Evolution of Geochemical and Mineralogical Parameters during In Situ Remediation of a Marine Shore Tailings Deposit by the Implementation of a Wetland Cover

Abstract
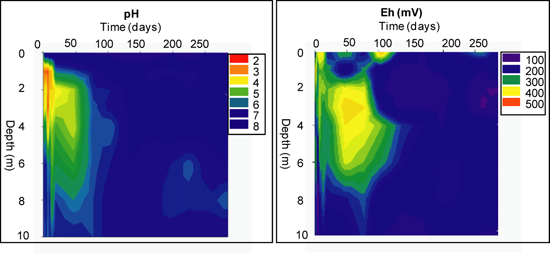
:
1. Introduction
2. Experimental Section
2.1. Preparation of the Remediation Cell
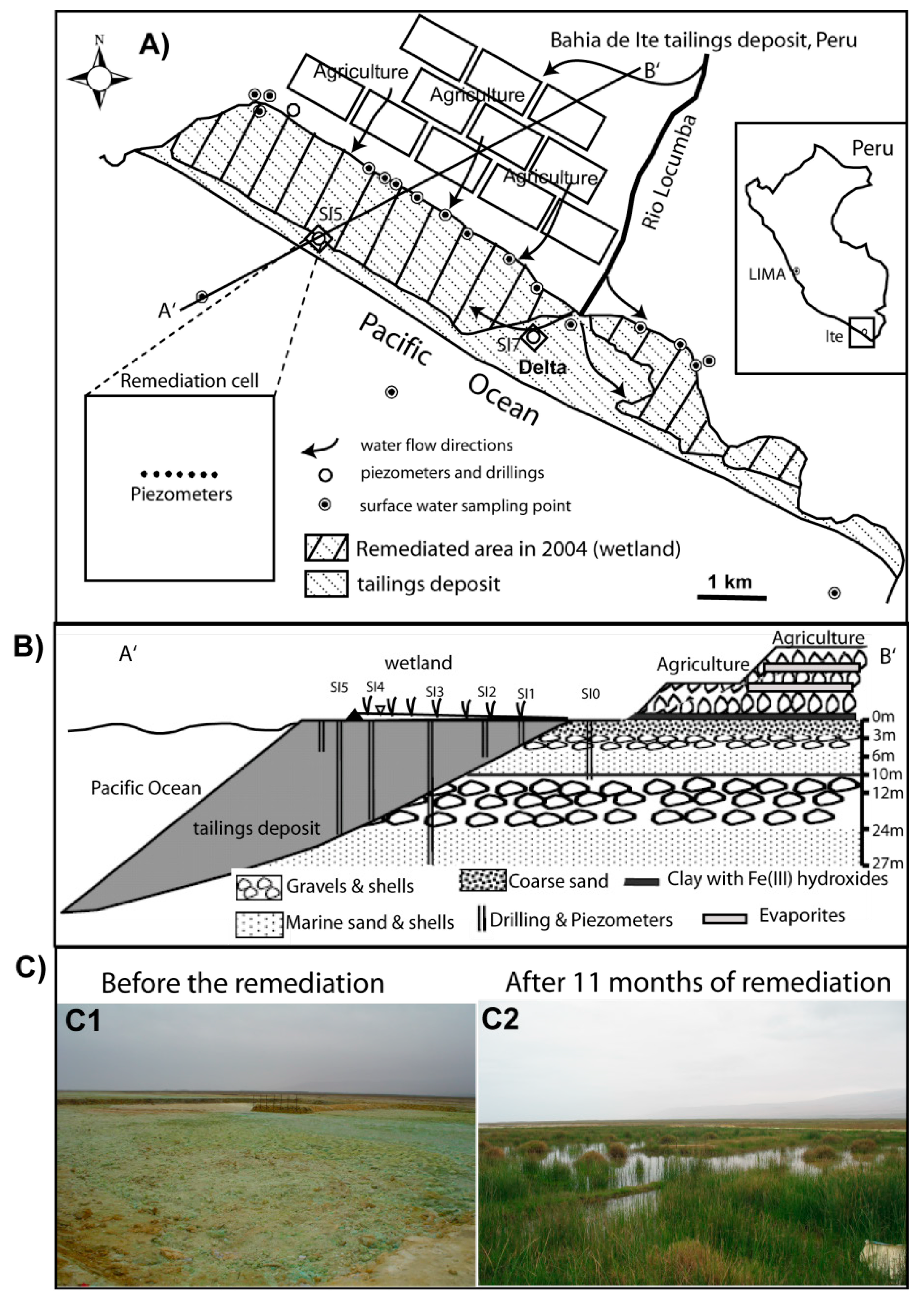

{kind=link}
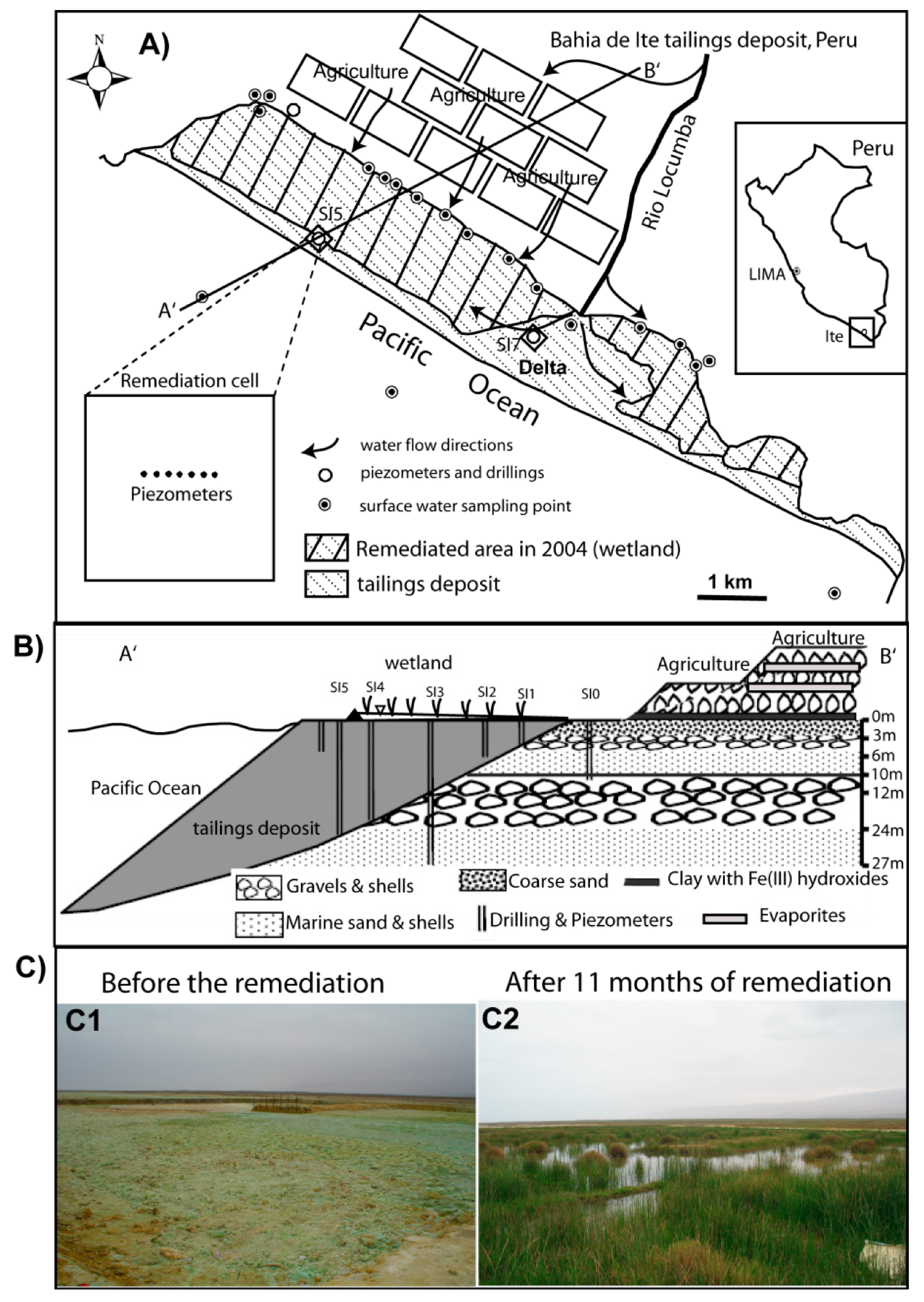{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
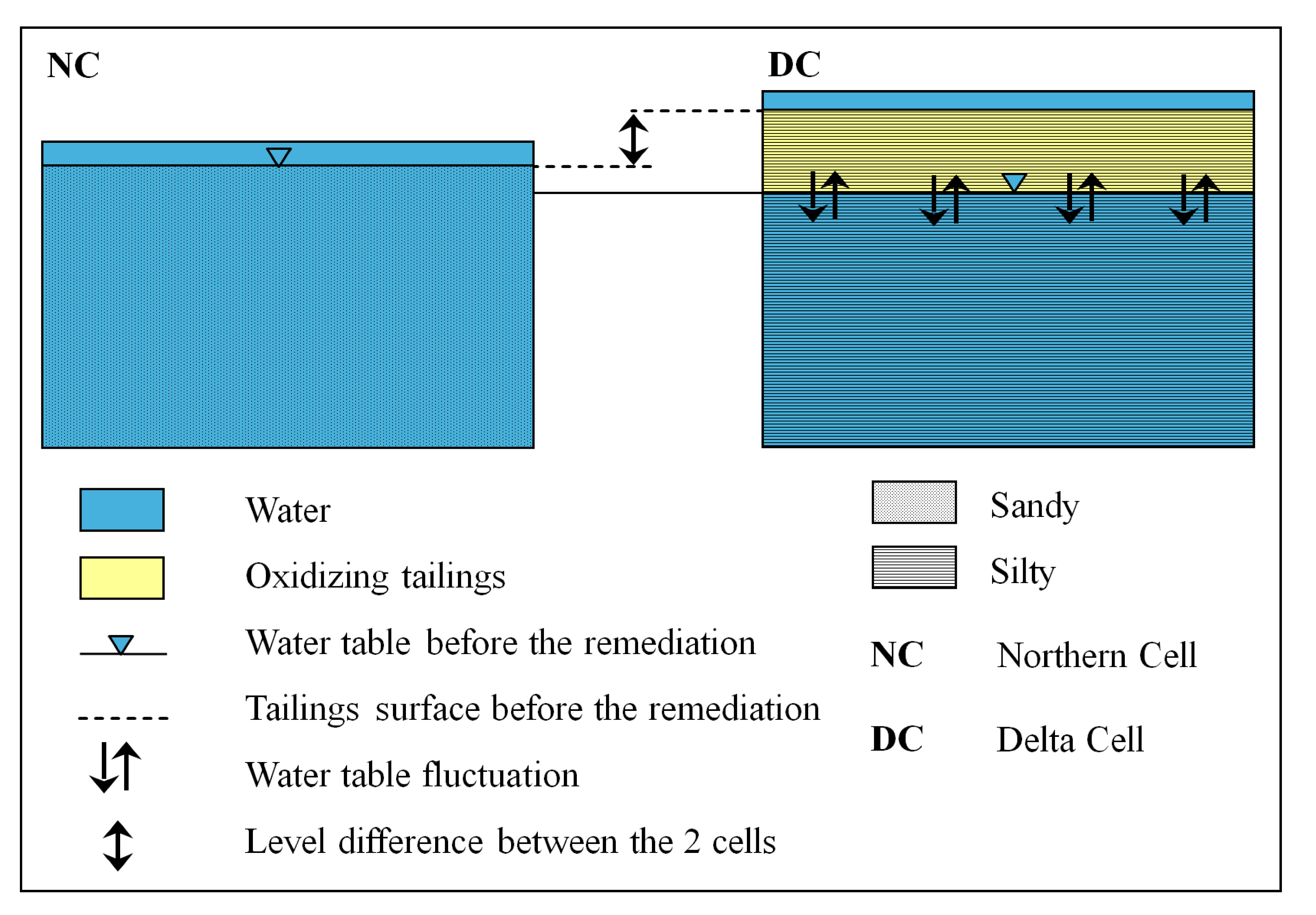{kind=link}
| Characteristics | Northern Cell (NC) | Delta Cell (DC) | |
|---|---|---|---|
| Grain size | Sandy | Clayey–silty | |
| Groundwater level | Groundwater level at 1 m | Groundwater level at 1.5 m | |
| Location | Close to shore line | Close to river inflow | |
| Altitude | Sea water level | 2 m above sea water level | |
| Characteristics of water used for remediation | Origin | From the old wetland | From Locumba river |
| pH | 7.6 | 7.3 | |
| Eh | 341 | 420 | |
| Alkalinity (mg/L CaCO3) | 241 | 133 | |
| DOC (mg/L) | 8.36 | 2.6 | |
| Fe total (mg/L) | 0.08 | <0.005 | |
| FeII (mg/L) | <0.1 | <0.1 | |
| Al (mg/L) | 0.05 | 0.1 | |
| Cu (mg/L) | 0.12 | 0.03 | |
| Ni (mg/L) | <0.02 | <0.02 | |
| Mn (mg/L) | 0.39 | <0.01 | |
| Mg (mg/L) | 100 | 37.4 | |
| K (mg/L) | 56 | 29.1 | |
| Zn (mg/L) | <0.04 | <0.04 | |
| As (mg/L) | 0.07 | 0.27 | |
| Mo (mg/L) | 0.06 | <0.04 | |
| Cl (mg/L) | 2080 | 433 | |
| Ca (mg/L) | 274 | 141 | |
| Na (mg/L) | 1555 | 300 | |
| SO4 (mg/L) | 1636 | 429 | |
2.2. Sampling
2.3. Mineralogical Analysis
2.4. Grain Size Analysis
2.5. Aquatic Geochemistry
2.6. Sequential Extractions
3. Results and Discussion
3.1. Stratigraphy and Mineralogy before Remediation
3.2. Geochemical Evolution of the Wetland
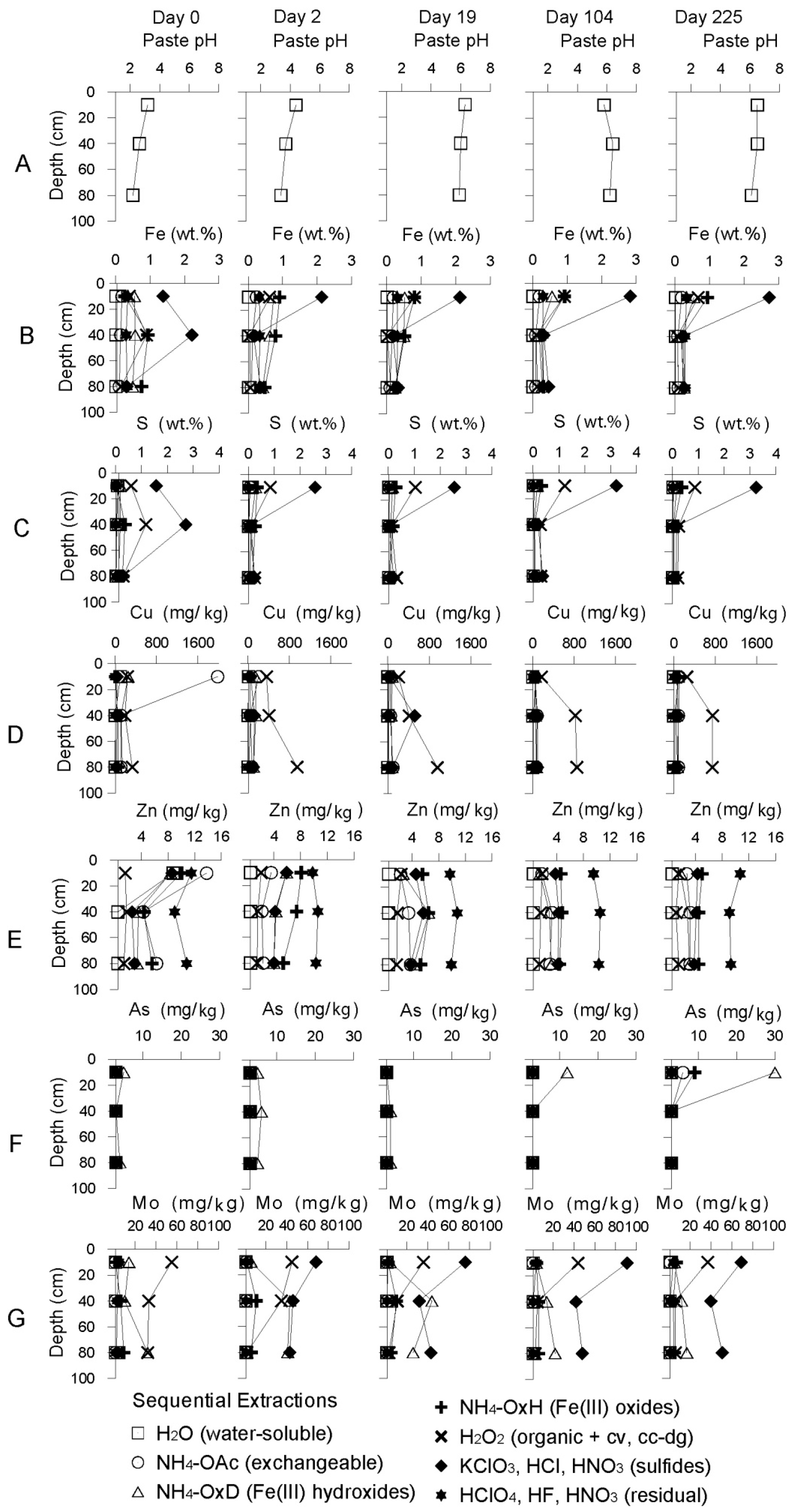
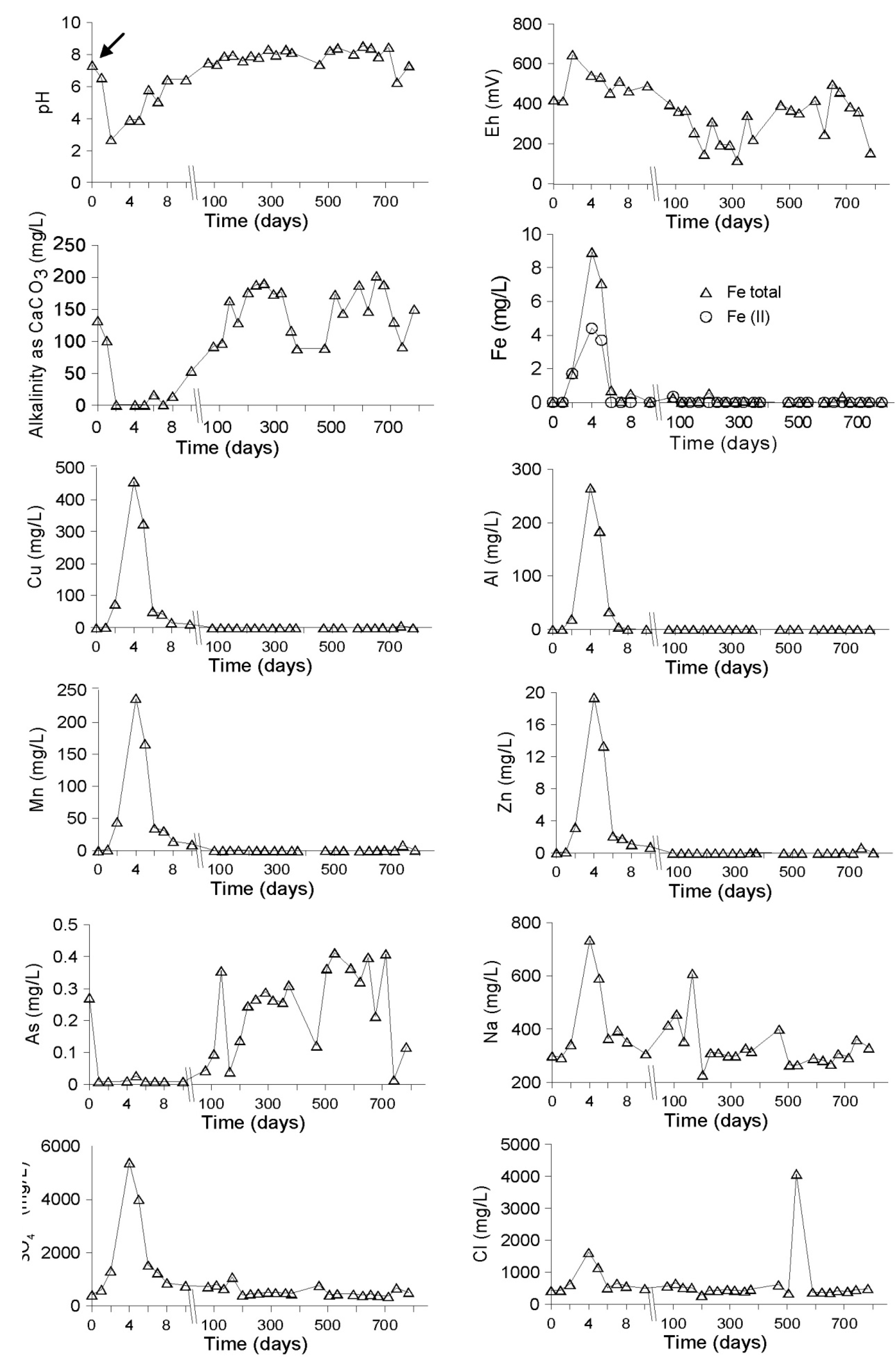
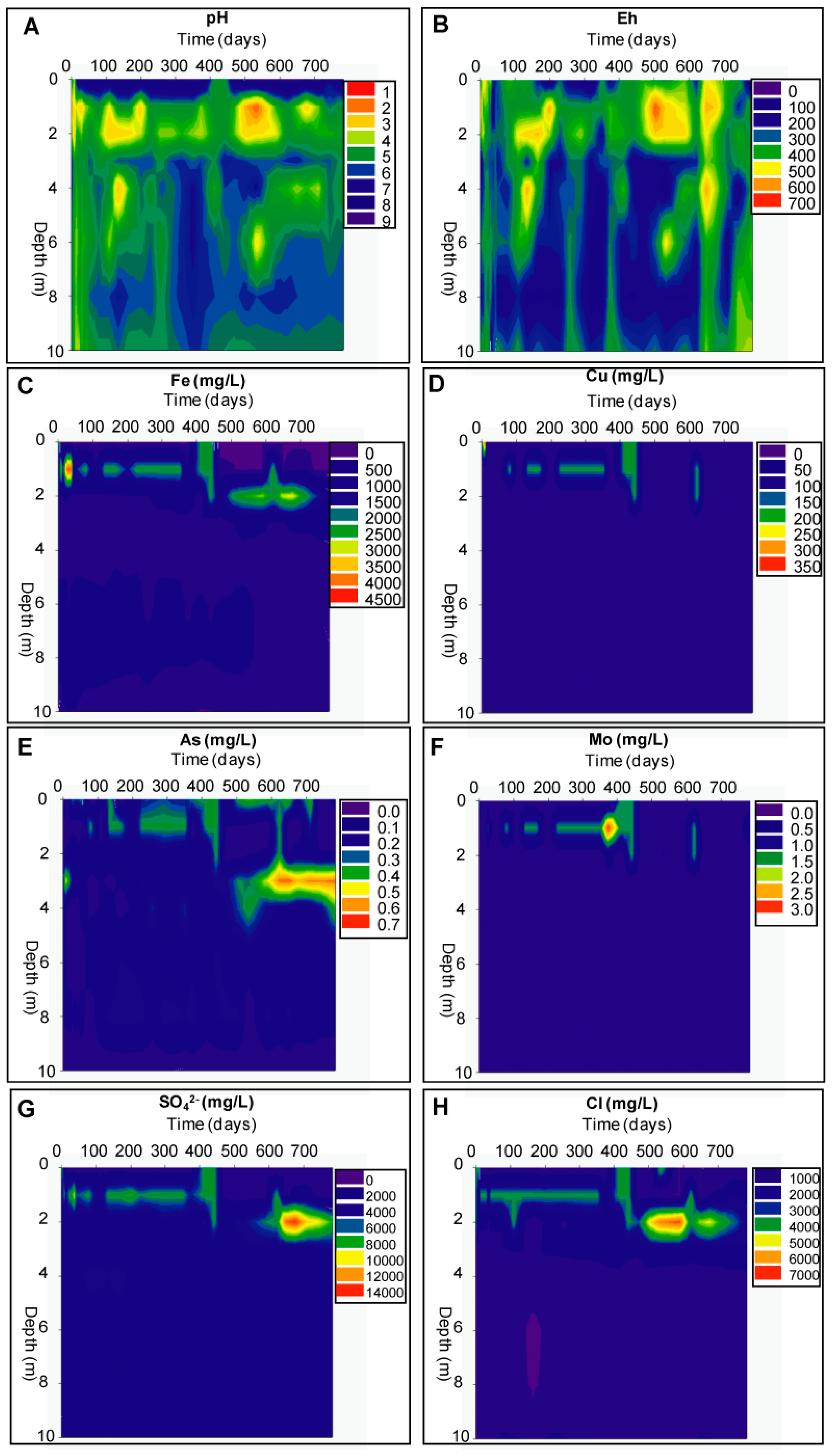
3.2.1. The Sandy Northern Remediation Cell (NC)
Starting Conditions before Remediation
| Samples | Depth | pH | EC | Eh | Alkalinity | Al | As | Ca | Cl | Co |
|---|---|---|---|---|---|---|---|---|---|---|
| (m) | (ms/cm) | (mV) | (mg/L CaCO3) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | ||
| SI5P1 | 1 | 3.1 | 7.63 | 358 | <1.5 | 104 | 0.02 | 332 | 2125 | 0.89 |
| SI5P2 | 2 | 4.0 | 7.20 | 342 | <1.5 | 97.2 | 0.15 | 400 | 2085 | 0.74 |
| SI5P3 | 3 | 4.4 | 8.00 | 253 | <1.5 | 51.1 | 0.12 | 591 | 2245 | 0.53 |
| SI5P4 | 4 | 5.4 | 6.15 | 274 | 30.2 | 0.25 | <0.02 | 343 | 1830 | 0.04 |
| SI5P6 | 6 | 6.5 | 6.25 | 286 | 105 | <0.05 | <0.02 | 223 | 1875 | <0.02 |
| SI5P8 | 8 | 6.4 | 7.55 | 222 | 116.5 | <0.05 | <0.02 | 243 | 2655 | <0.02 |
| SI5P10 | 10 | 6.5 | 10.6 | 256 | 112 | <0.05 | 0.05 | 307 | 3860 | 0.06 |
| Samples | Depth | Cu | Fe | K | Mg | Mn | Mo | Na | SO4 | Zn |
| (m) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | |
| SI5P1 | 1 | 329 | 176 | 21.0 | 308 | 80.1 | 0.05 | 1748 | 4167 | 12.9 |
| SI5P2 | 2 | 229 | 154 | 57.5 | 267 | 64.2 | 0.35 | 1674 | 3807 | 10.4 |
| SI5P3 | 3 | 140 | 242 | 71.2 | 358 | 23.7 | 0.32 | 1814 | 4581 | 5.98 |
| SI5P4 | 4 | 3.57 | 10.6 | 53.3 | 197 | 1.23 | 0.07 | 1449 | 2124 | 0.22 |
| SI5P6 | 6 | 0.17 | 1.13 | 55.9 | 137 | 0.37 | <0.04 | 1774 | 2162 | 0.05 |
| SI5P8 | 8 | 0.03 | 5.7 | 61.0 | 129 | 0.88 | <0.04 | 2245 | 2174 | 0.05 |
| SI5P10 | 10 | 0.10 | 54.1 | 75.6 | 157 | 5.57 | <0.04 | 3167 | 2861 | 0.75 |
Evolution of the Aquatic Geochemistry in the Surface Waters of the Northern Remediation Cell (NC)
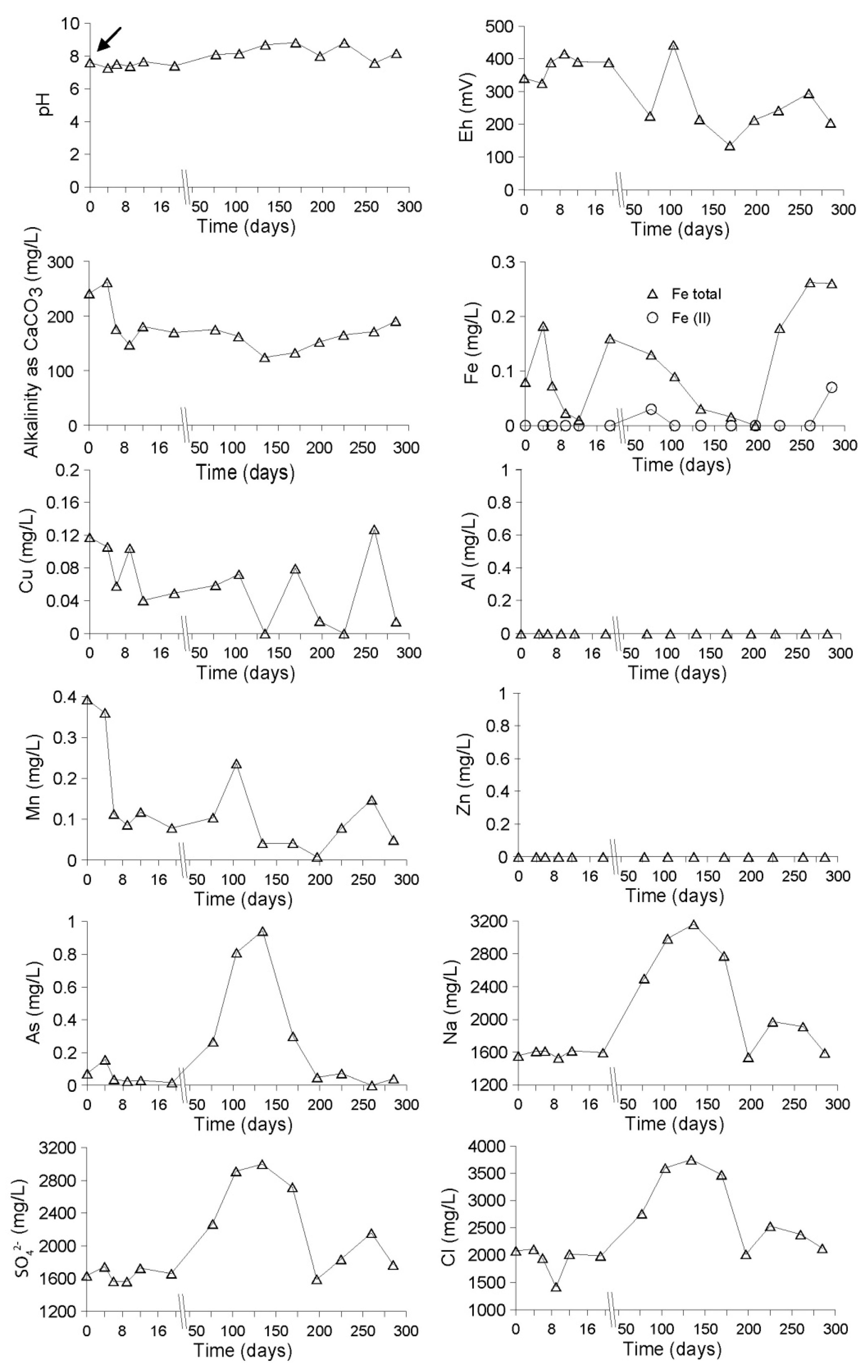
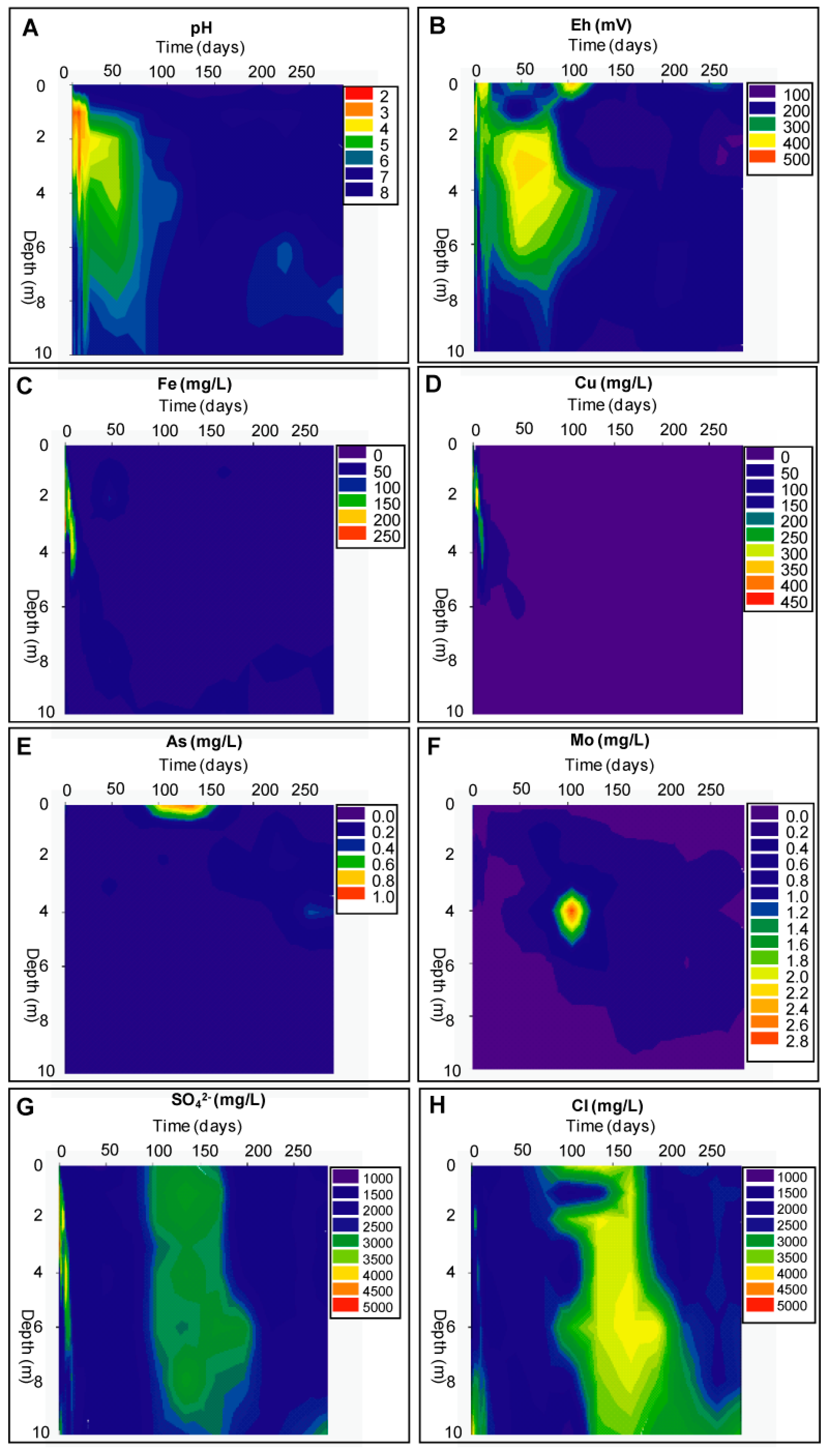
Geochemical Evolution in the NC Tailings Stratigraphy over Time
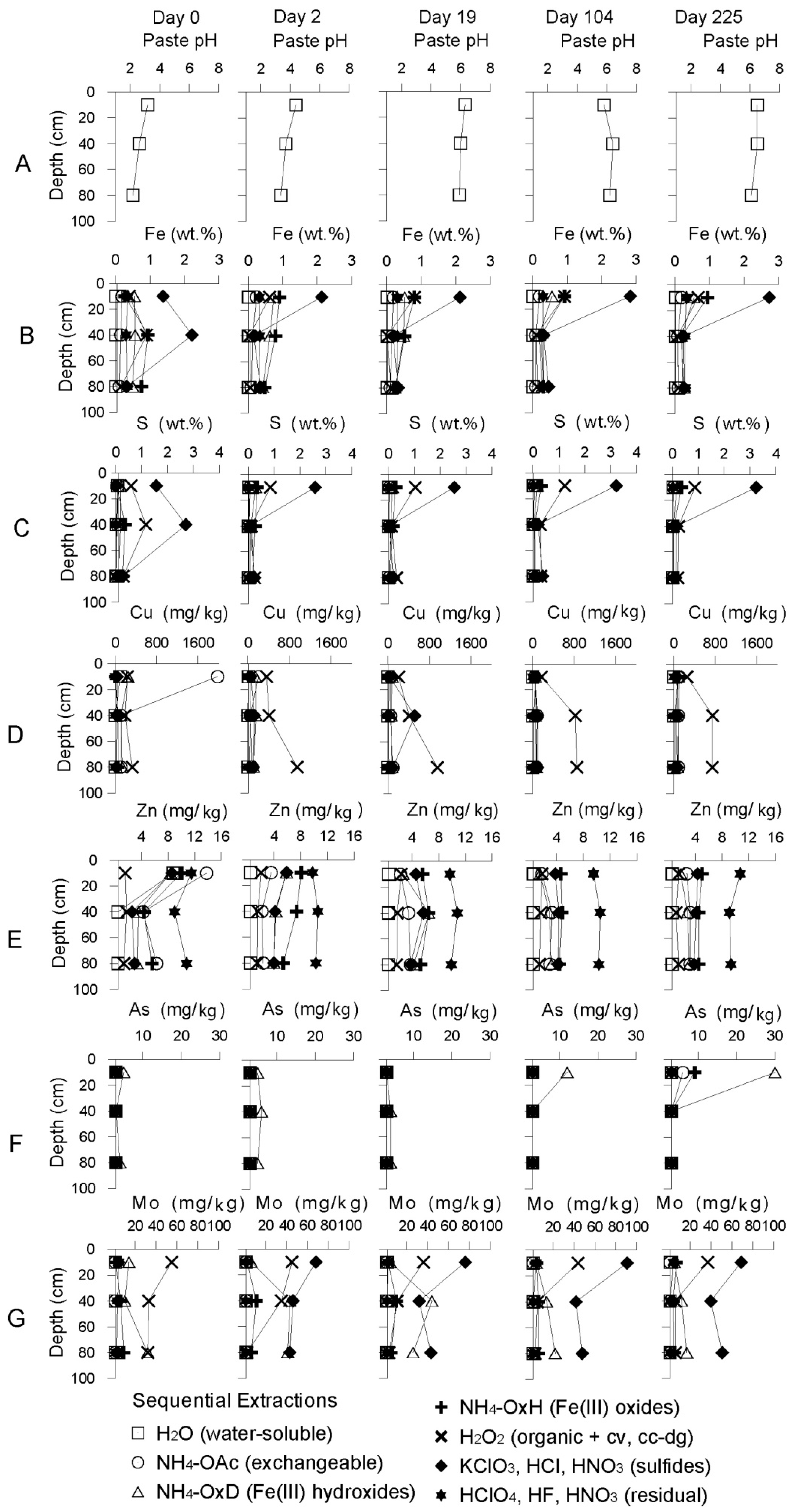
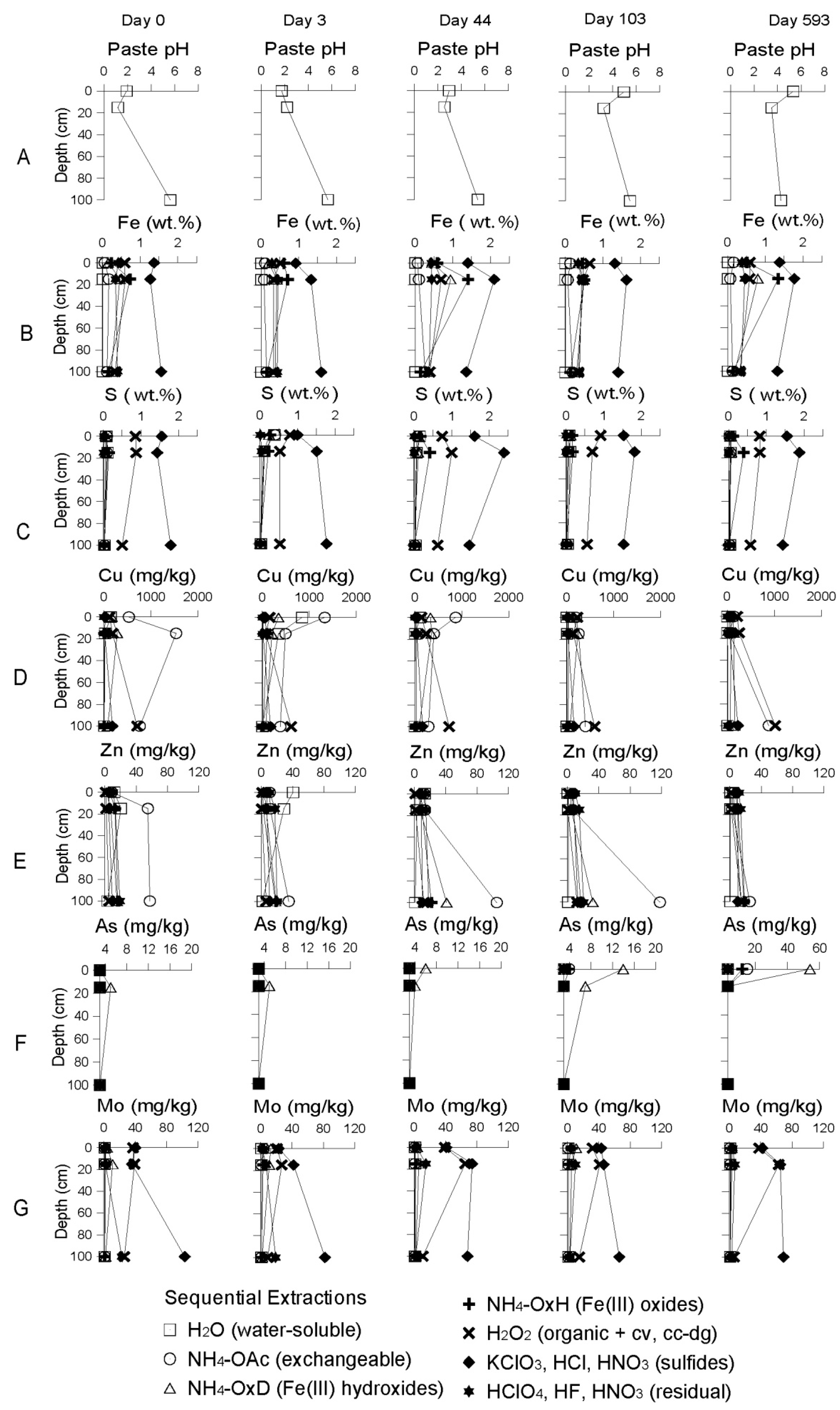
3.2.2. The Delta Area Remediation Cell (DC)
Starting Conditions before the Remediation
| Samples | Depth | pH | EC | Eh | Alkalinity | Al | As | Ca | Cl | Co |
|---|---|---|---|---|---|---|---|---|---|---|
| (m) | (ms/cm) | (mV) | (mg/L CaCO3) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | ||
| SI7SI | 0.2 | 3.0 | 23.50 | 578 | <1.5 | 4448 | 0.15 | 980 | N/A | 9.80 |
| SI7SII | 0.55 | 3.1 | 5.11 | 566 | <1.5 | 1653 | 0.10 | 466 | N/A | 5.26 |
| SI7P1 | 1 | 3.1 | 2.49 | 533 | <1.5 | 3.72 | 0.02 | 552 | N/A | 0.37 |
| SI7P2 | 2 | 4.2 | 6.05 | 410 | <1.5 | 0.87 | 0.13 | 551 | N/A | 0.15 |
| SI7P3 | 3 | 6.2 | 7.01 | 105 | 165 | <0.05 | 0.25 | 632 | 1,009 | BDL |
| SI7P4 | 4 | 6.0 | 6.60 | 243 | 105.15 | <0.05 | 0.15 | 539 | 545 | 0.44 |
| SI7P6 | 6 | 5.4 | 5.00 | 305 | 13.8 | <0.05 | 0.16 | 380 | 476 | 0.68 |
| SI7P8 | 8 | 5.7 | 5.20 | 265 | 62.95 | 1.91 | 0.10 | 404 | 510 | 0.73 |
| SI7P10 | 10 | 5.2 | 6.11 | 359 | 38.7 | <0.05 | <0.002 | 517 | 965 | 1.37 |
| Samples | Depth | Cu | Fe | K | Mg | Mn | Mo | Na | SO4 | Zn |
| (m) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | (mg/L) | |
| SI7SI | 0.2 | 1,035 | 2,299 | 3.58 | 6,138 | 716 | <0.04 | 1,650 | 33,270 | 213 |
| SI7SII | 0.55 | 335 | 2,001 | 0.91 | 1,631 | 132 | <0.04 | 319 | 19,560 | 10.1 |
| SI7P1 | 1 | 0.26 | 703 | 34.2 | 276 | 62.5 | <0.04 | 692 | 3,483 | 1.20 |
| SI7P2 | 2 | 1.37 | 615 | 87.9 | 120 | 13.2 | <0.04 | 733 | 2,554 | 2.53 |
| SI7P3 | 3 | <0.005 | 146 | 78.9 | 256 | 12.8 | 0.05 | 634 | 2,635 | 0.07 |
| SI7P4 | 4 | <0.005 | 383 | 89.9 | 393 | 54.8 | <0.04 | 399 | 3,612 | 10.1 |
| SI7P6 | 6 | 0.02 | 598 | 52.5 | 204 | 28.8 | <0.04 | 325 | 2,835 | 5.43 |
| SI7P8 | 8 | 0.28 | 634 | 58.7 | 193 | 33.9 | <0.04 | 377 | 2,873 | 3.16 |
| SI7P10 | 10 | <0.005 | 412 | 107 | 345 | 69.4 | <0.04 | 727 | 3,483 | 7.63 |
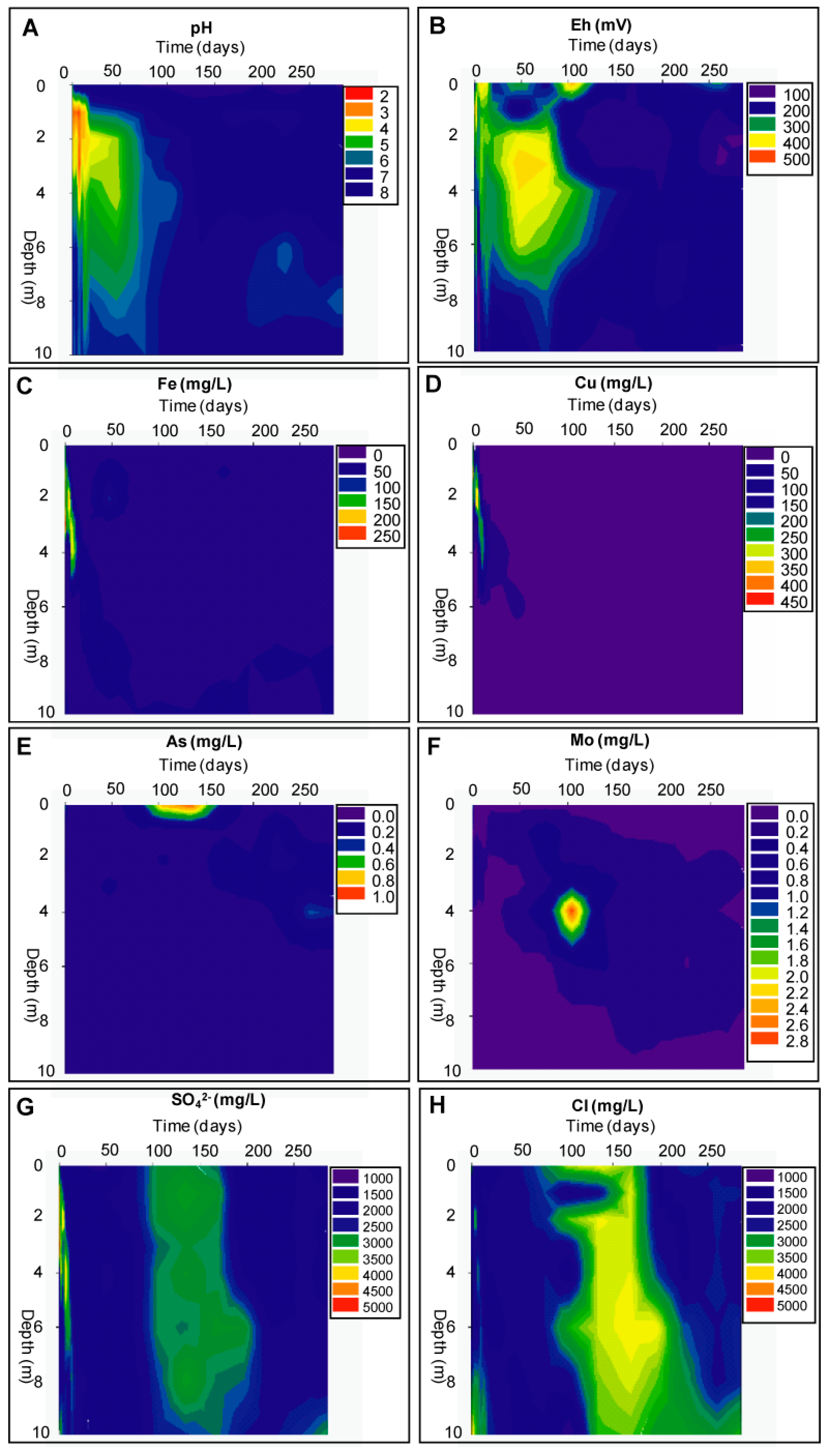
Evolution of the Aquatic Geochemistry in the Surface Waters of the Remediation Cell DC
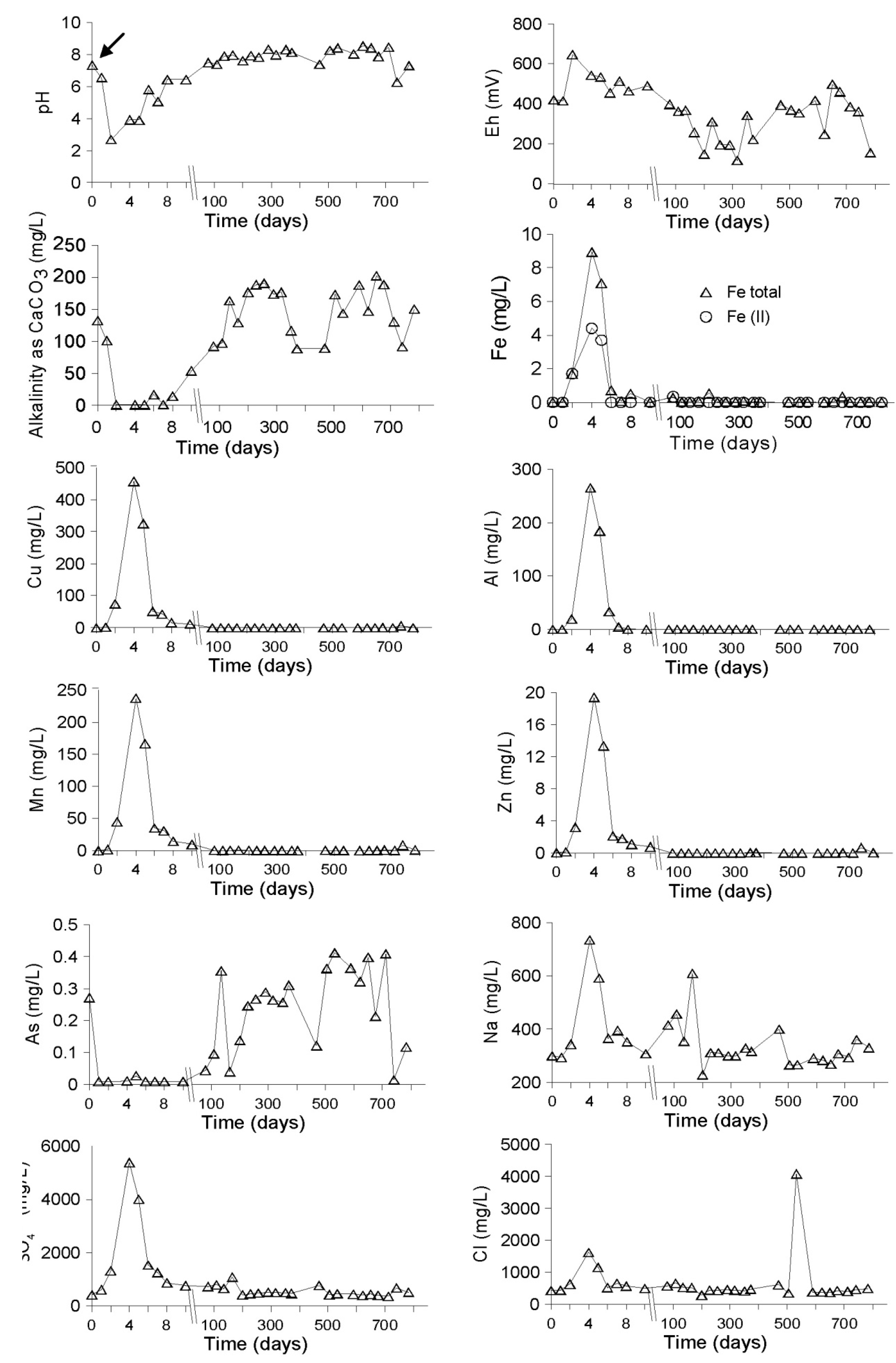
Geochemical Evolution in the DC Tailings Stratigraphy over Time
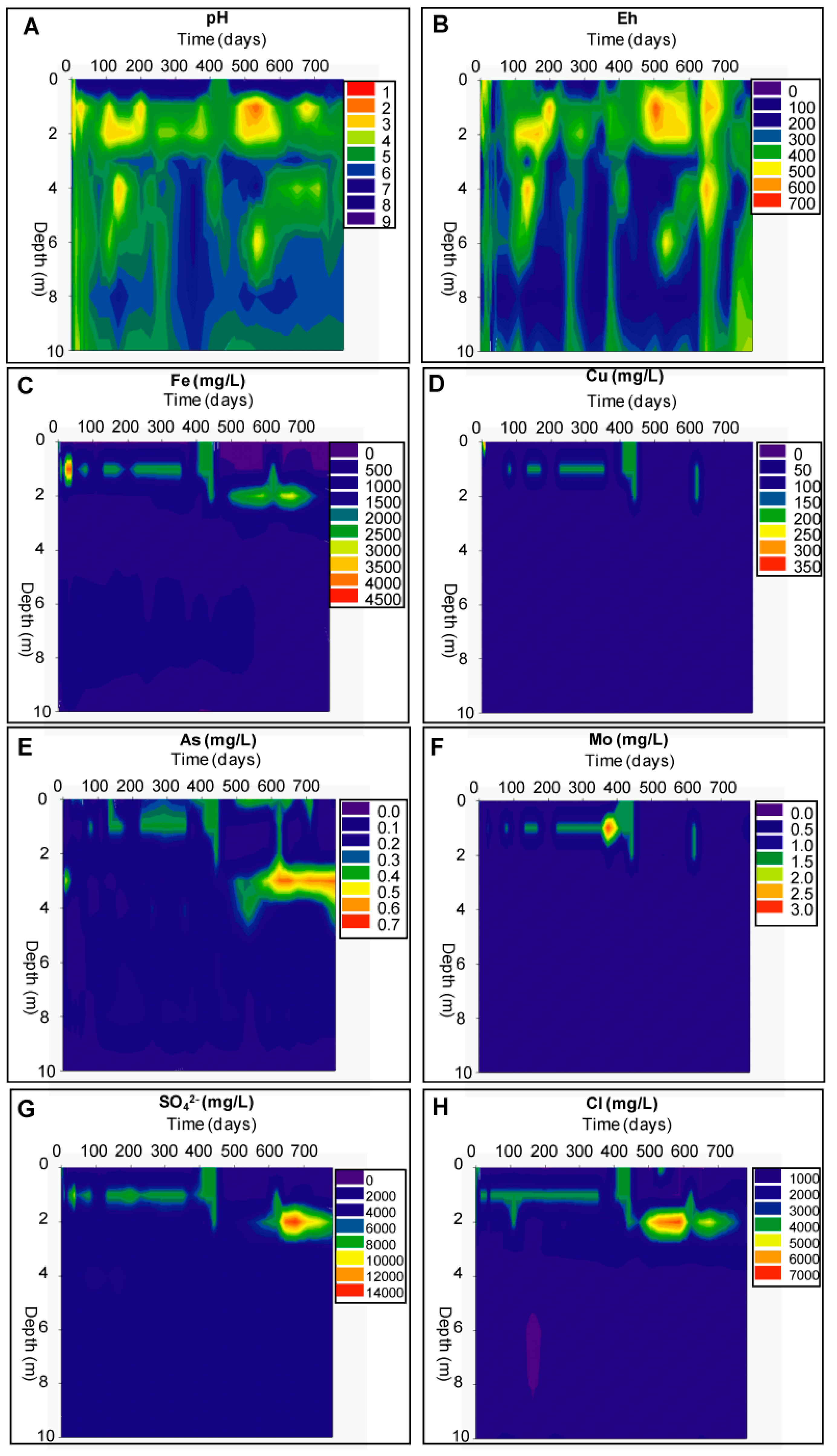
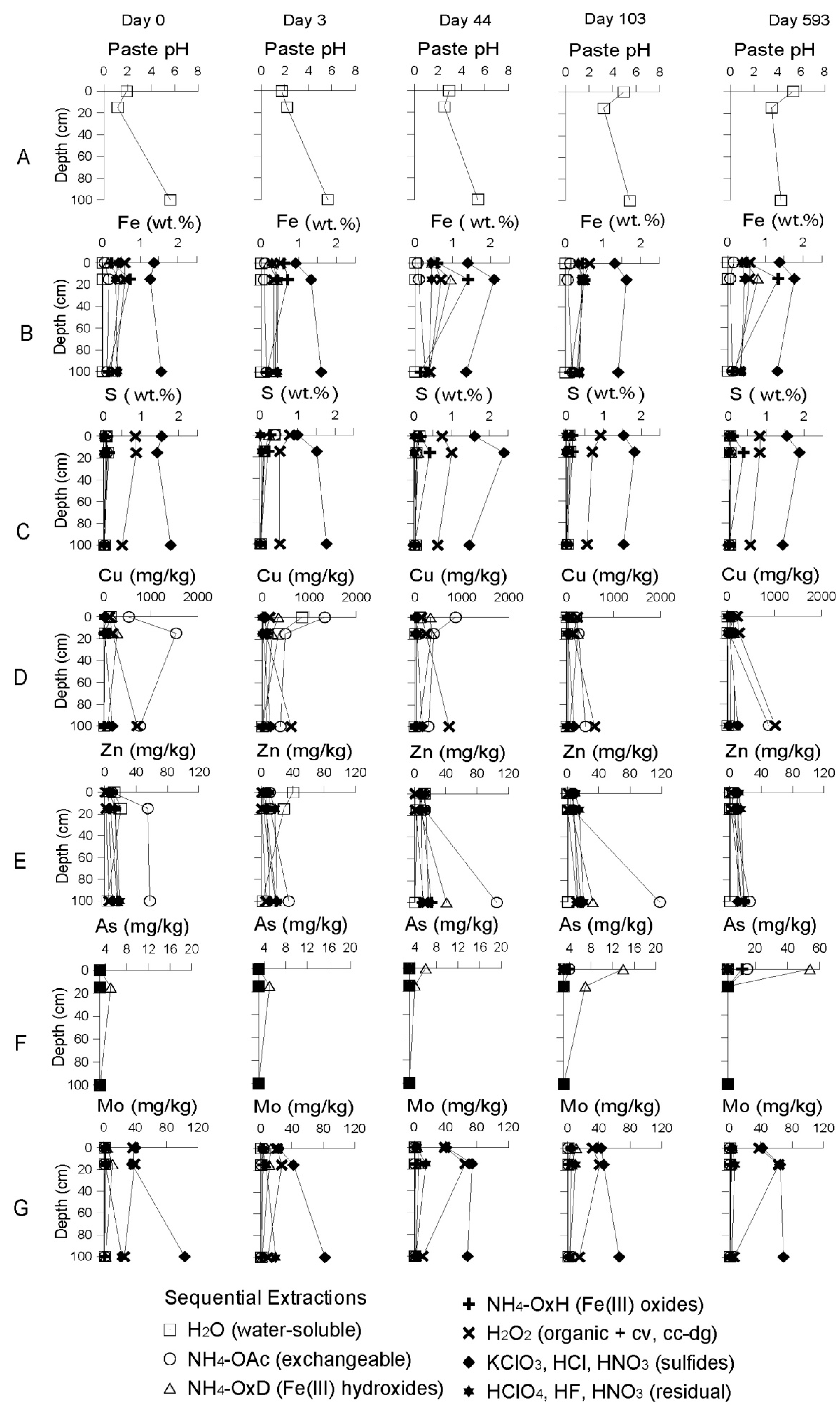
4. Conclusions
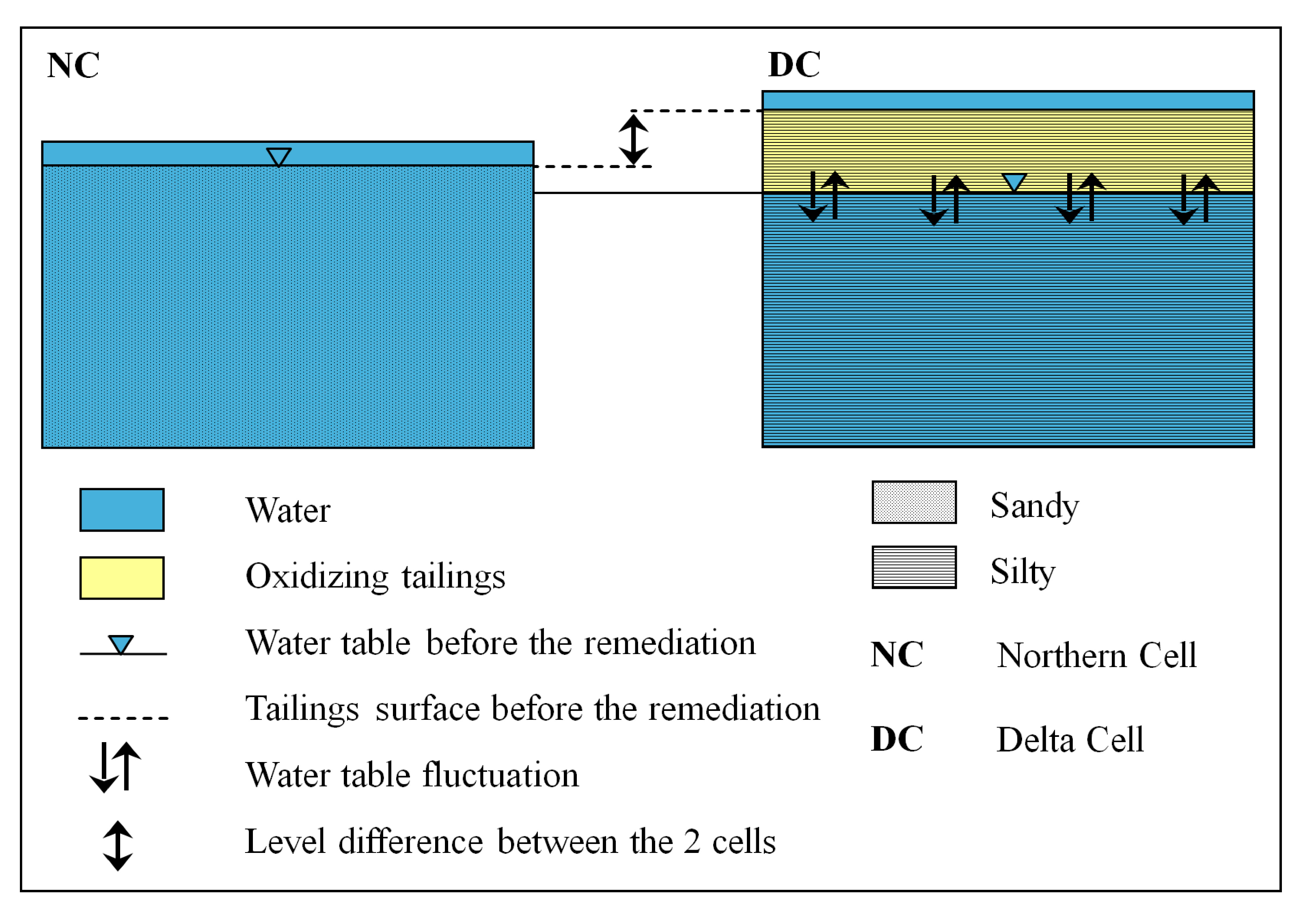
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nordstrom, D.K.; Alpers, C.N. Geochemistry of Acid Mine Waste. In The Environmental Geochemistry of Ore Deposits. Part A: Processes, Techniques, and Health Issues; Plumlee, G.S., Logsdon, M.J., Eds.; Reviews in Economic Geology Volume 6A; Society of Economic Geologist: Littleton, CO, USA, 1999; pp. 133–160. [Google Scholar]
- Dold, B.; Fontboté, L. Element cycling and secondary mineralogy in porphyry copper tailings as a function of climate, primary mineralogy, and mineral processing. J. Geochem. Explor. 2001, 74, 3–55. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Acid mine drainage remediation options: A review. Sci. Total Environ. 2005, 338, 3–14. [Google Scholar] [CrossRef]
- Machemer, S.D.; Reynolds, J.S.; Laudon, L.S.; Wildeman, T.R. Balance of S in a constructed wetland built to treat acid mine drainage, Idaho Springs, Colorado, USA. Appl. Geochem. 1993, 8, 587–603. [Google Scholar] [CrossRef]
- Sheoran, A.S.; Bhandari, S. Treatment of mine water by a microbial mat: Bench-scale experiments. Mine Water Environ. 2005, 24, 38–42. [Google Scholar] [CrossRef]
- Catalan, L.J.J.; Yanful, E.K.; St-Arnaud, L. Field assessment of metal and sulfate fluxes during flooding of pre-oxidized mine tailings. Adv. Environ. Res. 2000, 4, 295–306. [Google Scholar]
- Das, M.; Maiti, S.K. Comparison between availability of heavy metals in dry and wetland tailing of an abandoned copper tailing pond. Environ. Monit. Assess. 2008, 137, 343–350. [Google Scholar] [CrossRef]
- Moreno, L.; Neretnieks, I. Long-term environmental impact of tailings deposits. Hydrometallurgy 2006, 83, 176–183. [Google Scholar] [CrossRef]
- Widerlund, A.; Ebenå, G.; Landin, J. Potential biogeochemical and ecological development of a flooded tailings impoundment at the Kristineberg Zn-Cu mine, northern Sweden. Sci. Total Environ. 2004, 333, 249–266. [Google Scholar] [CrossRef]
- Dold, B.; Diaby, N.; Spangenberg, J.E. Remediation of a marine shore tailings deposit and the importance of water-rock interaction on element cycling in the coastal aquifer. Environ. Sci. Technol. 2011, 45, 4876–4883. [Google Scholar] [CrossRef]
- Elliott, P.; Ragusa, S.; Catcheside, D. Growth of sulfate-reducing bacteria under acidic conditions in an upflow anaerobic bioreactor as a treatment system for acid mine drainage. Water Res. 1998, 32, 3724–3730. [Google Scholar] [CrossRef]
- Fortin, D.; Roy, M.; Rioux, J.P.; Thibault, P.J. Occurrence of sulfate-reducing bacteria under a wide range of physico-chemical conditions in Au and Cu-Zn mine tailings. FEMS Microbiol. Ecol. 2000, 33, 197–208. [Google Scholar]
- Luptakova, A.; Kusnierova, M. Bioremediation of acid mine drainage contaminated by SRB. Hydrometallurgy 2005, 77, 97–102. [Google Scholar] [CrossRef]
- Praharaj, T.; Fortin, D. Indicators of microbial sulfate reduction in acidic sulfide-rich mine tailings. Geomicrobiol. J. 2004, 21, 457–467. [Google Scholar] [CrossRef]
- Canmet and Coastech Research Inc. Acid Rock Prediction Manual; Canada Centre for Mineral and Energy Technology: Ottawa, ON, Canada, 1991. [Google Scholar]
- Coggans, C.J.; Blowes, D.W.; Robertson, W.D.; Jambor, J.L. The Hydrogeochemistry of a Nickel-Mine Tailings Impoundment—Copper Cliff, Ontario. In The Environmental Geochemistry of Ore Deposits. Part A: Processes, Techniques, and Health Issues; Plumlee, G.S., Logsdon, M.J., Eds.; Reviews in Economic Geology Volume 6A; Society of Economic Geologist: Littleton, CO, USA, 1999; pp. 447–478. [Google Scholar]
- Dold, B. Speciation of the most soluble phases in a sequential extraction procedure adapted for geochemical studies of copper sulfide mine waste. J. Geochem. Explor. 2003, 80, 55–68. [Google Scholar]
- Dold, B. Element flows associated with marine shore mine tailings deposits. Environ. Sci. Technol. 2006, 40, 752–758. [Google Scholar]
- Hammarstrom, J.M.; Seal, R.R., II; Meier, A.L.; Kornfeld, J.M. Secondary sulfate minerals associated with acid drainage in the eastern US: Recycling of metals and acidity in surficial environments. Chem. Geol. 2005, 215, 407–431. [Google Scholar]
- Smuda, J.; Dold, B.; Friese, K.; Morgenstern, P.; Glaesser, W. Mineralogical and geochemical study of element mobility at the sulfide-rich Excelsior waste rock dump from the polymetallic Zn-Pb-(Ag-Bi-Cu) deposit, Cerro de Pasco, Peru. J. Geochem. Explor. 2007, 92, 97–110. [Google Scholar]
- Nealson, K.H.; Saffarini, D. Iron and Manganese in Anaerobic Respiration: Environmental Significance, Physiology, and Regulation. Annu. Rev. Microbiol. 1994, 48, 311–343. [Google Scholar]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batchreaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; U.S. Geological Survey: Reston, VA, USA, 1999. [Google Scholar]
- Woulds, C.; Ngwenya, B.T. Geochemical processes governing the performance of a constructed wetland treating acid mine drainage, Central Scotland. Appl. Geochem. 2004, 19, 1773–1783. [Google Scholar]
- Webster, J.G.; Swedlund, P.J.; Webster, K.S. Trace metal adsorption onto an acid mine drainage iron(III)oxyhydry sulfate. Environ. Sci. Technol. 1998, 32, 1362–1368. [Google Scholar]
- Carlsson, E.; Öhlander, B.; Holmström, H. Geochemistry of the infiltrating water in the vadose zone of a remediated tailings impoundment, Kristineberg mine, northern Sweden. Appl. Geochem. 2003, 18, 659–674. [Google Scholar]
- Holmstrom, H.; Salmon, U.J.; Carlsson, E.; Petrov, P.; Ohlander, B. Geochemical investigations of sulfide-bearing tailings at Kristineberg, northern Sweden, a few years after remediation. Sci. Total Environ. 2001, 273, 111–133. [Google Scholar] [CrossRef]
- Peltier, E.F.; Webb, S.M.; Gaillard, J.F. Zinc and lead sequestration in an impacted wetland system. Adv. Environ. Res. 2003, 8, 103–112. [Google Scholar]
- Wilkie, J.A.; Hering, J.G. Adsorption of arsenic onto hydrous ferric oxide: Effects of adsorbate/adsorbent ratios and co-occurring solutes. Colloids Surf. A Physicochem. Eng. Asp. 1996, 107, 97–110. [Google Scholar] [CrossRef]
- Fox, P.M.; Doner, H.E. Accumulation, release, and solubility of arsenic, molybdenum, and vanadium in wetland sediments. J. Environ. Qual. 2003, 32, 2428–2435. [Google Scholar] [CrossRef]
- Bone, S.E.; Eagle, M.; Charette, M.A. Geochemical cycling of arsenic in a coastal aquifer. Environ. Sci. Technol. 2006, 40, 3273–3278. [Google Scholar] [CrossRef]
- Miyata, N.; Tani, Y.; Sakata, M.; Iwahori, K. Microbial manganese oxide formation and interaction with toxic metal ions. J. Biosci. Bioeng. 2007, 104, 1–8. [Google Scholar] [CrossRef]
- O’Day, P.A.; Vlassopoulos, D.; Root, R.; Rivera, N. The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 13703–13708. [Google Scholar] [CrossRef]
- Goldberg, S.; Forster, H.S.; Godfrey, C.L. Molybdenum adsorption on oxides, clay minerals, and soils. Soil Sci. Soc. Am. J. 1996, 60, 425–432. [Google Scholar] [CrossRef]
- O’Sullivan, A.D.; Moran, B.M.; Otte, M.L. Accumulation and fate of contaminants (Zn, Pb, Fe and S) in substrates of wetlands constructed for treating mine wastewater. Water Air Soil Pollut. 2004, 157, 345–364. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Diaby, N.; Dold, B. Evolution of Geochemical and Mineralogical Parameters during In Situ Remediation of a Marine Shore Tailings Deposit by the Implementation of a Wetland Cover. Minerals 2014, 4, 578-602. https://doi.org/10.3390/min4030578
Diaby N, Dold B. Evolution of Geochemical and Mineralogical Parameters during In Situ Remediation of a Marine Shore Tailings Deposit by the Implementation of a Wetland Cover. Minerals. 2014; 4(3):578-602. https://doi.org/10.3390/min4030578
Chicago/Turabian StyleDiaby, Nouhou, and Bernhard Dold. 2014. "Evolution of Geochemical and Mineralogical Parameters during In Situ Remediation of a Marine Shore Tailings Deposit by the Implementation of a Wetland Cover" Minerals 4, no. 3: 578-602. https://doi.org/10.3390/min4030578
APA StyleDiaby, N., & Dold, B. (2014). Evolution of Geochemical and Mineralogical Parameters during In Situ Remediation of a Marine Shore Tailings Deposit by the Implementation of a Wetland Cover. Minerals, 4(3), 578-602. https://doi.org/10.3390/min4030578