Bringing the Heavy Chain to Light: Creating a Symmetric, Bivalent IgG-Like Bispecific

 , , , ,
, , , , 
Abstract
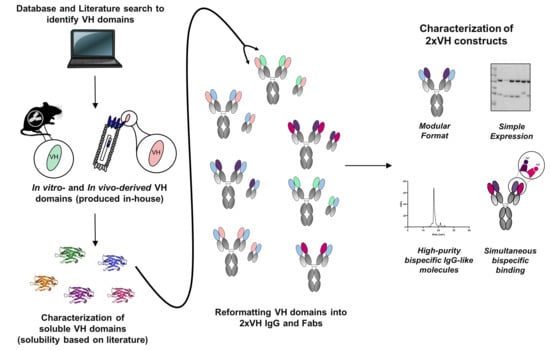
1. Introduction
2. Materials and Methods
2.1. Selection of VH Domains from the Literature and the Protein Databank (PDB)
2.2. Expression and Purification of VH Domains
2.3. Expression and Purification of 2xVH Fabs and IgG Molecules
2.4. SPR Analysis
2.5. Analytical SEC
2.6. NanoDSF
2.7. ELISA
2.8. 2xVH Fab Crystallization
3. Results
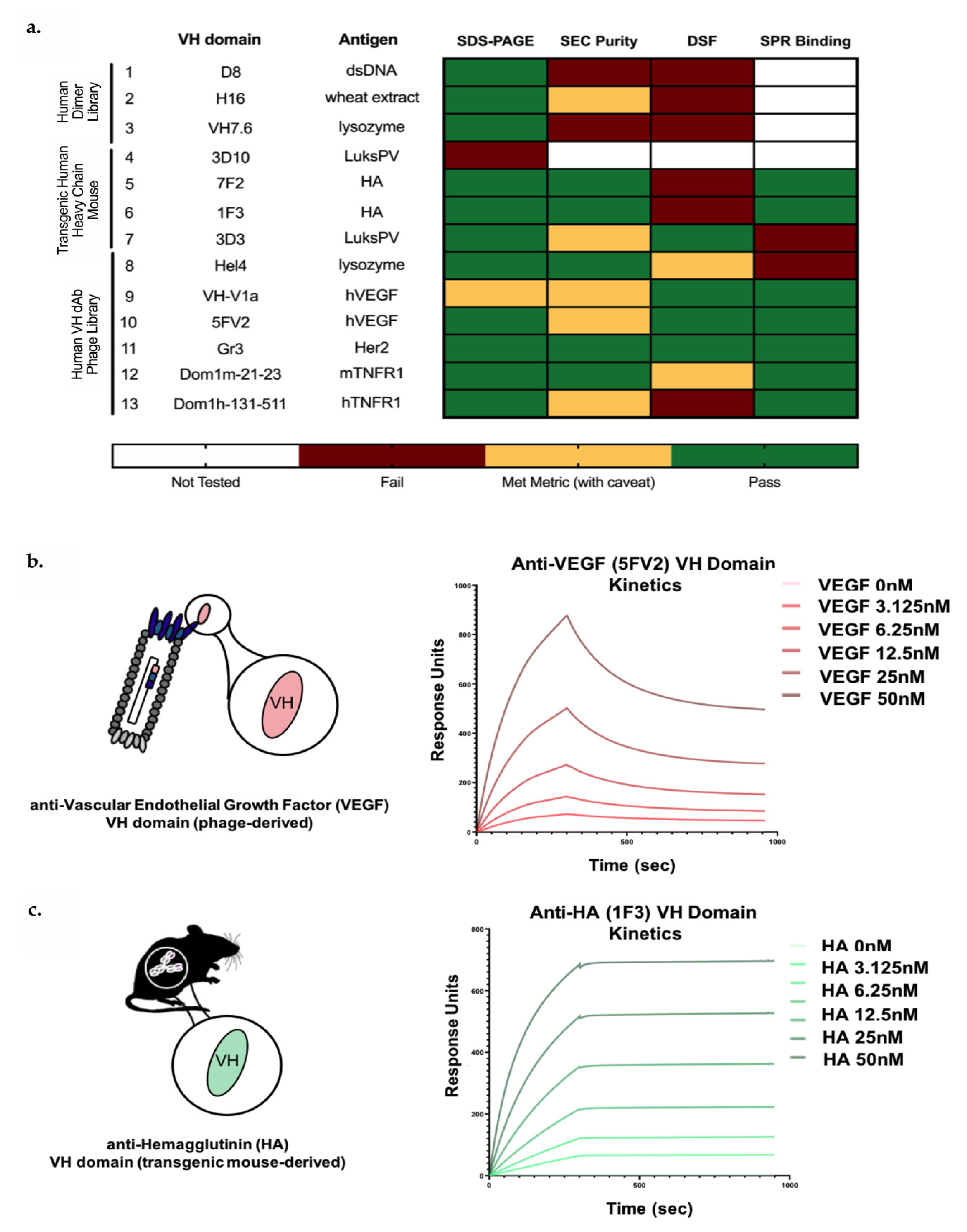
3.1. Identification of In Vitro and In Vivo-Derived Candidate VH Domains
3.2. Biophysical Characterization of sVH Candidates Shows Source-Dependent Differences in Expression, Purity and Thermal Stability
3.3. Candidate In Vitro- and In Vivo-Derived sVH Domains Show nM and Sub-nM Binding
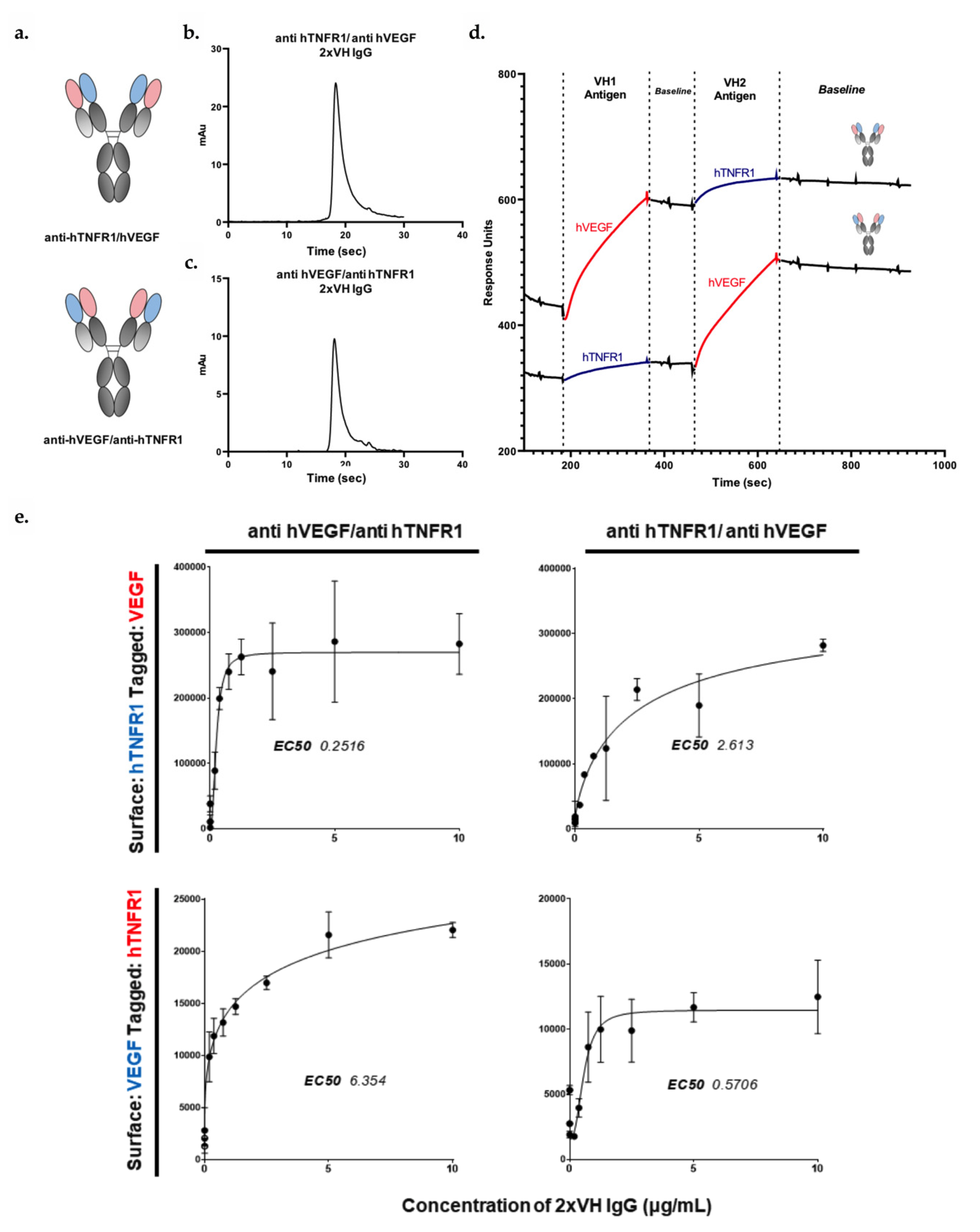
3.4. Generation and Characterization of 2xVH Molecules with High Yield and Purity
3.5. 2xVH IgG and Fab Can Co-Engage Two Antigens: A Case Study of Anti-HA/Anti-hVEGF
3.6. VH Domain Placement Does Not Impact Binding to Target Antigen in 2xVH Format: A Case Study of Clinically Relevant Anti-hVEGF/Anti-hTNFR1
3.7. Anti-Her2/Anti-Lysozyme: VH/VH Interfaces Are Larger than Canonical VH/VL Interfaces
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsumoto, K.; Isozaki, Y.; Yagami, H.; Tomita, M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy 2019, 11, 119–127. [Google Scholar] [CrossRef]
- Akabane, H. Issues of the biopharmaceutical industry and recommendations for further development. Ser. Res. Pap. 2018, 71, 1–79. [Google Scholar]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Antibody Therapeutics Approved or in Regulatory Review in the EU or US. Available online: https://www.antibodysociety.org/resources/approved-antibodies/ (accessed on 14 July 2020).
- Schett, G.; Elewaut, D.; Mcinnes, I.B.; Dayer, J.; Neurath, M.F. How cytokine networks fuel inflammation. Nat. Med. 2013, 19, 822–826. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Faivre, S.; Djelloul, S.; Raymond, E. New Paradigms in Anticancer Therapy: Targeting Multiple Signaling Pathways With Kinase Inhibitors. Semin. Oncol. 2006, 33, 407–420. [Google Scholar] [CrossRef]
- Zhao, Q. Bispecific Antibodies for Autoimmune and Inflammatory Diseases: Clinical Progress to Date. BioDrugs 2020, 34, 111–119. [Google Scholar] [CrossRef]
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Krah, S.; Kolmar, H.; Becker, S.; Zielonka, S. Engineering IgG-Like Bispecific Antibodies—An Overview. Antibodies 2018, 7, 28. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Hladnik, L.; Augustin, K.; DeFrates, S. Advancements in Therapy for Acute Lymphoblastic Leukemia: Blinatumomab. J. Adv. Pract. Oncol. 2016, 7, 76–82. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Jackman, J.; Chen, Y.; Huang, A.; Moffat, B.; Scheer, J.M.; Leong, S.R.; Lee, W.P.; Zhang, J.; Sharma, N.; Lu, Y.; et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J. Biol. Chem. 2010, 285, 20850–20859. [Google Scholar] [CrossRef]
- Tustian, A.D.; Endicott, C.; Adams, B.; Mattila, J.; Bak, H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs 2016, 8, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 2015, 7, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Y. Pharmacokinetics of Bispecific Antibody. Curr. Pharmacol. Rep. 2017, 3, 126–137. [Google Scholar] [CrossRef]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef]
- Morrison, S.J.; Spradling, A.C.; Charnay, P.; Burns, D.K.; Parada, L.F.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- Coloma, M.J.; Morrison, S.L. Design and production of novel tetravalent bispecific antibodies. Nat. Biotechnol. 1997, 15, 159–163. [Google Scholar] [CrossRef]
- Marvin, J.S.; Zhu, Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol. Sin. 2005, 26, 649–658. [Google Scholar] [CrossRef]
- Bostrom, J.; Yu, S.-F.; Kan, D.; Appleton, B.A.; Lee, C.V.; Billeci, K.; Man, W.; Peale, F.; Ross, S.; Wiesmann, C.; et al. Variants of the Antibody Herceptin That Interact with HER2 and VEGF at the Antigen Binding Site. Science 2009, 323, 1610–1614. [Google Scholar] [CrossRef]
- Everett, K.L.; Kraman, M.; Wollerton, F.P.G.; Zimarino, C.; Kmiecik, K.; Gaspar, M.; Pechouckova, S.; Allen, N.L.; Doody, J.F.; Tuna, M. Generation of Fcabs targeting human and murine LAG-3 as building blocks for novel bispecific antibody therapeutics. Methods 2019, 154, 60–69. [Google Scholar] [CrossRef]
- Ridgway, J.B.B.; Presta, L.G.; Carter, P. “Knobs-into-holes” engineering of antibody C(H)3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Bönisch, M.; Sellmann, C.; Maresch, D.; Halbig, C.; Becker, S.; Toleikis, L.; Hock, B.; Rüker, F. Novel CH1:CL interfaces that enhance correct light chain pairing in heterodimeric bispecific antibodies. Protein Eng. Des. Sel. 2017, 30, 685–696. [Google Scholar] [CrossRef]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Liu, Z.; Leng, E.C.; Gunasekaran, K.; Pentony, M.; Shen, M.; Howard, M.; Stoops, J.; Manchulenko, K.; Razinkov, V.; Liu, H.; et al. A novel antibody engineering strategy for making monovalent bispecific heterodimeric IgG antibodies by electrostatic steering mechanism. J. Biol. Chem. 2015, 290, 7535–7562. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. Antibody VH Domains as Small Recognition Units. Bio/Technology 1995, 13, 475–479. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Dekker, S.; Hendriks, R.W.; Panayotou, G.; Van Remoortere, A.; San, J.K.A.; Grosveld, F.; Drabek, D. Generation of heavy-chain-only antibodies in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 15130–15135. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.K.; Grosveld, F.G.; Janssens, R.W.; Drabek, D. Binding Molecules. WIPO Patent WO2006008548A3, 26 January 2006. [Google Scholar]
- Laventie, B.J.; Rademaker, H.J.; Saleh, M.; De Boer, E.; Janssens, R.; Bourcier, T.; Subilia, A.; Marcellin, L.; Van Haperen, R.; Lebbink, J.H.G.; et al. Heavy chain-only antibodies and tetravalent bispecific antibody neutralizing Staphylococcus aureus leukotoxins. Proc. Natl. Acad. Sci. USA 2011, 108, 16404–16409. [Google Scholar] [CrossRef]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161–1165. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, Z.; Feng, Y.; Xiao, X.; Dimitrov, D.S. Construction of a Large Phage-Displayed Human Antibody Domain Library With a Scaffold Based on a Newly Identified Highly Soluble, Stable Heavy Chain Variable Domain. J. Mol. Biol. 2008, 382, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.Y.; Lu, Y.W.; Liu, Z.; Stevens, J.; Murawsky, C.M.; Wilson, V.; Hu, Z.; Richards, W.G.; Michaels, M.L.; Zhang, J.; et al. A biparatopic agonistic antibody that mimics fibroblast growth factor 21 ligand activity. J. Biol. Chem. 2018, 293, 5909–5919. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Kaas, Q.; Ruiz, M.; Lefranc, M. IMGT/3Dstructure-DB and IMGT/StructuralQuery, a database and a tool for immunoglobulin, T cell receptor and MHC structural data. Nucleic Acids Res. 2004, 32, D208–D210. [Google Scholar] [CrossRef]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef]
- Durocher, Y. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, e9. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Eisenbraun, B.; Key, J.; Sanschagrin, P.C.; Timony, M.A.; Ottaviano, M.; Sliz, P. Collaboration gets the most out of software. eLife 2013, 2013, e01456. [Google Scholar] [CrossRef]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Jirholt, P.; Ohlin, M.; Borrebaeck, C.A.K.; Söderlind, E. Exploiting sequence space: Shuffling in vivo formed complementarity determining regions into a master framework. Gene 1998, 215, 471–476. [Google Scholar] [CrossRef]
- Nilvebrant, J.; Tessier, P.; Sidhu, S. Engineered Autonomous Human Variable Domains. Curr. Pharm. Des. 2016, 22, 6527–6537. [Google Scholar] [CrossRef] [PubMed]
- Jespers, L.; Schon, O.; James, L.C.; Veprintsev, D.; Winter, G. Crystal structure of HEL4, a soluble, refoldable human VH single domain with a germ-line scaffold. J. Mol. Biol. 2004, 337, 893–903. [Google Scholar] [CrossRef]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef]
- Famm, K.; Hansen, L.; Christ, D.; Winter, G. Thermodynamically Stable Aggregation-Resistant Antibody Domains through Directed Evolution. J. Mol. Biol. 2008, 376, 926–931. [Google Scholar] [CrossRef]
- Enever, C.; Pupecka-Swider, M.; Sepp, A. Stress selections on domain antibodies: “What doesn’t kill you makes you stronger”. Protein Eng. Des. Sel. 2015, 28, 59–66. [Google Scholar] [CrossRef]
- Walker, A.; Chung, C.W.; Neu, M.; Burman, M.; Batuwangala, T.; Jones, G.; Tang, C.M.; Steward, M.; Mullin, M.; Tournier, N.; et al. Novel interaction mechanism of a domain antibody-based inhibitor of human vascular endothelial growth factor with greater potency than ranibizumab and bevacizumab and improved capacity over aflibercept. J. Biol. Chem. 2016, 291, 5500–5511. [Google Scholar] [CrossRef]
- Goodall, L.J.; Ovecka, M.; Rycroft, D.; Friel, S.L.; Sanderson, A.; Mistry, P.; Davies, M.L.; Stoop, A.A. Pharmacokinetic and pharmacodynamic characterisation of an anti-mouse TNF receptor 1 domain antibody formatted for in vivo half-life extension. PLoS ONE 2015, 10, e0137065. [Google Scholar] [CrossRef][Green Version]
- Baral, T.N.; Chao, S.Y.; Li, S.; Tanha, J.; Arbabi-Ghahroudi, M.; Zhang, J.; Wang, S. Crystal structure of a human single domain antibody dimer formed through V H-V H non-covalent interactions. PLoS ONE 2012, 7, e30149. [Google Scholar] [CrossRef]
- Ma, X.; Barthelemy, P.A.; Rouge, L.; Wiesmann, C.; Sidhu, S.S. Design of synthetic autonomous VH domain libraries and structural analysis of a VH domain bound to vascular endothelial growth factor. J. Mol. Biol. 2013, 425, 2247–2259. [Google Scholar] [CrossRef]
- Drabek, D.; Janssens, R.; de Boer, E.; Rademaker, R.; Kloess, J.; Skehel, J.; Grosveld, F. Expression cloning and production of human heavy-chain-only antibodies from murine transgenic plasma cells. Front. Immunol. 2016, 7, 3–12. [Google Scholar] [CrossRef]
- Craig, R.K.; Grosveld, F.G.; Janssens, R.W.; Drabek, D.; Chen, T.; De Boer, E. Soluble Heavy-Chain Only Antibodies. U.S. Patent US 9365655 B2, 14 January 2016. [Google Scholar]
- Sepúlveda, J.; Jin, H.; Sblattero, D.; Bradbury, A.; Burrone, O.R. Binders based on dimerised immunoglobulin VH domains. J. Mol. Biol. 2003, 333, 355–365. [Google Scholar] [CrossRef]
- Jin, H.; Sepúlveda, J.; Burrone, O.R. Specific recognition of a dsDNA sequence motif by an immunoglobulin VH homodimer. Protein Sci. 2009, 13, 3222–3229. [Google Scholar] [CrossRef]
- Jin, H.; Sepúlveda, J.; Burrone, O.R. Selection and characterisation of binders based on homodimerisation of immunoglobulin VH domains. FEBS Lett. 2003, 554, 323–329. [Google Scholar] [CrossRef]
- Hong, P.; Koza, S.; Bouvier, E.S.P. A review size-exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef]
- Friguet, B.; Chaffotte, A.F.; Djavadi-ohaniance, L.; Goldberg, M.E. Measurements of the true affinity constant in solution of antige-antibody complexes by ELISA—J. Immunol. Methods.pdf. J. Immunol. Methods 1985, 77, 305–319. [Google Scholar] [CrossRef]
- Boss, M.A.; Kenten, J.H.; Wood, C.R.; Emtage, J.S. Assembly of functional antibodies from immunoglobulin heavy and light chains synthesised in E. coli. Nucleic Acids Res. 1984, 12, 3791–3806. [Google Scholar] [CrossRef]
- Cabilly, S.; Riggs, A.D.; Pande, H.; Shively, J.E.; Holmes, W.E.; Rey, M.; Perry, L.J.; Wetzel, R.; Heyneker, H.L. Generation of antibody activity from immunoglobulin polypeptide chains produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 3273–3277. [Google Scholar] [CrossRef]
- Hakim, R.; Benhar, I. “Inclonals”: IgGs and IgG-enzyme fusion proteins produced in an E. coli expression-refolding system. mAbs 2009, 1, 281–287. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, K.W.; Kim, B.M.; Cho, M.L.; Lee, S.H. The effect of vascular endothelial growth factor on osteoclastogenesis in rheumatoid arthritis. PLoS ONE 2015, 10, e0124909. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Koch, A. Targeting Angiogenesis in Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2008, 4, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Luo, J.; Muzammil, S.; Sweet, R.; Almagro, J.C.; Gilliland, G.L. Structural diversity in a human antibody germline library. mAbs 2016, 8, 1045–1063. [Google Scholar] [CrossRef]
- Mullard, A. Bispecific antibody pipeline moves beyond oncology. Nat. Rev. Drug Discov. 2017, 16, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Hector, A.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally Designed Bispecific Antibodies Using Negative State Repertoires. Structure 2016, 24, 641–651. [Google Scholar] [CrossRef]
- Froning, K.J.; Leaver-Fay, A.; Wu, X.; Phan, S.; Gao, L.; Huang, F.; Pustilnik, A.; Bacica, M.; Houlihan, K.; Chai, Q.; et al. Computational design of a specific heavy chain/κ light chain interface for expressing fully IgG bispecific antibodies. Protein Sci. 2017, 26, 2021–2038. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | PDB | Contact Surface VH (Å2) or VH1 | Contact Surface VL (Å2) or VH2 | Interface (Å2) |
|---|---|---|---|---|
| H3-23:L1-39 | 5I19 | 795 | 817 | 806 |
| H3-23:L3-11 | 5I1A | 822 | 834 | 828 |
| H3-23:L3-20 | 5I1C | 670 | 698 | 684 |
| H3-23:L4-1 | 5I1D | 743 | 770 | 757 |
| 2xVH anti-Her2, Lysozyme | 7JKB | 1200 | 1143 | 1171 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramasubramanian, A.; Tennyson, R.; Magnay, M.; Kathuria, S.; Travaline, T.; Jain, A.; Lord, D.M.; Salemi, M.; Sullivan, C.; Magnay, T.; et al. Bringing the Heavy Chain to Light: Creating a Symmetric, Bivalent IgG-Like Bispecific. Antibodies 2020, 9, 62. https://doi.org/10.3390/antib9040062
Ramasubramanian A, Tennyson R, Magnay M, Kathuria S, Travaline T, Jain A, Lord DM, Salemi M, Sullivan C, Magnay T, et al. Bringing the Heavy Chain to Light: Creating a Symmetric, Bivalent IgG-Like Bispecific. Antibodies. 2020; 9(4):62. https://doi.org/10.3390/antib9040062
Chicago/Turabian StyleRamasubramanian, Anusuya, Rachel Tennyson, Maureen Magnay, Sagar Kathuria, Tara Travaline, Annu Jain, Dana M. Lord, Megan Salemi, Caitlin Sullivan, Tristan Magnay, and et al. 2020. "Bringing the Heavy Chain to Light: Creating a Symmetric, Bivalent IgG-Like Bispecific" Antibodies 9, no. 4: 62. https://doi.org/10.3390/antib9040062
APA StyleRamasubramanian, A., Tennyson, R., Magnay, M., Kathuria, S., Travaline, T., Jain, A., Lord, D. M., Salemi, M., Sullivan, C., Magnay, T., Hu, J., Bric-Furlong, E., Rival, P., Zhou, Y., Hoffmann, D., Brondyk, W., Radošević, K., & Chowdhury, P. S. (2020). Bringing the Heavy Chain to Light: Creating a Symmetric, Bivalent IgG-Like Bispecific. Antibodies, 9(4), 62. https://doi.org/10.3390/antib9040062