Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors

Abstract
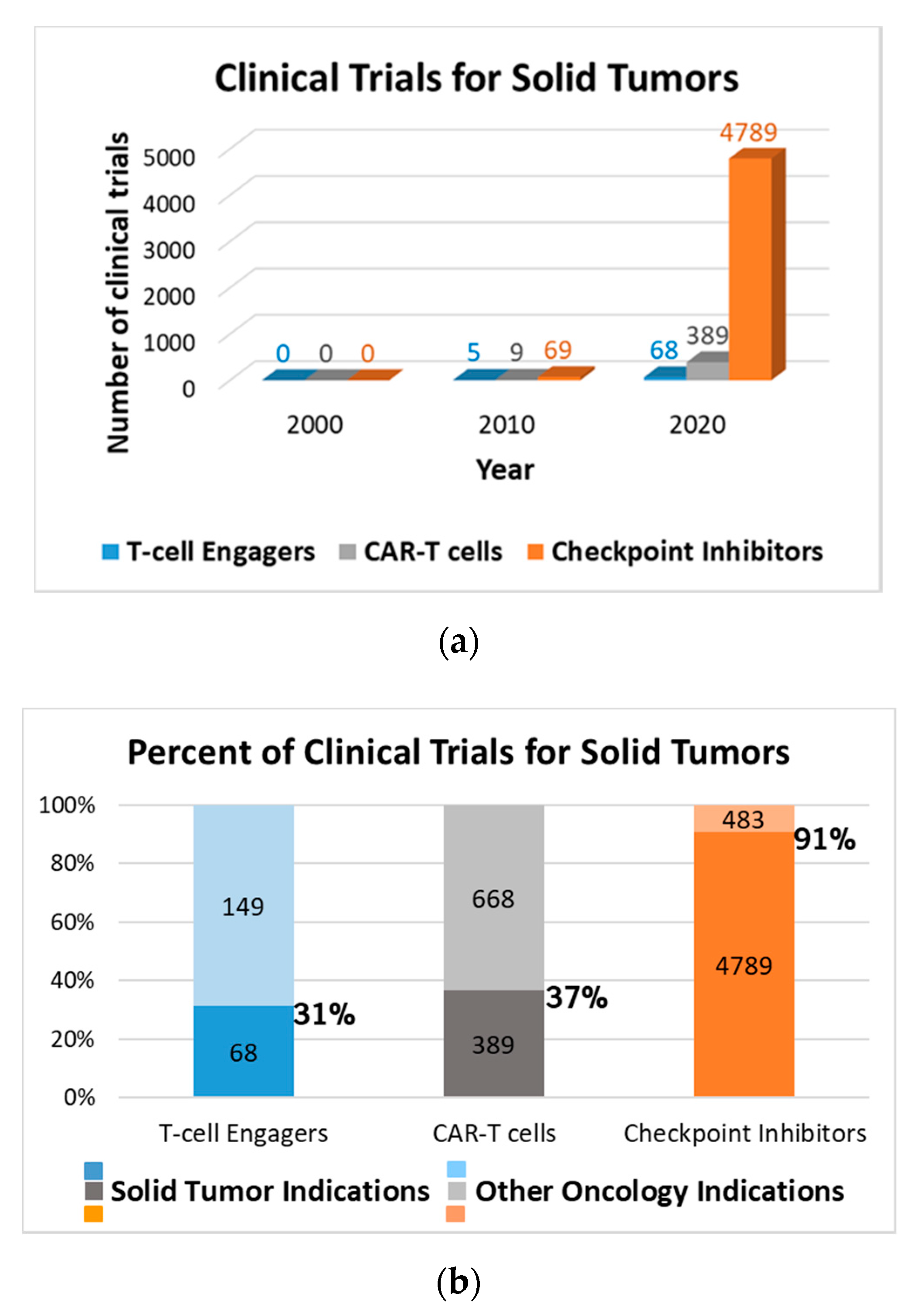
1. Introduction
2. Clinical Use
3. Challenges
3.1. Targeting Strategies for Solid Tumors
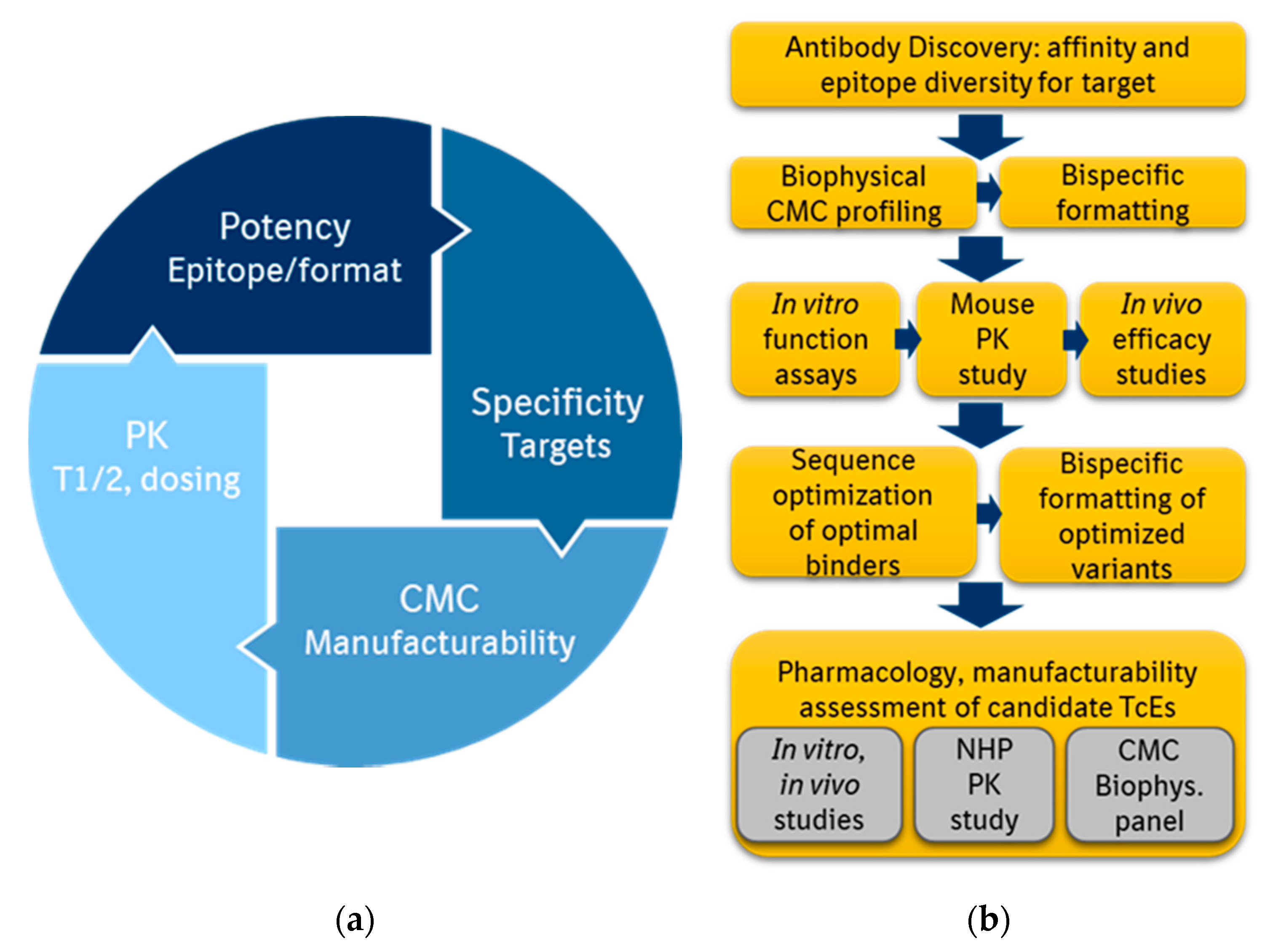
3.2. Identifying Optimal Target Binders
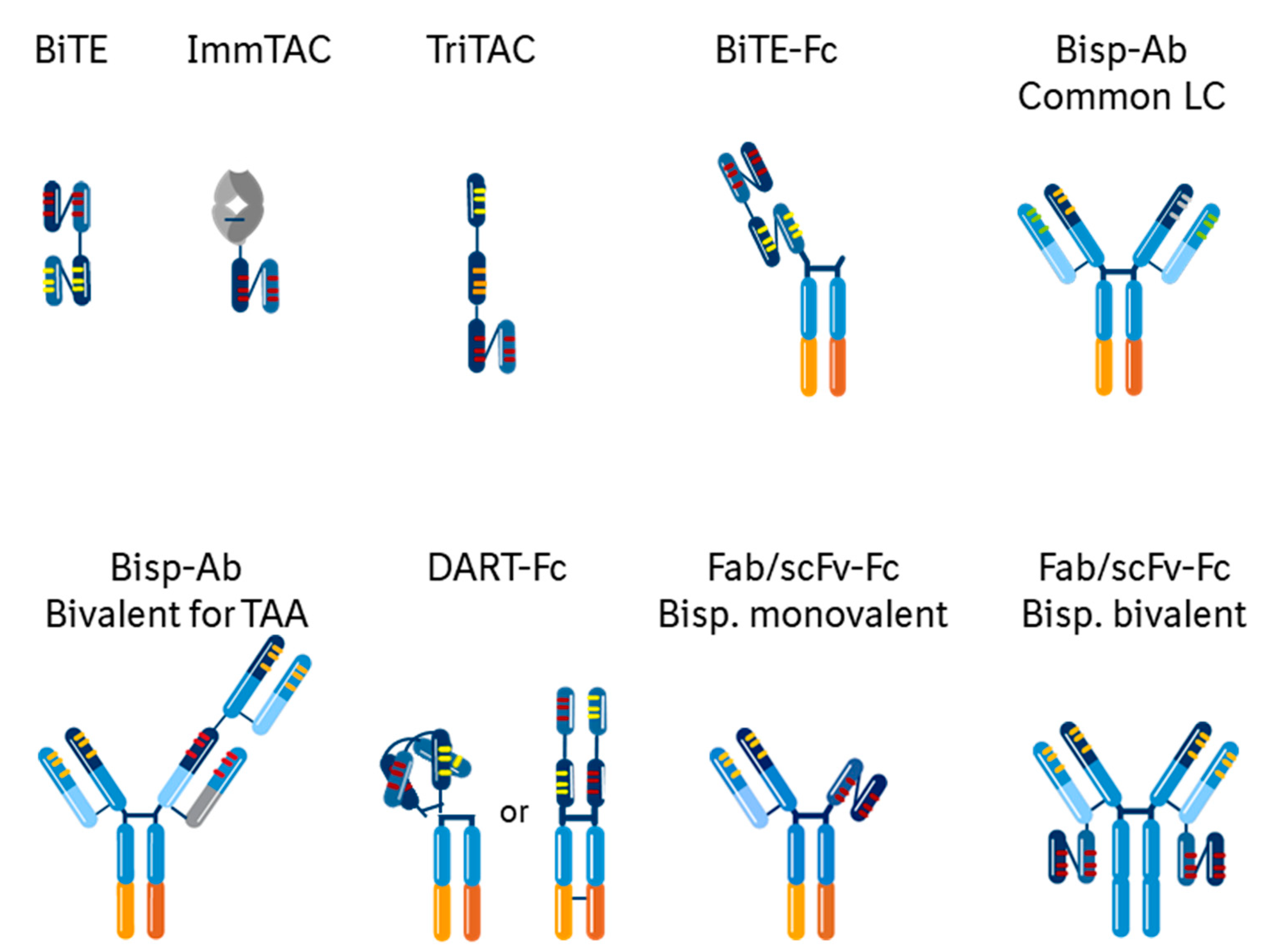
3.3. Multispecific Engineering Approaches
4. Proposed Discovery Strategies and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Brown, J.A.; Dorfman, D.M.; Ma, F.-R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of Programmed Death-1 Ligands on Dendritic Cells Enhances T Cell Activation and Cytokine Production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunology 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.P.; Kershaw, M.H. CARs versus BiTEs: A Comparison between T Cell–Redirection Strategies for Cancer Treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Pulte, D.; Vallejo, J.; Przepiorka, D.; Nie, L.; Farrell, A.T.; Goldberg, K.B.; McKee, A.E.; Pazdur, R. FDA Supplemental Approval: Blinatumomab for Treatment of Relapsed and Refractory Precursor B-Cell Acute Lymphoblastic Leukemia. Oncologist 2018, 23, 1366–1371. [Google Scholar] [CrossRef]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Hipp, S.; Tai, Y.-T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2016, 31, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti–B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.; Balana, C.; Van Linde, M.E.; Sayehli, C.; Fiedler, W.M.; Wermke, M.; Massard, C.; Ang, A.; Kast, J.; Stienen, S.; et al. Novel anti-EGFRvIII bispecific T cell engager (BiTE) antibody construct in glioblastoma (GBM): Trial in progress of AMG 596 in patients with recurrent or newly diagnosed disease. J. Clin. Oncol. 2019, 37, TPS2071. [Google Scholar] [CrossRef]
- Hummel, H.-D.; Kufer, P.; Grüllich, C.; Deschler-Baier, B.; Chatterjee, M.; Goebeler, M.-E.; Miller, K.; De Santis, M.; Loidl, W.C.; Buck, A.; et al. Phase I study of pasotuxizumab (AMG 212/BAY 2010112), a PSMA-targeting BiTE (Bispecific T-cell Engager) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 124. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.-E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor Efficacy of a Bispecific Antibody That Targets HER2 and Activates T Cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Berg, J.V. Rationale for Combining Bispecific T Cell Activating Antibodies with Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell Redirection for the Treatment of Metastatic Uveal Melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.F.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Cancer immunology: The search for specificity--G. H. A. Clowes Memorial lecture. Cancer Res. 1981, 41, 361–375. [Google Scholar]
- Schietinger, A.; Philip, M.; Schreiber, K. Specificity in cancer immunotherapy. Semin. Immunol. 2008, 20, 276–285. [Google Scholar] [CrossRef]
- Sengupta, S.; Sun, S.Q.; Huang, K.-L.; Oh, C.; Bailey, M.H.; Varghese, R.; Wyczalkowski, M.A.; Ning, J.; Tripathi, P.; McMichael, J.F.; et al. Integrative omics analyses broaden treatment targets in human cancer. Genome Med. 2018, 10, 1–20. [Google Scholar] [CrossRef]
- Owen, D.H.; Giffin, M.J.; Bailis, J.M.; Smit, M.-A.D.; Carbone, D.P.; He, K. DLL3: An emerging target in small cell lung cancer. J. Hematol. Oncol. 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results From the Phase II TRINITY Study. Clin. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef]
- Hipp, S.; Voynov, V.; Drobits-Handl, B.; Giragossian, C.; Trapani, F.; Nixon, A.E.; Scheer, J.M.; Adam, P.J. A Bispecific DLL3/CD3 IgG-like T-cell Antibody induces anti-tumor responses in Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5258–5268. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.-A.D.; Borghaei, H.; Owonikoko, T.K.; Hummel, H.-D.; Johnson, M.L.; Champiat, S.; Salgia, R.; Udagawa, H.; Boyer, M.J.; Govindan, R. Phase 1 study of AMG 757, a half-life extended bispecific T cell engager (BiTE) antibody construct targeting DLL3, in patients with small cell lung cancer (SCLC). J. Clin. Oncol. 2019, 37, TPS8577. [Google Scholar] [CrossRef]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2019, 38, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Jakobsen, K.B. ImmTACs: Novel bi-specific agents for targeted cancer therapy. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef]
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef]
- Pessano, S.; Oettgen, H.; Bhan, A.K.; Terhorst, C. The T3/T cell receptor complex: Antigenic distinction between the two 20-kd T3 (T3-delta and T3-epsilon) subunits. EMBO J. 1985, 4, 337–344. [Google Scholar] [CrossRef]
- Salmerón, A.; Sánchez-Madrid, F.; Ursa, M.A.; Fresno, M.; Alarcón, B. A conformational epitope expressed upon association of CD3-epsilon with either CD3-delta or CD3-gamma is the main target for recognition by anti-CD3 monoclonal antibodies. J. Immunol. 1991, 147, 3047–3052. [Google Scholar]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. mAbs 2019, 11, 639–652. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Rev. Cancer 2019, 1, 86–98. [Google Scholar] [CrossRef]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; A Baeuerle, P.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Fisher, T.S.; Hooper, A.T.; Lucas, J.; Clark, T.H.; Rohner, A.K.; Peano, B.; Elliott, M.W.; Tsaparikos, K.; Wang, H.; Golas, J.; et al. A CD3-bispecific molecule targeting P-cadherin demonstrates T cell-mediated regression of established solid tumors in mice. Cancer Immunol. Immunother. 2017, 67, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.; Gruen, M.; Wuchter, C.; Schriever, F.; Kufer, P.; Dreier, T.; Hanakam, F.; Baeuerle, P.A.; Bommert, K.; Karawajew, L.; et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia 2003, 17, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018, 10, 5775. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Exposito, R.; Semiannikova, M.; Griffiths, B.; Khan, K.; Barber, L.J.; Woolston, A.; Spain, G.; Von Loga, K.; Challoner, B.; Patel, R.; et al. CEA expression heterogeneity and plasticity confer resistance to the CEA-targeting bispecific immunotherapy antibody cibisatamab (CEA-TCB) in patient-derived colorectal cancer organoids. J. Immunother. Cancer 2019, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Ueda, O.; Wada, N.A.; Kinoshita, Y.; Hino, H.; Kakefuda, M.; Ito, T.; Fujii, E.; Noguchi, M.; Sato, K.; Morita, M.; et al. Entire CD3epsilon, delta, and gamma humanized mouse to evaluate human CD3-mediated therapeutics. Sci. Rep. 2017, 7, 45839. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sano, Y.; Komatsu, S.I.; Kamata-Sakurai, M.; Kaneko, A.; Kinoshita, Y.; Shiraiwa, H.; Azuma, Y.; Tsunenari, T.; Kayukawa, Y.; et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci. Transl. Med. 2017, 9, 4291. [Google Scholar] [CrossRef]
- Benonisson, H.; Altıntaş, I.; Sluijter, M.; Verploegen, S.; Labrijn, A.F.; Schuurhuis, D.H.; Houtkamp, M.A.; Verbeek, J.S.; Schuurman, J.; Van Hall, T. CD3-Bispecific Antibody Therapy Turns Solid Tumors into Inflammatory Sites but Does Not Install Protective Memory. Mol. Cancer Ther. 2018, 18, 312–322. [Google Scholar] [CrossRef]
- Puré, E.; Lo, A. Can Targeting Stroma Pave the Way to Enhanced Antitumor Immunity and Immunotherapy of Solid Tumors? Cancer Immunol. Res. 2016, 4, 269–278. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef]
- Dépis, F.; Hatterer, E.; Ballet, R.; Daubeuf, B.; Cons, L.; Glatt, S.; Reith, W.; Kosco-Vilbois, M.; Dean, Y. Characterization of a surrogate murine antibody to model anti-human CD3 therapies. mAbs 2013, 5, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Leo, O.; Foo, M.; Sachs, D.H.; Samelson, L.E.; Bluestone, J.A. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc. Natl. Acad. Sci. USA 1987, 84, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Tiller, T.; Schuster, I.; Deppe, D.; Siegers, K.; Strohner, R.; Herrmann, T.; Berenguer, M.; Poujol, D.; Stehle, J.; Stark, Y.; et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs 2013, 5, 445–470. [Google Scholar] [CrossRef] [PubMed]
- Valadon, P.; Pérez-Tapia, S.M.; Nelson, R.S.; Guzmán-Bringas, O.U.; Arrieta-Oliva, H.I.; Gómez-Castellano, K.M.; Pohl, M.A.; Almagro, J.C. ALTHEA Gold Libraries™: Antibody libraries for therapeutic antibody discovery. mAbs 2019, 11, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef]
- Kim, D.M.; Yao, X.; Vanam, R.P.; Marlow, M.S. Measuring the effects of macromolecular crowding on antibody function with biolayer interferometry. mAbs 2019, 11, 1319–1330. [Google Scholar] [CrossRef]
- Liu, Y.; Caffry, I.; Wu, J.; Geng, S.B.; Jain, T.; Sun, T.; Reid, F.; Cao, Y.; Estep, P.; Yu, Y.; et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. mAbs 2013, 6, 483–492. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef]
- Kumar, S.; Plotnikov, N.V.; Rouse, J.C.; Singh, S.K. Biopharmaceutical Informatics: Supporting biologic drug development via molecular modelling and informatics. J. Pharm. Pharmacol. 2017, 70, 595–608. [Google Scholar] [CrossRef]
- Voynov, V.; Chennamsetty, N.; Kayser, V.; Helk, B.; Trout, B.L. Predictive tools for stabilization of therapeutic proteins. mAbs 2009, 1, 580–582. [Google Scholar] [CrossRef]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Plueckthun, A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Pastan, I.; Lee, B. Design of interchain disulfide bonds in the framework region of the Fv fragment of the monoclonal antibody B3. Proteins Struct. Funct. Bioinform. 1994, 19, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Ridgway, J.B.; A Wells, J.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef]
- McAleese, F.; Eser, M. RECRUIT-TandAbs®: Harnessing the immune system to kill cancer cells. Futur. Oncol. 2012, 8, 687–695. [Google Scholar] [CrossRef]
- Moore, P.A.; Shah, K.; Yang, Y.; Alderson, R.; Roberts, P.; Long, V.; Liu, D.; Li, J.C.; Burke, S.; Ciccarone, V.; et al. Development of MGD007, a gpA33 x CD3-Bispecific DART Protein for T-Cell Immunotherapy of Metastatic Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 1761–1772. [Google Scholar] [CrossRef]
- Austin, R.; Aaron, W.; Baeuerle, P.; Barath, M.; Jones, A.; Jones, S.D.; Law, C.-L.; Kwant, K.; Lemon, B.; Muchnik, A.; et al. Abstract 1781: HPN536, a T cell-engaging, mesothelin/CD3-specific TriTAC for the treatment of solid tumors. Immunology 2018, 78, 1781. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 1–15. [Google Scholar] [CrossRef]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a bispecific antibody with a common light chain: Identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.M.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2019, 26, 984–989. [Google Scholar] [CrossRef]
- Boustany, L.M.; Wong, L.; White, C.W.; Diep, L.; Huang, Y.; Liu, S.; Richardson, J.H.; Kavanaugh, W.M.; Irving, B.A. Abstract A164: EGFR-CD3 bispecific Probody™ therapeutic induces tumor regressions and increases maximum tolerated dose > 60-fold in preclinical studies. In Proceedings of the Abstracts: AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Philadelphia, PA, USA, 26–30 October 2017; p. A164. [Google Scholar]
- Kamata-Sakurai, M.; Narita, Y.; Hori, Y.; Nemoto, T.; Uchikawa, R.; Honda, M.; Hironiwa, N.; Taniguchi, K.; Shida-Kawazoe, M.; Metsugi, S.; et al. Antibody to CD137 activated by extracellular adenosine triphosphate is tumor selective and broadly effective in vivo without systemic immune activation. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Xu, D.; Alegre, M.-L.; Varga, S.S.; Rothermel, A.L.; Collins, A.M.; Pulito, V.L.; Hanna, L.S.; Dolan, K.P.; Parren, P.W.; Bluestone, J.A.; et al. In Vitro Characterization of Five Humanized OKT3 Effector Function Variant Antibodies. Cell. Immunol. 2000, 200, 16–26. [Google Scholar] [CrossRef]
- Dustin, M.L.; Baldari, C.T. The Immune Synapse: Past, Present, and Future. Adv. Struct. Safety Stud. 2017, 1584, 1–5. [Google Scholar] [CrossRef]
- Huppa, J.B.; Gleimer, M.; Sumen, C.; Davis, M.M. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat. Immunol. 2003, 4, 749–755. [Google Scholar] [CrossRef]
- Cartwright, A.N.R.; Griggs, J.; Davis, D.M. The immune synapse clears and excludes molecules above a size threshold. Nat. Commun. 2014, 5, 5479. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Karle, A.; Meissburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved Pharmacokinetics of Recombinant Bispecific Antibody Molecules by Fusion to Human Serum Albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef] [PubMed]
- Stork, R.; Campigna, E.; Robert, B.; Müller, D.; Kontermann, R.E. Biodistribution of a Bispecific Single-chain Diabody and Its Half-life Extended Derivatives. J. Biol. Chem. 2009, 284, 25612–25619. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Technology and Key Features | Examples (Targets): Phase, Indication, Trial Number, Status (Other Information) |
|---|---|
| BiTE: -Two tandem scFvs; -short half-life (hours) | * AMG 110/MT-110/solitomab (EpCAMxCD3): -PhI, Solid tumors, NCT00635596, Completed * MEDI-565/AMG 211/MT-111 (CEAxCD3): -PhI, Gastrointestinal Adenocarcinomas, NCT01284231, Completed; -PhI, Advanced Gastrointestinal Cancer, NCT02291614, Completed (Terminated) * Pasotuxizumab/AMG 212/MT-112/BAY 2010112 (PSMAxCD3): -PhI, Prostate Cancer, NCT01723475, Completed (Terminated) * AMG 596 (EGFRvIIIxCD3): -PhI, Glioblastoma, NCT03296696, Recruiting (alone or in combination with AMG 404 (anti-PD-1)) |
| ImmTAC: -Bispecific of a TCR domain and anti-CD3 scFv; -short half-life (hours) | * Tebentafusp/IMCgp100 (gp100xCD3): -Early PhI, Advanced Melanoma, NCT01209676, Completed; -PhI, Malignant Melanoma, NCT01211262, Completed; -PhII, Malignant Melanoma, NCT02889861, Terminated; -PhI/II, Malignant Melanoma, NCT02535078, Recruiting (combination with Durvalumab (anti-PD-L1) and/or Tremelimumab (anti-CTLA-4)); -PhI/II, Uveal Melanoma, NCT02570308, Active; -PhII, Uveal Melanoma, NCT03070392, Recruiting; * IMCnieso (NY-ESO-1- and/or LAGE-1AxCD3): -PhI/II, Advanced Solid Tumors, NCT03515551, Recruiting * IMC-C103C (MAGE-A4xCD3): -PhI/II, Advanced Solid Tumors, NCT03973333, Recruiting (alone and in combination with Atezolizumab (anti-PD-L1)) * IMC-F106C (PRAMExCD3): -PhI/II, Advanced Solid Tumors, NCT04262466, Recruiting (alone and in combination with CPIs) |
| TriTAC: -Trispecific construct: TAA-HSA-CD3, with anti-HSA binder for half-life extension | * HPN424 (PSMAxCD3): -PhI, Advanced Prostate Cancer, NCT03577028, Recruiting * HPN536 (MesothelinxCD3): -PhI/II, Advanced Cancers, NCT03872206, Recruiting |
| BiTE with Fc: -Two tandem scFvs linked to an Fc domain for half-life extension to several days, for less frequent dosing | * AMG 160 (PSMAxCD3): -PhI, Prostate Cancer, NCT03792841, Recruiting * AMG 199 (MUC17xCD3): -PhI, Gastric and Gastroesophageal Junction Cancers, NCT04117958, Recruiting * AMG 757 (DLL3xCD3): -PhI, Small Cell Lung Cancer, NCT03319940, Recruiting |
| Bispecific Antibody with common Light Chain: -Fab domain binders -common light chain -Fc domain for half-life extension -Fc mutations for heterodimerization of heavy chains | * ERY974 (GPC3xCD3): -PhI, Advanced Solid Tumors, NCT02748837, Completed; -PhI, JapicCTI-194805, Recruiting * REGN4018 (MUC16xCD3): -PhI/II, Recurrent Ovarian Cancer, NCT03564340, Recruiting (alone or in combination with Cemiplimab (anti-PD-1) |
| DuoBody Bispecific Antibody: -Fab domain binders -Fc domain for half-life extension -mutations in Fc -process for bispecific antibody generation from two regular IgGs after purification | * JNJ-63898081 (PSMAxCD3): -PhI, Advanced Stage Solid Tumors, NCT03926013, Recruiting |
| Bispecific TcE with Fc and bivalent for TAA: -Fc domain for half-life extension; -Knob-in-Hole technology in Fc for heterodimerization -CrossMab technology for correct LC-HC pairing in a bispecific -Two sites to bind TAA for improved therapeutic window. | * Cibisatamab/RO6958688/RG7802 (CEAxCD3): -PhI, Solid Tumors, NCT02324257, Completed; -PhI, Advanced Solid Tumors, JapicCTI-173764, Completed; -PhI, Solid Tumors, NCT02650713, Completed (in combination with Atezolizumab (anti-PD-L1)); -PhI/II, Non-small Cell Lung Cancer, NCT03337698, Recruiting; -PhI, Colorectal Cancer, NCT03866239, Active (in combination with Atezolizumab (anti-PD-L1) after pretreatment with Obinutuzumab (anti-CD20)) |
| DART-Fc: -Fab or Fv domain binders with linkers -Fc domain for half-life extension -Monovalent or bivalent for targets | * PF-06671008 (CDH3xCD3): -PhI, Advanced Solid tumors, NCT02659631, Terminated * MGD007 (gpA33xCD3): -PhI, Colorectal Cancer, NCT02248805, Completed; -PhI/II, Metastatic Colorectal Cancer, NCT03531632, Active (in combination with MGA012 (anti-PD-1)) * MGD009 (B7-H3xCD3): -PhI, Solid Tumors, NCT02628535, Terminated; -PhI, Solid Tumors, NCT03406949, Recruiting (in combination with MGA012 (anti-PD-1)) * PF07062119 (GUCY2CxCD3): -PhI, Advanced or Metastatic Gastrointestinal Tumors, NCT04171141, Recruiting |
| Fab/scFv-Fc Bispecific monovalent (XmAb): -one binder is Fab; the other is scFv -Fc domain for half-life extension -engineered CH3 domain for heterodimerization | * Tidutamab/XmAb18087 (SSTR2xCD3): -PhI, Neuroendocrine and Gastrointestinal Stromal Tumors, NCT03411915, Recruiting * GBR 1302/ISB 1302 (HER2xCD3): -PhI, HER2+ Solid Tumors, NCT02829372, Terminated -PhI/II, Breast Cancer, NCT03983395, Recruiting * AMG 509 (STEAP1xCD3): -PhI, Prostate Cancer, NCT04221542, Recruiting * M701 (EpCAMxCD3): -PhI, Ascites, Solid Tumors, ChiCTR1900024144, Recruiting * M802 (HER2xCD3): -PhI, HER2+ Solid Tumors, ChiCTR1900024128, Recruiting |
| scFv-Fc-scFv bispecific bivalent: -scFv domain binders -Fc domain for half-life extension -Bispecific and bivalent for targets | * ES414/APVO414/MOR209 (PSMAxCD3): -PhI, Prostate Cancer, NCT02262910, Completed (Terminated) |
| Fab/scFv-Fc bispecific bivalent: -scFv for CD3 attached to the C-terminus of the light chain of IgG -Fc domain for half-life extension | * Hu3F8-BsAb (GD2xCD3): -PhI/II, Neuroblastoma, Osteosarcoma, Other Solid Tumors, NCT03860207, Recruiting; |
| Other | * BTRC4017A/RG6194 (Her2xCD3): -PhI, HER2+ Solid Tumors, NCT03448042, Recruiting * GEM3PSCA (PSCAxCD3): -PhI, Solid Tumors, NCT03927573, Recruiting * REGN5678 (PSMAxCD28): -PhI, Prostate Cancer, NCT03972657, Recruiting (in combination with Cemiplimab (anti-PD-1)) * CCW702/ABBV-154 (PSMAxCD3): -PhI, Prostate Cancer, NCT04077021, Recruiting * AMV564 (CD33xCD3): -PhI, Advanced Solid Tumors, NCT04128423, Recruiting * A-337 (EpCAMxCD3): -PhI, Advanced Solid Tumors, ACTRN12617001181392, Terminated |
| Subject | Key Considerations |
|---|---|
| TME Biology | Effectiveness of TcE modality for solid tumors Biomarkers and functional requirements of therapeutic molecule |
| Target Identification | Uniqueness of target for a therapeutic concept Expression profile in tumor vs. healthy cells and tissues |
| Lead Identification | Fab vs. non-Fab platforms for discovery of diverse set of binders Epitope, affinity, cross-reactivity, biophysical stability requirements |
| Multispecific formatting | Format that enables desired potency, safety, manufacturability and PK Evaluation of different binders in format for both function and CMC |
| CMC properties | Inherent molecule stability for optimal potency and safety Good manufacturability and developability for fast path to the clinic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voynov, V.; Adam, P.J.; Nixon, A.E.; Scheer, J.M. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies 2020, 9, 65. https://doi.org/10.3390/antib9040065
Voynov V, Adam PJ, Nixon AE, Scheer JM. Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies. 2020; 9(4):65. https://doi.org/10.3390/antib9040065
Chicago/Turabian StyleVoynov, Vladimir, Paul J. Adam, Andrew E. Nixon, and Justin M. Scheer. 2020. "Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors" Antibodies 9, no. 4: 65. https://doi.org/10.3390/antib9040065
APA StyleVoynov, V., Adam, P. J., Nixon, A. E., & Scheer, J. M. (2020). Discovery Strategies to Maximize the Clinical Potential of T-Cell Engaging Antibodies for the Treatment of Solid Tumors. Antibodies, 9(4), 65. https://doi.org/10.3390/antib9040065