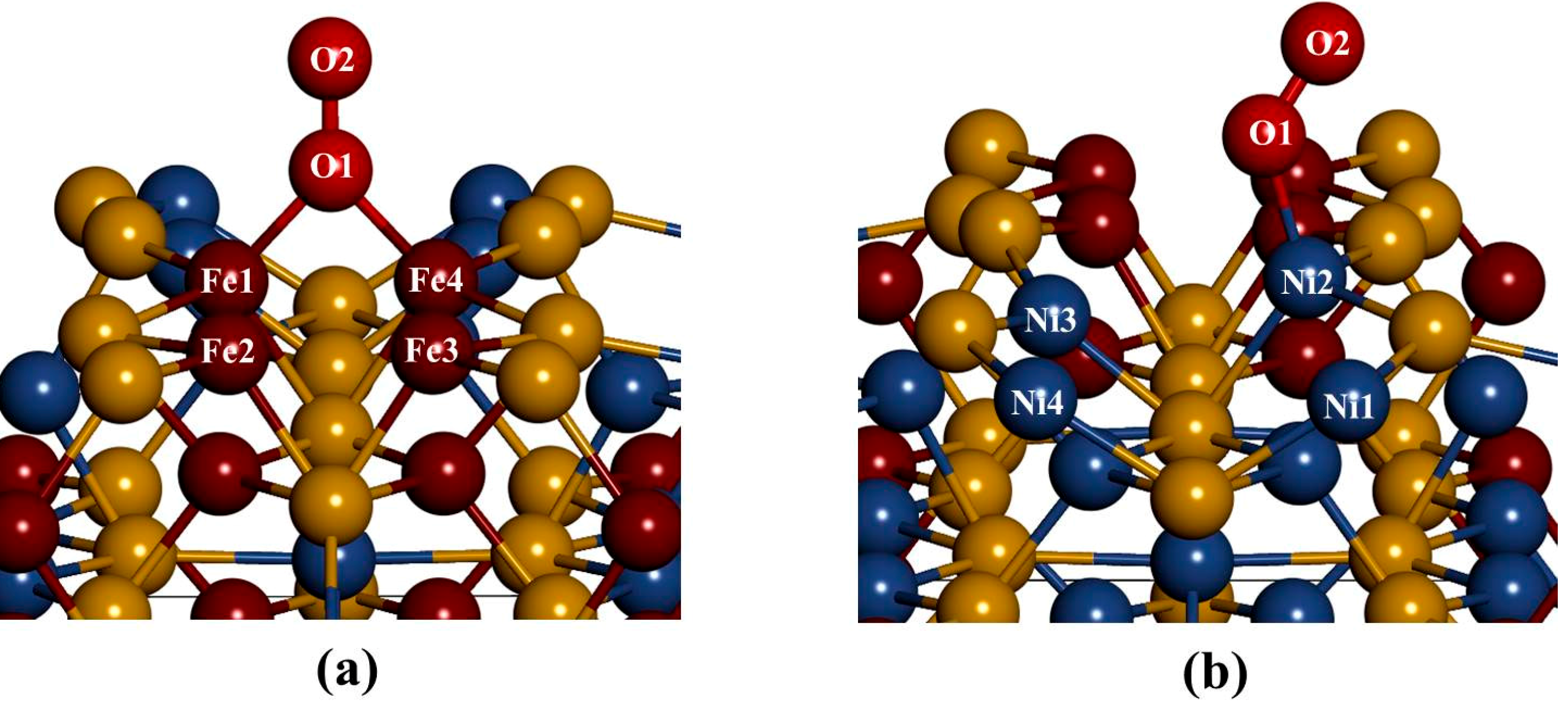
Figure 3), we noted that the S(l) and S(f) atoms change from four-coordinated and five-coordinated as in the bulk to three-coordinated and four-coordinated on the surface slab, respectively. The Fe(T) and Ni(T) atoms change from four-coordinated as in the bulk to three-coordinated on the surface slab. After the surface relaxation we noted that the top layer 1 relaxes into three layers. The surface results in sulphur termination as the sulphur (S2) atoms relaxes outwards forming layer 1. The Fe atoms and S1, S3 and S4 atoms form layer 2, while Ni atoms form layer 3.
The vertical displacement of these atoms results in S1 and S3 relaxing inwards as S4 relaxes outwards. The Fe atoms are noted to have the Fe1 and Fe4 relaxing outwards minimally, as Fe2 and Fe3 remain un-displaced.
Figure 3c clearly shows that the top bond length between the Ni atoms is weakened (stretches to 3.693 and 3.433 Å). The Ni atoms are observed to relax deep compared to the Fe atoms, indicating that the surface layer is composed of mainly Fe atoms. This suggests the Fe preferential oxidation character of the pentlandite [
8].
Figure 3.
The (110) surface: Show side view of the layers, (a) before relaxation and (b) after relaxation and (c) top view after relaxation with bond distances between the top four layers.
Figure 3.
The (110) surface: Show side view of the layers, (a) before relaxation and (b) after relaxation and (c) top view after relaxation with bond distances between the top four layers.
Table 1 shows the vertical displacement of the top atoms on the surface after relaxation (negative and positive sign indicate inwards and outwards displacement, respectively). Only S2 and Fe1 have a higher outwards displacement, and Fe2 and Fe3 remains un-displaced, significantly changes the bond distances. The Ni atoms are noted to have the highest inwards displacement.
Table 1.
Atomic vertical displacements (Å) of the relaxed top three layers on slab model.
Table 1.
Atomic vertical displacements (Å) of the relaxed top three layers on slab model.
| Atom | Displacement |
|---|
| S1 | −0.003 |
| S2 | +0.009 |
| S3 | −0.006 |
| S4 | +0.002 |
| Ni1–Ni4 | −0.018 |
| N2–Ni3 | −0.012 |
| Fe1 | +0.008 |
| Fe2 | 0.000 |
| Fe3 | 0.000 |
| Fe4 | +0.003 |
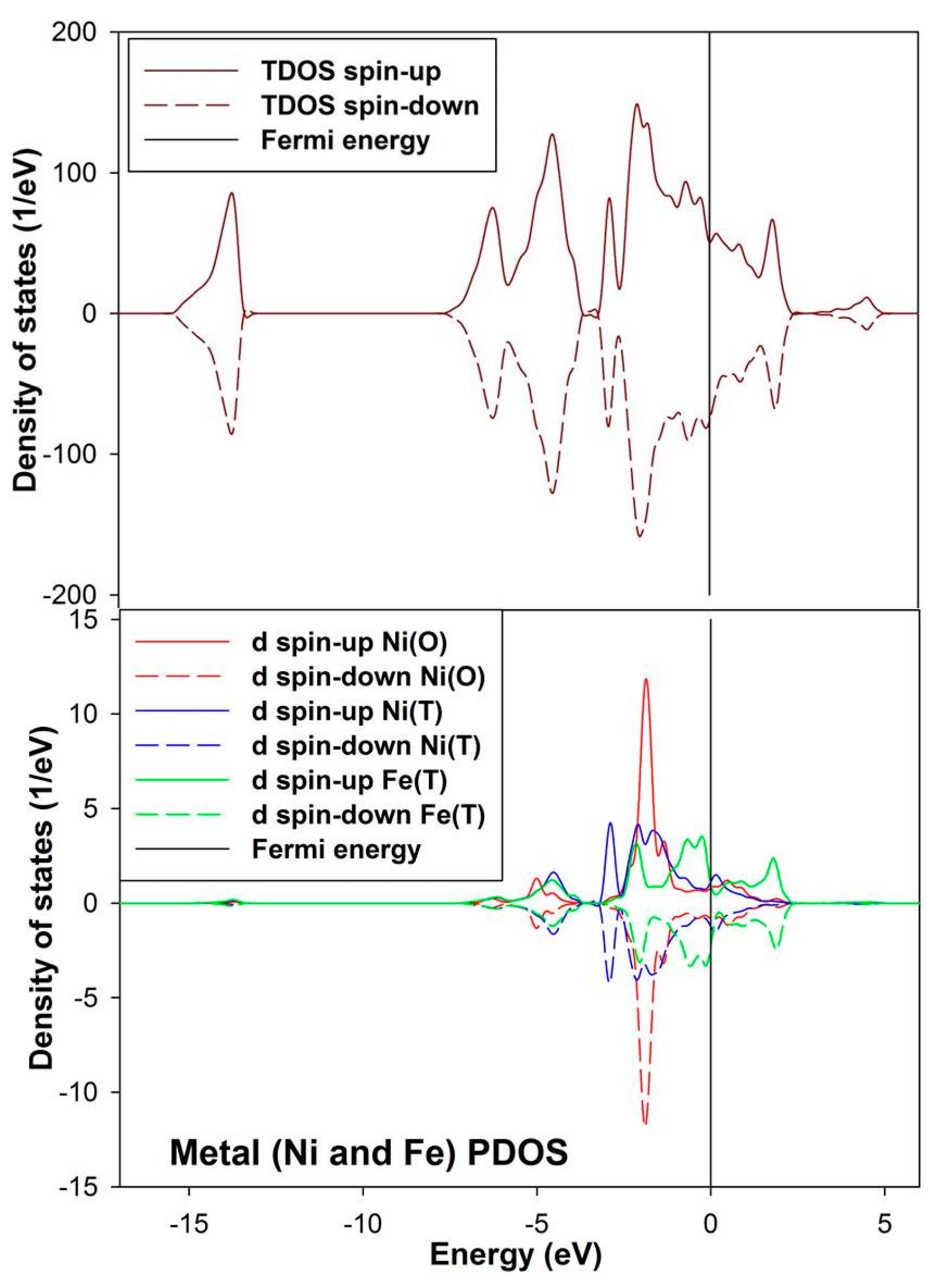
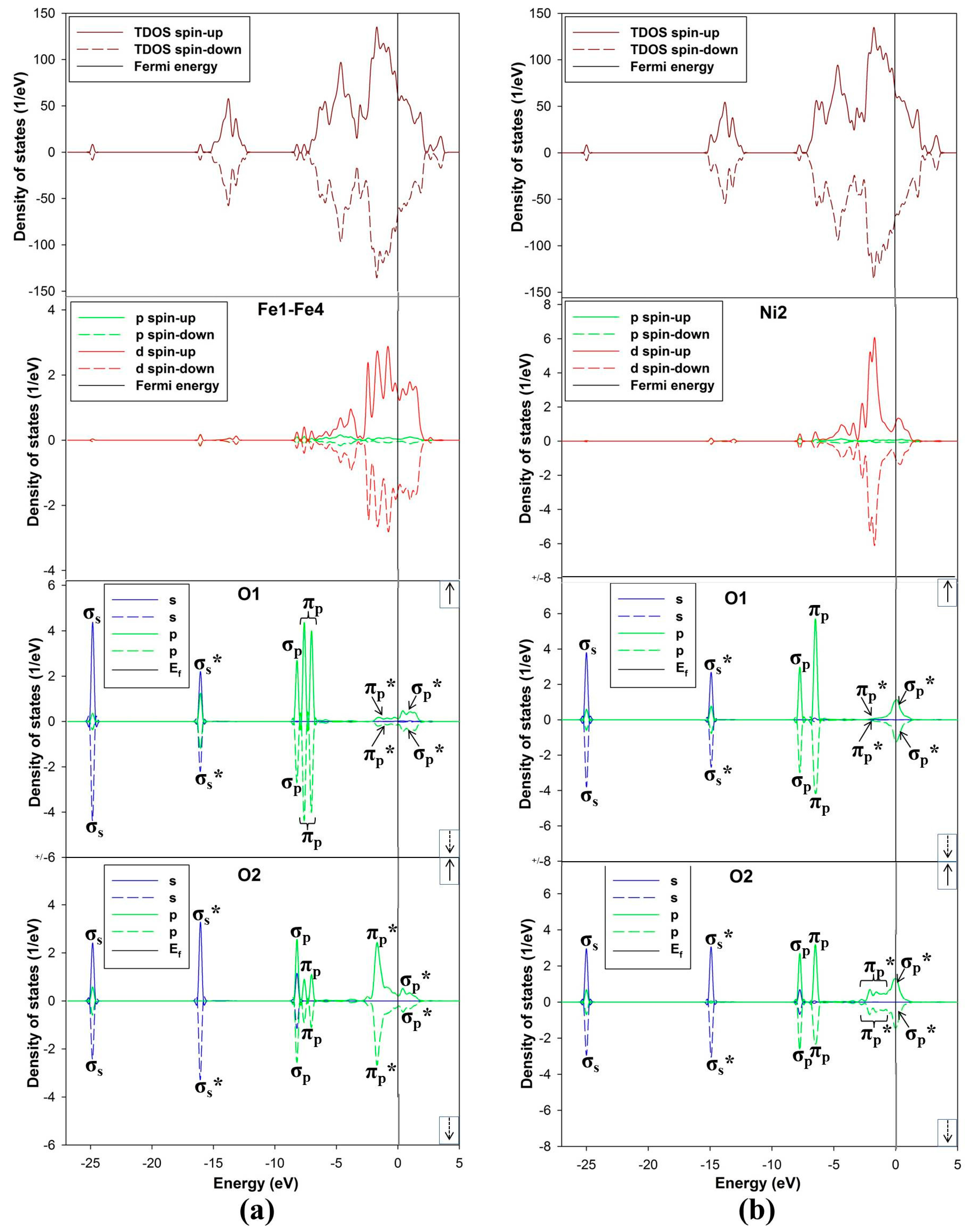
3.2. Electronic Structures of the Bulk and Non-Adsorbed Surface
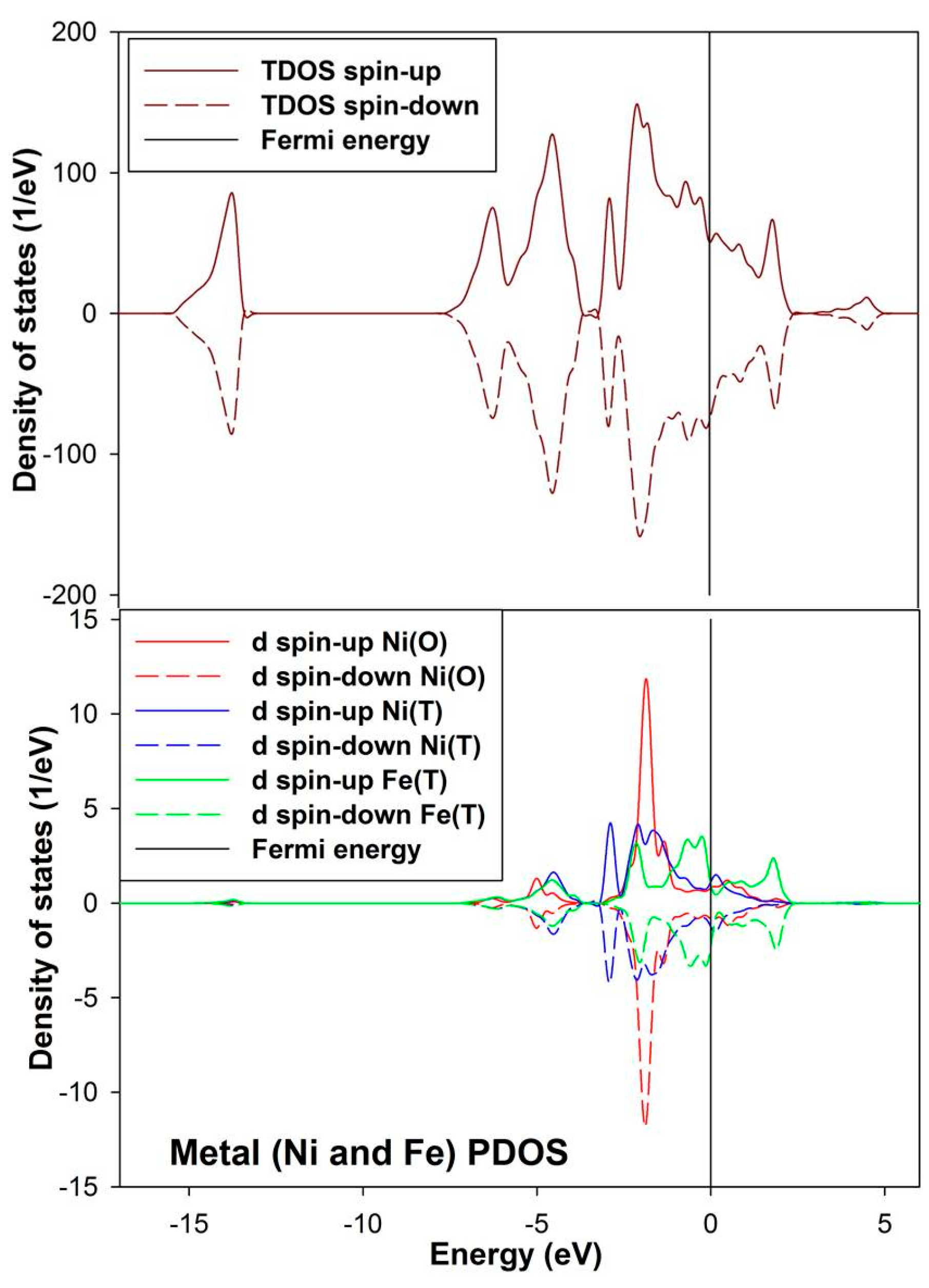
The density of state for the bulk convectional nickel-rich pentlandite is presented in
Figure 4. We noted from the total density of states (TDOS) that the spin up/down peaks are equally occupied and appear as mirror images suggesting a non-magnetic behavior, this confirms the character of the pentlandite mineral [
29]. The partial density of states (PDOS) clearly shows that the
d-orbital for Fe(T) are predominant and those of Ni(O) are lowest at the Fermi energy (E
F). We note that the octahedral Ni(O)
d-orbital form a sharp peak at about −2.0 eV. The Fe(T)
d-orbital peaks reside just below the E
F at the valence band (VB), while the Ni(T)
d-orbital contribution is very low at the E
F. At the conduction band (CB), the Ni atoms have very little contribution while the Fe atoms show a peak at around 2.0 eV.
Figure 4.
Projection total and partial density of states for the tetrahedral metals (Ni(T) and Fe(T)) and octahedral metals (Ni(O)) in the bulk conventional 4(Fe4Ni5S8) nickel-rich pentlandite.
Figure 4.
Projection total and partial density of states for the tetrahedral metals (Ni(T) and Fe(T)) and octahedral metals (Ni(O)) in the bulk conventional 4(Fe4Ni5S8) nickel-rich pentlandite.
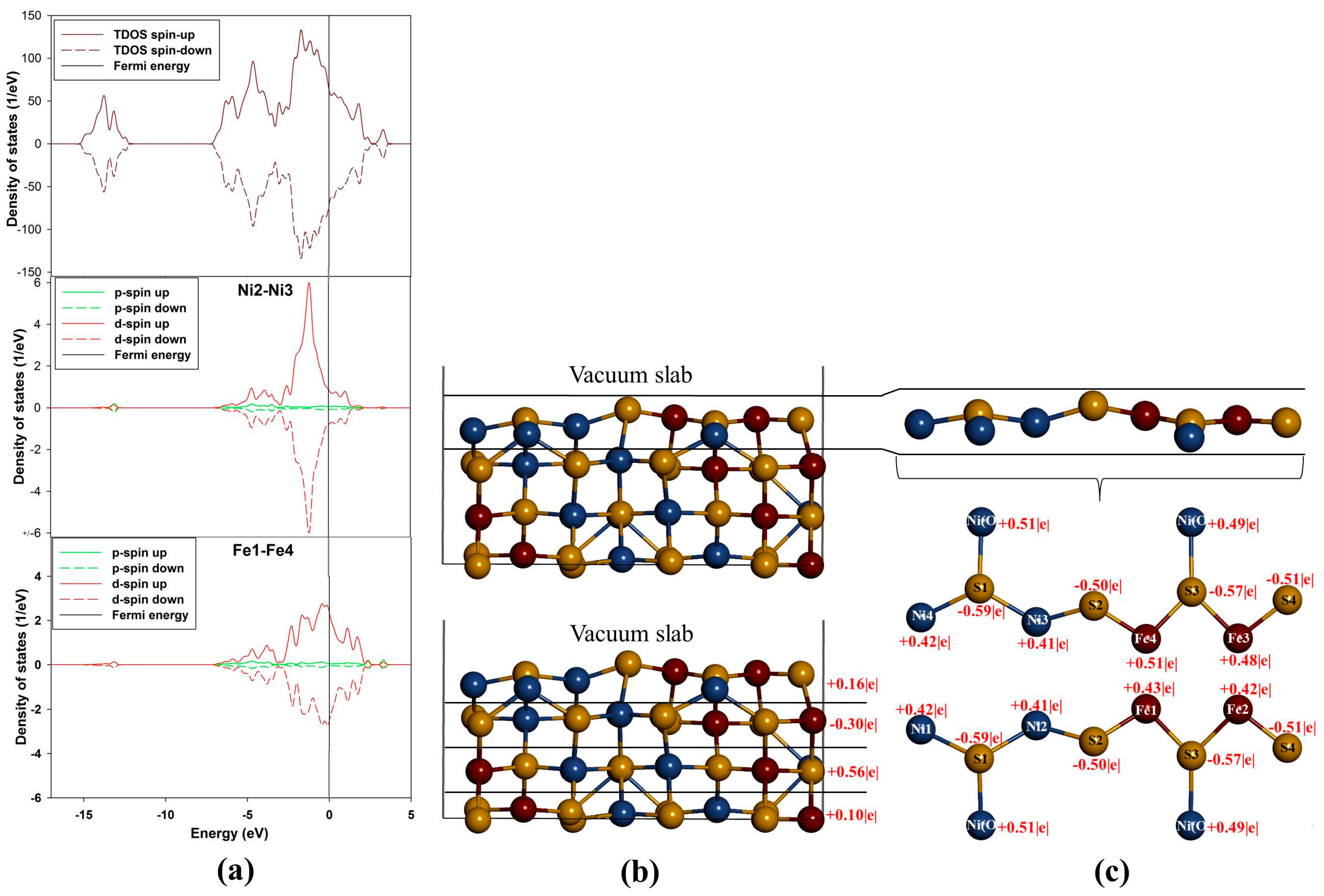
Figure 5a show density of states (DOS) of (110) surface and it is evident from the total density of states (TDOS) that the
d-orbital of the metals (M) are the centre of reactivity on the surface as they highly dominate more at the E
F. Moreover, the TDOS shows that the surface has a metallic behavior characteristic as there is no band gap at the E
F and still shows a non-magnetic character. Note that the partial density of states (PDOS) is plotted only for the top metals (
i.e., Fe and Ni) atoms adsorbed. The PDOS demonstrates that both the Ni and Fe atoms provide accessible bands at the E
F. This indicates that the electrons are delocalized, and that the Ni atoms contribute considerably to this delocalization.
Now considering the PDOS (
Figure 5a), we note different feature with respect to both top Ni and Fe atoms. Note that we show the PDOS of the Ni and Fe atoms where adsorption will take place. Firstly, the behavior of Ni2–Ni3
d-orbital is observed to move closer to the E
F and its contribution is characterized by a sharp
d-orbital peak near E
F at the (VB) (compared to the Ni(O) in the bulk) with very little contribution at the E
F and at the (CB). Secondly, the Fe1–Fe4 PDOS are noted to have five
d-orbital peaks, three at the VB and two at the CB. We observed that the
d-orbital moves across the E
F, where the E
F cuts the highest states peak, and we also noted two broad peaks,
i.e., the highest states peak (at about 0.05 eV) and the second highest states peak (at about 2.0 eV). It is clear that the bulk
d-orbital contribution for both tetrahedral and octahedral metals is different than the surface metals. The bulk tetrahedral coordinated metals show predominance of states in the VB, while states are shifted towards the E
F for the surface metals.
Figure 5.
Projection density of states and Bader charges for the top adsorbed Ni and Fe atoms: (a) total and partial density of states; (b) Bader charges for each layer and (c) Bader charges for the top layer atoms.
Figure 5.
Projection density of states and Bader charges for the top adsorbed Ni and Fe atoms: (a) total and partial density of states; (b) Bader charges for each layer and (c) Bader charges for the top layer atoms.
The Fe atoms on (110) surface are observed to have different Bader charges (
Figure 5c). This difference indicates that the Fe atoms are not charge ordered and there is alternation of the charges on the Fe atoms. Furthermore, this suggests that the presence of Ni atoms breaks the charge symmetry of the Fe atoms to a small degree, such that the Fe atoms with Fe
+0.42|e| have 0.09|e| less than the Fe
+0.51|e|. Such observations of the charge symmetry breakage have been observed on doping of FeS with Ni by Devey
et al. [
30]. Now, for the case of Ni atoms, we observed that Ni1–Ni4 have the same charges and so are Ni2–Ni3 atoms. Thus, by analogy it can be deduced that there is a high level of covalent nature predicted for the Ni–S and Fe–S bonds in Fe
4Ni
5S
8 pentlandite. The Bader charges of the metals shown in
Figure 5c indicate that the Fe atoms carry more positive charges than the Ni atoms. The three outermost layers are positively charged, while the second layer is electronegative (
Figure 5b).
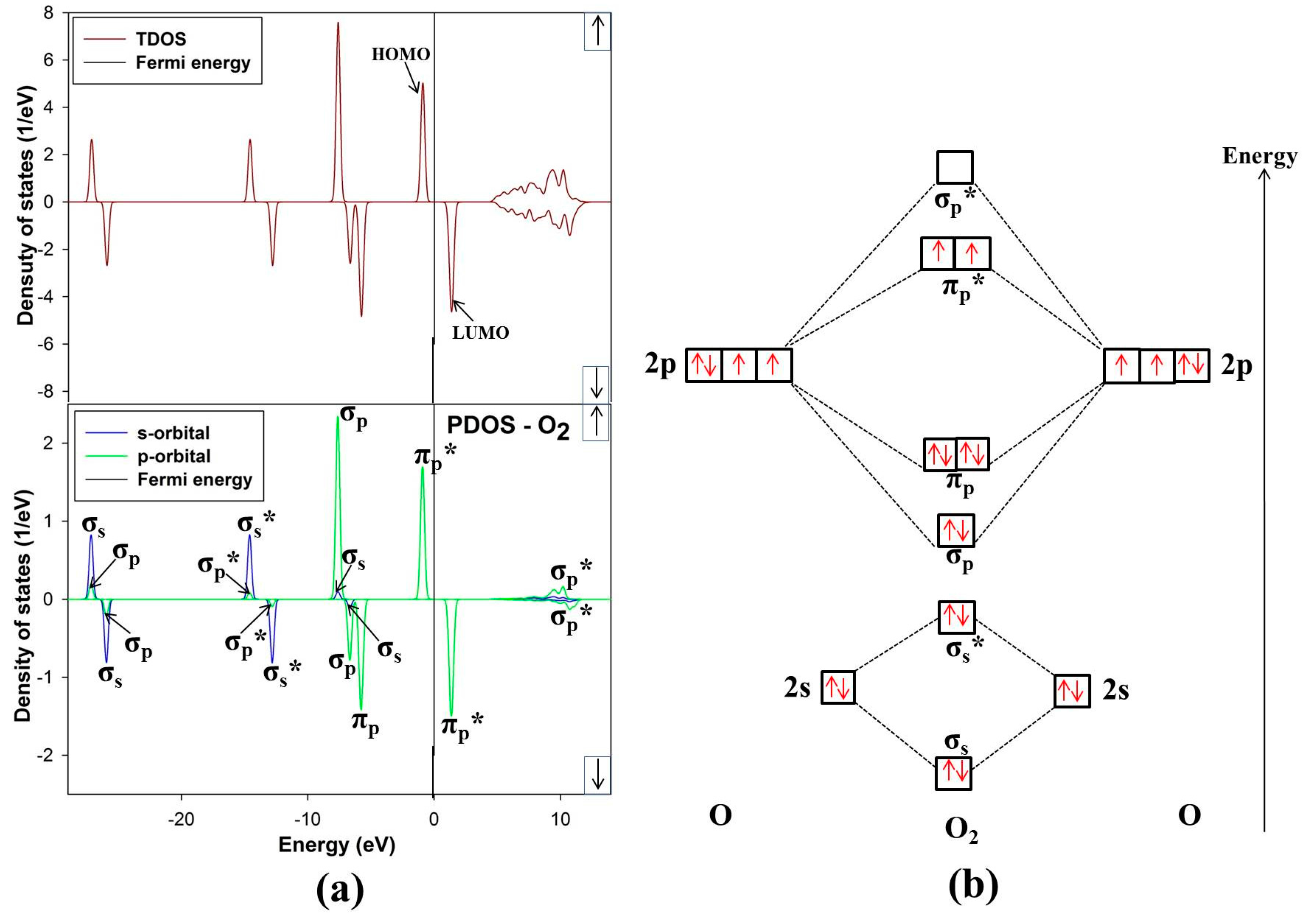
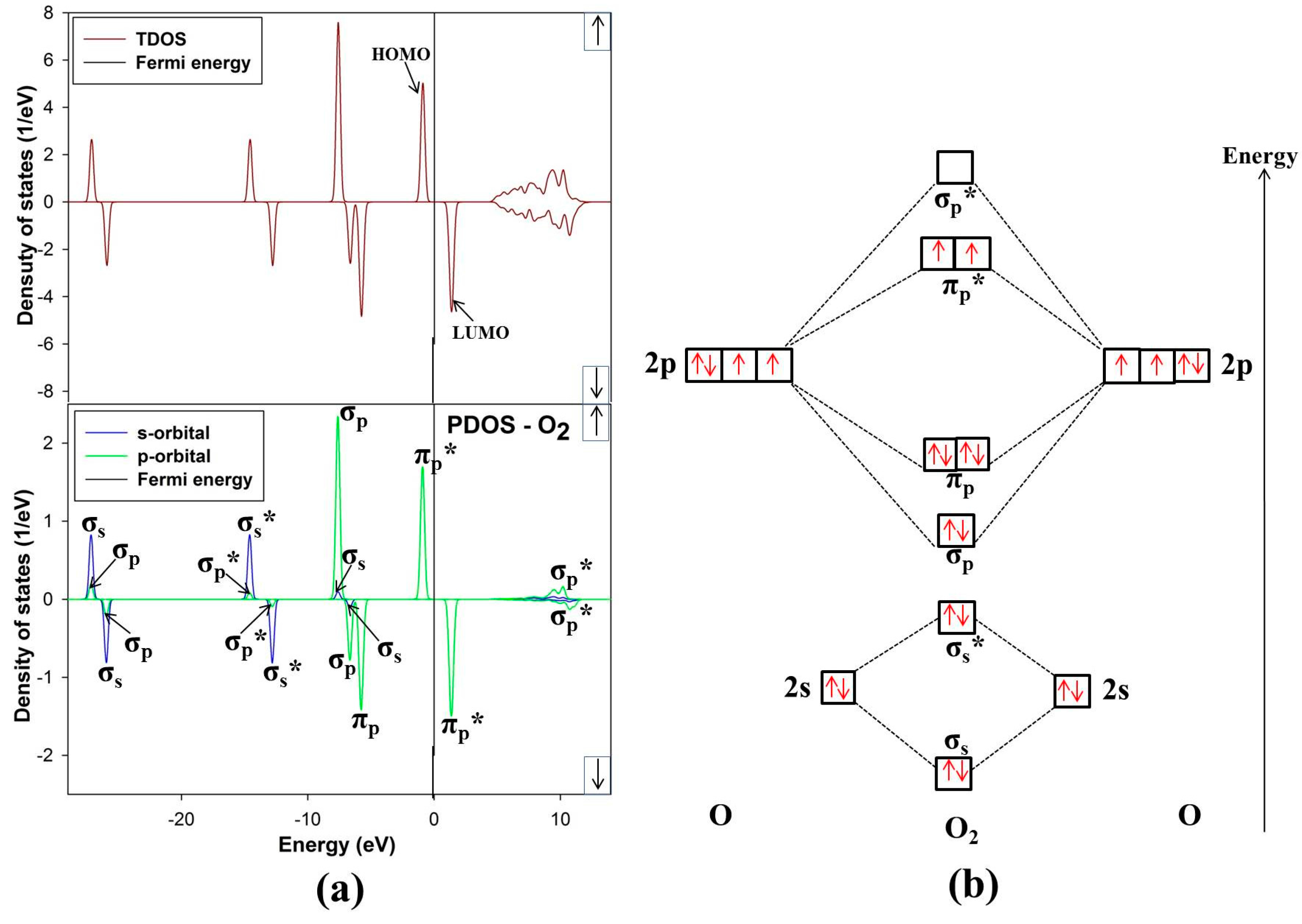
3.3. Free O2 Molecule
The ground state of free O
2 molecule is experimentally found to be a spin triplet state, and the equilibrium bond length is 1.21 Å [
24], this compares well with our predicted value 1.211 Å. In order to get more information about the electronic structure of the oxygen molecule, we analyze the density of states (DOS) of free O
2 molecule in
Figure 6. The energy level of the occupied orbitals of free oxygen molecule shows a band gap between the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO), referred to as HOMO-LUMO (H-L) gap. Our predicted H-L gap is 1.3 eV, this is low compared to the H-L gap of 2.27 eV using PBE [
31]. However, it has been reported that HSEO6 and PBEO hybrid functionals widen the H-L gap [
31]. The current H-L gap suggests that the small value is significant and may suggest that the O
2 will react strongly with the surface. This correlates with previous literature that the chemical reactivity of a molecule is dependent on the H-L gap [
32].
Figure 6.
(a) Total and partial density of states and (b) molecular electron configuration of free O2 molecule.
Figure 6.
(a) Total and partial density of states and (b) molecular electron configuration of free O2 molecule.
The energy levels of the molecular orbitals for O
2 especially that of the
p-orbital LUMO peak, play an important role in the initial adsorption for O
2 on the nickel-rich pentlandite surface. This is so because O
2 accepts electrons from the mineral surface through the LUMO π
p* antibonding [
26].
Figure 6 of free-O
2 clearly illustrates the versatility of O
2. We note that not only σ and π orbitals; bonding and antibonding orbitals are present but also a significant spin splitting of the levels. This clearly shows the significance of Hund’s first rule, since the antibonding spin-up π
p* orbitals are occupied and the spin-down ones are empty reflecting that O
2 molecule in its ground state is a spin triplet. We also noted some
s-p hybridization, particularly for the 2
s derived σ
s and σ
s* orbitals, these observations have been previously reported [
33].
3.4. O2 Adsorption
Now we evaluate the effect of O
2 molecule adsorbed on the (110) surface, considering three distinct adsorption trajectories as shown in
Figure 7. The three sites fcc-hollow, Ni-top, and Fe-top are the possible active adsorption sites. The oxygen molecule is positioned vertical on the surface and the O–O bond length is optimized at each height. In this case the reaction co-ordinate is the height from surface to oxygen molecule. The adsorption strength on interaction of O
2 and the substrate is calculated as,
where E
system is the energy of the surface slab with adsorbate, E
slab is the energy of the slab and E
adsorbate is the energy of the free O
2 molecule. Note that a negative value shows a spontaneous exothermic reaction between the oxygen molecule and the surface, whereas a positive value reveals the opposite.
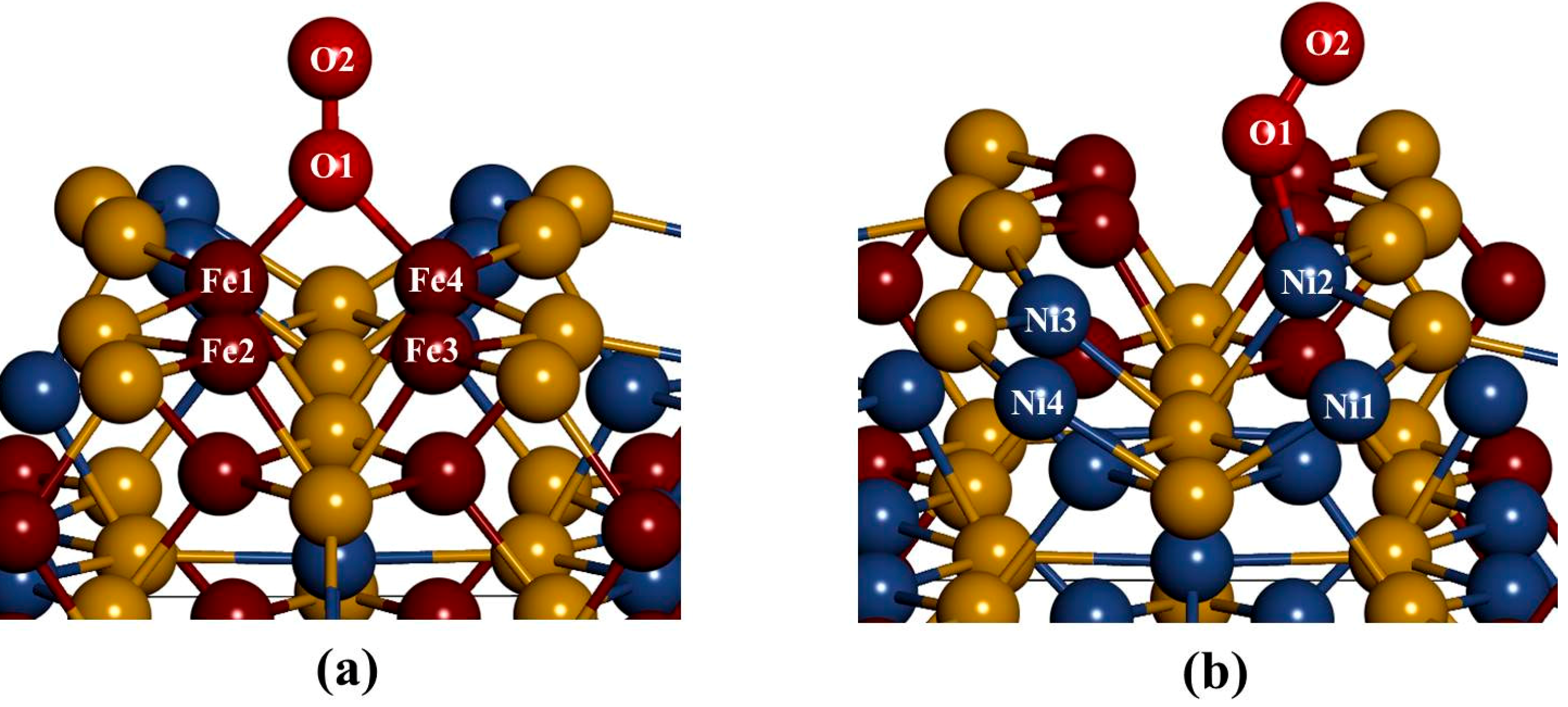
In considering the adsorption of O
2 at fcc-hollow site (
Figure 8a); we noted that the oxygen molecule forms a bridging bond with Fe atoms (Fe1–O
2–Fe4). This observation suggests the preferential oxidation of Fe atoms, and similar results have been previously reported by Merape
et al. [
8]. A bond angle of 84.68° was noted on the bridge bond and compares well with the previous studies of Wang
et al. [
34], where a bond angle of 81.9° was reported. The difference of 2.78 between these two bond angles may be due to the fact that unlike in the current study an oxygen atom was adsorbed instead of the oxygen molecule. This also had an effect on the Fe1–Fe4 bond length, which increased from 2.292 to 2.449 Å. The bond lengths of Fe1–O1 and Fe4–O1 are similar (1.818 Å), which is smaller than the radius between Fe–O of 1.920 Å. This indicated a strong electron overlap across Fe1–O1 and Fe4–O1.
Figure 7.
(a) Top view and (b) side view of the three different adsorption geometries investigated: fcc-hollow, Fe-top, and Ni-top.
Figure 7.
(a) Top view and (b) side view of the three different adsorption geometries investigated: fcc-hollow, Fe-top, and Ni-top.
The adsorption of O
2 on Fe-top site (
Figure 8a) showed similar behavior as observed for fcc-hollow site. However, the resulting bond lengths of Fe1–O1 and Fe4–O1 are different with 1.815 Å and 1.817 Å, respectively (
Table 2). Again these values are smaller than the radius between Fe–O of 1.920 Å, suggesting a strong electron overlap across Fe1–O1 and Fe4–O1. The bond angle of Fe1–O1–Fe4 was found to be 84.96° slightly higher than that of fcc-hollow. The oxygen bond length (O1–O2) for both fcc-hollow site and Fe-top site is found to increase to 1.316 Å, which indicates weakening of the O
2 bond. This corresponds to the report that the back donation of an electron from Fe
d-orbital to oxygen π
p* antibonding molecular orbitals generates the diminishing of the O–O bond and lowers the bond strength of oxygen molecule [
35].
Figure 8.
The relaxed geometries of O2 molecule on the surface: (a) fcc-hallow site and Fe-top site and (b) Ni-top site.
Figure 8.
The relaxed geometries of O2 molecule on the surface: (a) fcc-hallow site and Fe-top site and (b) Ni-top site.
Table 2.
Variation of bond distances between O and metal (M) atoms of the mineral (110) surfaces after adsorption.
Table 2.
Variation of bond distances between O and metal (M) atoms of the mineral (110) surfaces after adsorption.
| Bond Distances | Fcc-Hollow | Fe-Top | Ni-Top |
|---|
| Bonds | Fe–O | Fe–O | Ni–O |
| dₒ = rₒ + rmetal (Å) | 1.920 | 1.920 | 1.840 |
| dads (Å) | 1.818 | 1.815 and 1.817 | 1.801 |
| ∆d = dads − dₒ(Å) | −0.102 | −0.105 and −0.103 | −0.039 |
The adsorption on Ni-top (
Figure 8b) shows that the oxygen molecule bends and forms an orientation of a superoxo. Furthermore, we observed that O
2 moves the Ni atom to the outermost layer, forming a Ni2–O1 bond distance of 1.801 Å, which is smaller than the radius between Ni–O of 1.840 Å. This suggested a strong electron overlap across Ni2–O1. We also found an O1–O2 bond distance of 1.284 Å and a Ni2–O1–O2 bond angle of 128.78°. This observation is in line with the previous studies by Gutsev
at al. [
36]. A Ni superoxo isomer, Ni-O bond lengths of 1.728 Å and O–O of 1.284 Å and a Ni–O–O bond angle of 125.3° were reported. Similarly, Citra
et al. [
37], found Ni–O (1.721 Å), O–O (1.302 Å) and Ni–O–O (123.0°). Also Uzunova
et al. [
38], found Ni–O and O–O of 1.722 Å and 1.272 Å, respectively and a Ni–O–O bond angle of 126.3°. These findings compared well with the current results and confirm a neutral superoxo isomer species forming on the surface.
Table 2 present the change in distances (∆
d) between O atom (of O
2 molecule) and adsorbed-metal atom of the mineral surface (
dads) compared with the sum of the atomic radius of O atom and adsorbed-metal atom (
dₒ). We noted that the distance between O and Fe atoms decrease (−0.102 Å), indicating strong interaction between O
2 molecule and top layer Fe atoms. The distance between O atom and Ni atom also slightly changes (−0.039 Å). These results indicate that Fe oxidize rapidly than Ni atoms.
Table 3 list the surface and adsorption energies of the nickel-rich pentlandite (110) surface. Adsorption of O
2 is found to be an exothermic process (the reaction happens spontaneously). The adsorption strength of O
2 on the surface is found to be stronger on the Fe-top (−1.902 eV) than fcc-hollow site (−1.891 eV) and Ni-top (−0.040 eV). Clearly the Fe-top is more exothermic than fcc-hollow by 0.011 eV, suggesting that some energy is lost in the chemisorption process on fcc-hollow site. It is thus evident that the adsorption energy of Fe atoms is more spontaneous than on Ni atoms, confirming the preferential oxidation of Fe.
Table 3.
Calculated surface and adsorption energies.
Table 3.
Calculated surface and adsorption energies.
| Surface | Surface Energy | Adsorption (Ads.) Energies |
|---|
| E(surface) (eV/Å2) | E(Ads.) (eV) |
|---|
| (110) | 0.061 | fcc-hollow | Ni-top | Fe-top |
| −1.891 | −0.040 | −1.902 |
3.5. Electronic Structure of O2 Adsorbed Surface
The density of states has been calculated to describe the interaction between the O
2 molecule and the nickel-rich pentlandite mineral (110) surface (
Figure 9a,b). Note that we only discuss the DOS for adsorption on Fe-top and Ni-top, since the fcc-hollow showed similar behavior as the Fe-top site. The TDOS is characterized by three spin up/down broad peaks at the VB at −2.0, −5.0 and −14.0 eV, where the −2.0 eV peaks clearly emanate from 3
d hybridization with O
2 π
p* orbital. A clear distinction is noticed on the PDOS depicting effects of O
2 adsorption on metal atoms. The LUMO π
p* moves to the VB, clearly illustrated that the adsorption of O
2 on the surface is initiated by accepting electrons from the mineral surface into the LUMO π
p* antibonding orbital. This charge transfer is in the form of back-donation of electrons from the mineral surface
d-orbital to the π
p* anti-bonding orbital, confirming report by Blyholder
et al. [
39].
In
Figure 9a, the PDOS for Fe display three dominant spin up/down peaks near E
F at the VB, while only one peak for Ni2
d-orbital contribution is observed in
Figure 9b. This may suggest strong hybridization of Fe 3
d with O 2
p orbital and less for Ni 3
d. It is also clear that the M-bonded oxygen (O1) and terminal oxygen (O2) PDOS are different for Fe and Ni interaction. The Fe–O
2 interaction shows a single peak of σ
p-orbital (equal states for both O1 and O2) and double degenerate π
p-orbital. The Ni–O
2 interaction shows a single peak of σ
p-orbital and a single degenerate π
p-orbital. More importantly for Fe–O
2, the O1 π
p-orbital peak is two degenerate and becomes reduced for the case of O2 π
p-orbital peak. The single σ
p-orbital peak becomes dominant for O2 PDOS. This behavior is different for Ni and O interaction (
Figure 9b), displaying singlet degenerate π
p and σ
p peaks in the VB. However, σ
p and π
p orbital shows strong overlap for O1.
The other contribution peaks are noticed at energies −25.0 and −16.0 eV for Fe-top and −25.0 and −15.0 for Ni-top, which are of σ
s and σ
s* character, respectively. Furthermore, O2 contribution for both case are also distinct, a sharp π
p* peak and broad π
p* (reduced states) peak are observed at about −2.0 eV for Fe and Ni, respectively. The PDOS on O1 is observed to have the π
p* peaks reduced to zero states at −2.0 eV, while the σ
p* peaks reside just above the E
F and is half-occupied at the E
F for Fe–O
2 and Ni–O
2, respectively. The half occupancy of the σ
p* orbital have been observed previously on O
2 adsorption on Al(111) surface [
40].
Figure 9.
The density of states (DOS) TDOS and PDOS: (a) adsorption on Fe-top site and (b) adsorption on Ni-top site.
Figure 9.
The density of states (DOS) TDOS and PDOS: (a) adsorption on Fe-top site and (b) adsorption on Ni-top site.
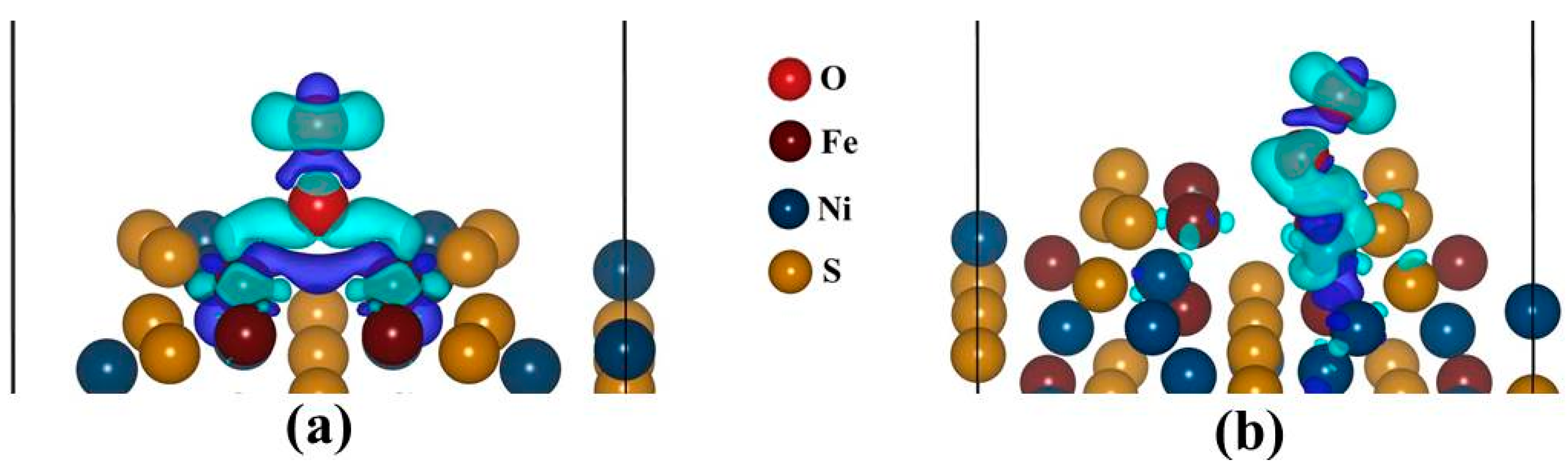
Figure 10a,b display the charge density difference, for the Fe-top and Ni-top adsorption sites. We observed that the Fe and Ni atoms donate electrons while O
2 accept. This is due to a formation of negative electronic cloud (charge depletion) while the O
2 has a positive electronic cloud (charge accumulation). The cylindrical shape around O2 atom represents the π
p* orbital, as observed from the PDOS and it signifies a superoxo bonding mode. Similar observation has been formed in the studies by Yoon
et al. [
41].
Figure 10.
The sketch of isosurface charge density difference: (a) Fe-top site (isosurface level = 0.004 eÅ−3) and (b) Ni-top site (isosurface level = 0.003 eÅ−3). Cyan represents positive electronic clouds which accept electrons and blue stands for negative electronic clouds which donate electrons.
Figure 10.
The sketch of isosurface charge density difference: (a) Fe-top site (isosurface level = 0.004 eÅ−3) and (b) Ni-top site (isosurface level = 0.003 eÅ−3). Cyan represents positive electronic clouds which accept electrons and blue stands for negative electronic clouds which donate electrons.
Now, we use Bader analysis to examine the electron transfer from the surface Fe and Ni atoms to the oxygen molecule, in the fcc-hollow, Fe-top and Ni-top sites. Interestingly, we found that in all cases the oxidized Fe and Ni atoms adopts more positive charge. For fcc-hollow: Fe1 = Fe4 = +0.72|e|, Fe-top: Fe1 = +0.72|e| and Fe4 = +0.71|e| and for Ni-top: Ni2 = +0.64|e|, which indicates that an electron has been donated to the O
2 molecule. This is due to a negative electronic cloud (charge depletion) on the Fe and Ni atoms while the O
2 has a positive electronic cloud (charge accumulation), as observed from the charge density difference in
Figure 10a,b. The cylindrical shape around O2 atom, represent the π
p* orbital and this signifies a superoxo bonding mode, similarly to the PDOS observation (
Figure 9a,b). The oxygen molecule possesses negative charges: for fcc-hollow (O1 = −0.48|e| and O2 = −0.18|e|), Fe-top (O1 = −0.48|e| and O2 = −0.15|e|) and for Ni-top (O1 = −0.25|e| and O2 = −0.12|e|). It is clear that the terminal oxygen (O2) has a smaller charge than the M-bonded oxygen atom (O1). This charge distribution behavior for superoxide has been previously reported [
38]. The charge on O
2 (
i.e., charge sum of O1 and O2), for lowest ground state are as: for fcc-hollow (−0.66|e|), for Fe-top (−0.63|e|) and for Ni-top (−0.37|e|). It clearly shows that O
2 on Fe atoms possesses more negative charge than on Ni atom. The negative charge on the oxygen atoms is also confirmed by experiment for superoxo or end-bonded O
2 [
42].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}