Targeting the MHC Ligandome by Use of TCR-Like Antibodies


Abstract
1. Introduction
2. Peptide Presentation on MHC
3. Antibodies with Specificity for pMHC Molecules
3.1. TCR-Like mAbs via Hybridoma Technology
3.2. TCR-Like mAbs via Phage Display
3.3. TCR-Like mAbs from Other Display Platforms and Methodologies
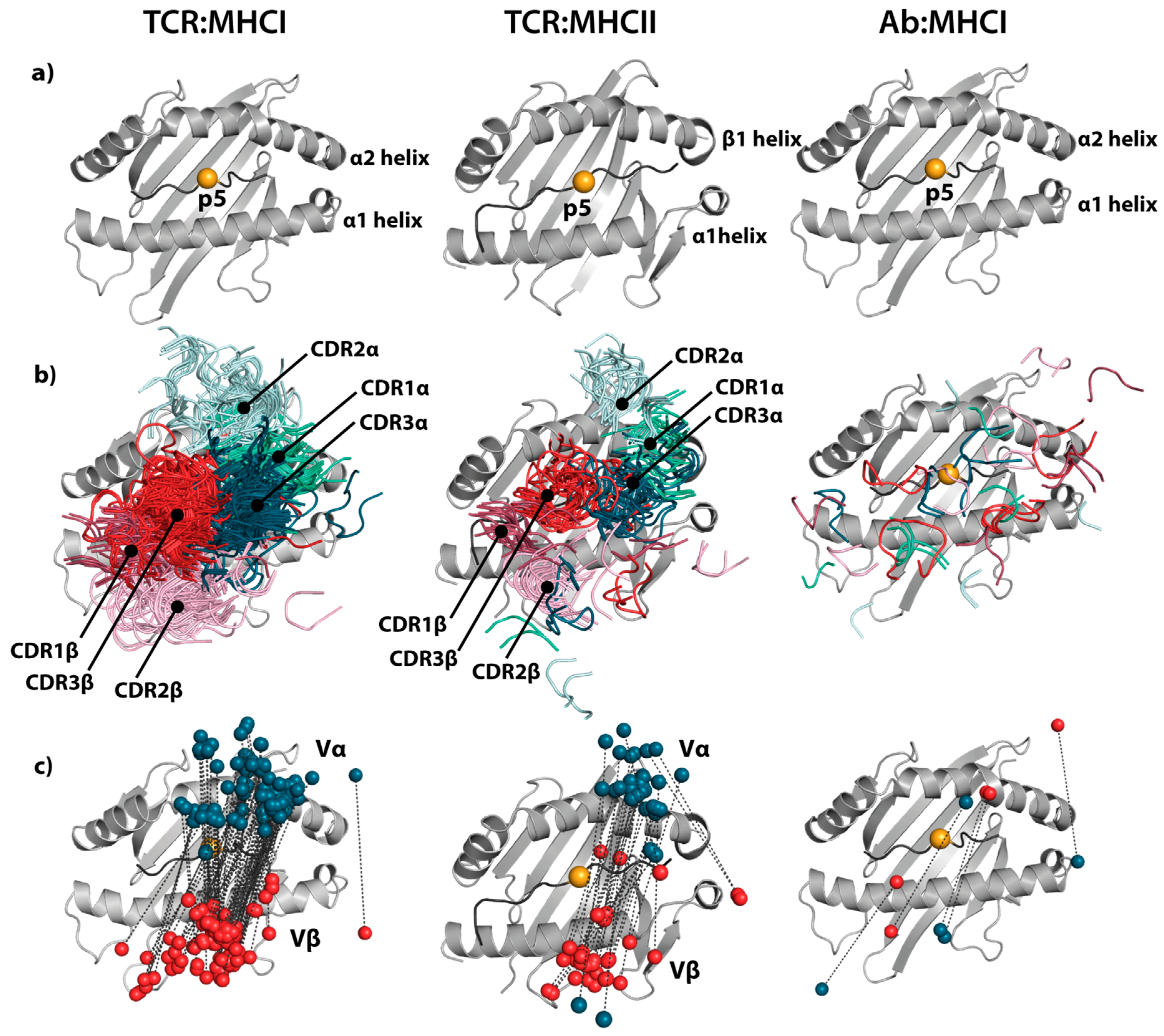
4. pMHC–mAb Structures
5. TCR-Like mAbs as Tools to Study Specific Peptide-Presentation
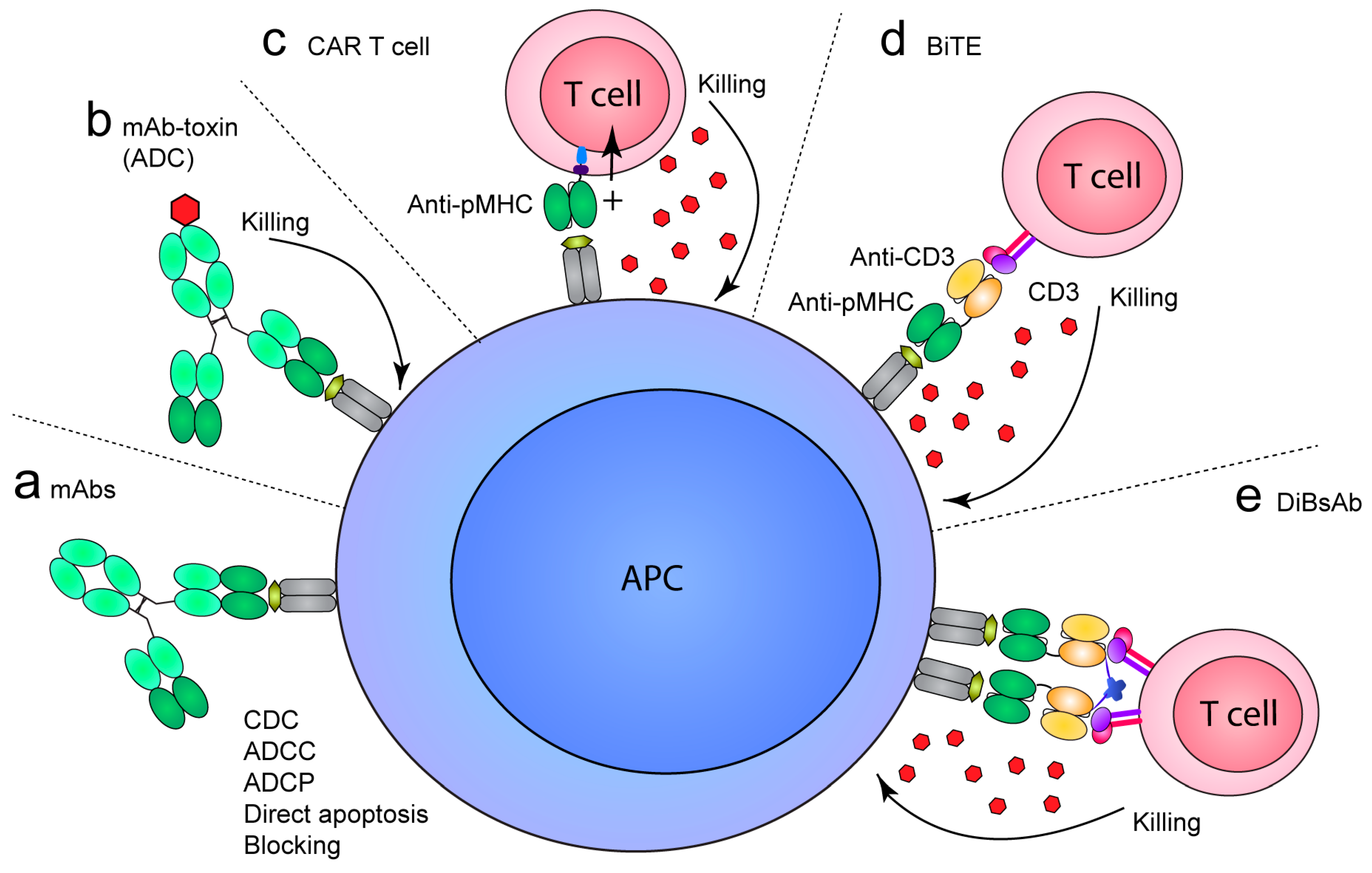
6. TCR-Like mAbs as Therapeutics
6.1. Cancer
6.2. Autoimmunity
7. Summary and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- La Gruta, N.L.; Gras, S.; Daley, S.R.; Thomas, P.G.; Rossjohn, J. Understanding the drivers of mhc restriction of t cell receptors. Nat. Rev. Immunol. 2018, 18, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How tcrs bind mhcs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta t cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.D.; Neuberger, M.S. Affinity dependence of the b cell response to antigen: A threshold, a ceiling, and the importance of off-rate. Immunity 1998, 8, 751–759. [Google Scholar] [CrossRef]
- Gunnarsen, K.S.; Høydahl, L.S.; Neumann, R.S.; Bjerregaard-Andersen, K.; Nilssen, N.R.; Sollid, L.M.; Sandlie, I.; Løset, G.Å. Soluble t-cell receptor design influences functional yield in an E. coli chaperone-assisted expression system. PLoS ONE 2018, 13, e0195868. [Google Scholar] [CrossRef] [PubMed]
- Laugel, B.; Boulter, J.M.; Lissin, N.; Vuidepot, A.; Li, Y.; Gostick, E.; Crotty, L.E.; Douek, D.C.; Hemelaar, J.; Price, D.A.; et al. Design of soluble recombinant t cell receptors for antigen targeting and t cell inhibition. J. Biol. Chem. 2005, 280, 1882–1892. [Google Scholar] [CrossRef]
- Subbramanian, R.A.; Moriya, C.; Martin, K.L.; Peyerl, F.W.; Hasegawa, A.; Naoi, A.; Chhay, H.; Autissier, P.; Gorgone, D.A.; Lifton, M.A.; et al. Engineered t-cell receptor tetramers bind mhc-peptide complexes with high affinity. Nat. Biotechnol. 2004, 22, 1429–1434. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Mempel, T.R.; Mazo, I.B.; Liu, B.; Artyomov, M.N.; Zheng, H.; Peixoto, A.; Flynn, M.P.; Senman, B.; Junt, T.; et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for t cell activation. Nat. Biotechnol. 2008, 9, 282–291. [Google Scholar]
- Zhu, X.; Belmont, H.J.; Price-Schiavi, S.; Liu, B.; Lee, H.I.; Fernandez, M.; Wong, R.L.; Builes, J.; Rhode, P.R.; Wong, H.C. Visualization of p53(264-272)/hla-a*0201 complexes naturally presented on tumor cell surface by a multimeric soluble single-chain t cell receptor. J. Immunol. 2006, 176, 3223–3232. [Google Scholar] [CrossRef]
- Holler, P.D.; Holman, P.O.; Shusta, E.V.; O’Herrin, S.; Wittrup, K.D.; Kranz, D.M. In vitro evolution of a t cell receptor with high affinity for peptide/mhc. Proc. Natl. Acad. Sci. USA 2000, 97, 5387–5392. [Google Scholar] [CrossRef]
- Weber, K.S.; Donermeyer, D.L.; Allen, P.M.; Kranz, D.M. Class ii-restricted t cell receptor engineered in vitro for higher affinity retains peptide specificity and function. Proc. Natl. Acad. Sci. USA 2005, 102, 19033–19038. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered t cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. Ny-eso-1-specific tcr-engineered t cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bennett, A.D.; Zheng, Z.; Wang, Q.J.; Robbins, P.F.; Yu, L.Y.; Li, Y.; Molloy, P.E.; Dunn, S.M.; Jakobsen, B.K.; et al. High-affinity tcrs generated by phage display provide cd4+ t cells with the ability to recognize and kill tumor cell lines. J. Immunol. 2007, 179, 5845–5854. [Google Scholar] [CrossRef]
- Holler, P.D.; Chlewicki, L.K.; Kranz, D.M. Tcrs with high affinity for foreign pmhc show self-reactivity. Nat. Biotechnol. 2003, 4, 55–62. [Google Scholar] [CrossRef]
- Stone, J.D.; Harris, D.T.; Kranz, D.M. Tcr affinity for p/mhc formed by tumor antigens that are self-proteins: Impact on efficacy and toxicity. Curr. Opin. Immunol. 2015, 33, 16–22. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a titin-derived hla-a1-presented peptide as a cross-reactive target for engineered mage a3-directed t cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced t cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is autoimmunity the achilles’ heel of cancer immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef]
- Hickman, E.S.; Lomax, M.E.; Jakobsen, B.K. Antigen selection for enhanced affinity t-cell receptor-based cancer therapies. J. Biomol. Screen. 2016, 21, 769–785. [Google Scholar] [CrossRef]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. Immtac-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal tcr-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human t-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of mhc class i and mhc class ii antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Mohan, J.F.; Unanue, E.R. Unconventional recognition of peptides by t cells and the implications for autoimmunity. Nat. Rev. Immunol. 2012, 12, 721–728. [Google Scholar] [CrossRef]
- Cruz, F.M.; Colbert, J.D.; Merino, E.; Kriegsman, B.A.; Rock, K.L. The biology and underlying mechanisms of cross-presentation of exogenous antigens on mhc-i molecules. Annu. Rev. Immunol. 2017, 35, 149–176. [Google Scholar] [CrossRef]
- Paul, S.; Karosiene, E.; Dhanda, S.K.; Jurtz, V.; Edwards, L.; Nielsen, M.; Sette, A.; Peters, B. Determination of a predictive cleavage motif for eluted major histocompatibility complex class ii ligands. Front. Immunol. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Shao, W.; Pedrioli, P.G.A.; Wolski, W.; Scurtescu, C.; Schmid, E.; Vizcaíno, J.A.; Courcelles, M.; Schuster, H.; Kowalewski, D.; Marino, F.; et al. The systemhc atlas project. Nucleic Acids Res. 2017, 46, D1237–D1247. [Google Scholar] [CrossRef]
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The role of mass spectrometry and proteogenomics in the advancement of hla epitope prediction. Proteomics 2018, 18, e1700259. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. Hla variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Solberg, O.D.; Mack, S.J.; Lancaster, A.K.; Single, R.M.; Tsai, Y.; Sanchez-Mazas, A.; Thomson, G. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: A meta-analytic review of 497 population studies. Hum. Immunol. 2008, 69, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.M.; Buhler, S.; Roessli, D.; Sanchez-Mazas, A.; HLA-net 2013 Collaboration. The hla-net gene[rate] pipeline for effective hla data analysis and its application to 145 population samples from europe and neighbouring areas. Tissue Antigens 2014, 83, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Wylie, D.E.; Sherman, L.A.; Klinman, N.R. Participation of the major histocompatibility complex in antibody recognition of viral antigens expressed on infected cells. J. Exp. Med. 1982, 155, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Froscher, B.G.; Klinman, N.R. Immunization with sv40-transformed cells yields mainly mhc-restricted monoclonal antibodies. J. Exp. Med. 1986, 164, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Duc, H.T.; Rucay, P.; Righenzi, S.; Halle-Pannenko, O.; Kourilsky, P. Monoclonal antibodies directed against t cell epitopes presented by class i mhc antigens. Int. Immunol. 1993, 5, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.S.; Stryhn, A.; Hansen, B.E.; Fugger, L.; Engberg, J.; Buus, S. A recombinant antibody with the antigen-specific, major histocompatibility complex-restricted specificity of t cells. Proc. Natl. Acad. Sci. USA 1996, 93, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Porgador, A.; Yewdell, J.W.; Deng, Y.; Bennink, J.R.; Germain, R.N. Localization, quantitation, and in situ detection of specific peptide-mhc class i complexes using a monoclonal antibody. Immunity 1997, 6, 715–726. [Google Scholar] [CrossRef]
- Chames, P.; Hufton, S.E.; Coulie, P.G.; Uchanska-Ziegler, B.; Hoogenboom, H.R. Direct selection of a human antibody fragment directed against the tumor t-cell epitope hla-a1-mage-a1 from a nonimmunized phage-fab library. Proc. Natl. Acad. Sci. USA 2000, 97, 7969–7974. [Google Scholar] [CrossRef] [PubMed]
- Hulsmeyer, M.; Chames, P.; Hillig, R.C.; Stanfield, R.L.; Held, G.; Coulie, P.G.; Alings, C.; Wille, G.; Saenger, W.; Uchanska-Ziegler, B.; et al. A major histocompatibility complex-peptide-restricted antibody and t cell receptor molecules recognize their target by distinct binding modes: Crystal structure of human leukocyte antigen (hla)-a1-mage-a1 in complex with fab-hyb3. J. Biol. Chem. 2005, 280, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Willemsen, R.A.; Rojas, G.; Dieckmann, D.; Rem, L.; Schuler, G.; Bolhuis, R.L.; Hoogenboom, H.R. Tcr-like human antibodies expressed on human ctls mediate antibody affinity-dependent cytolytic activity. J. immunol. 2002, 169, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, O.; Verma, B.; Lightfoot, S.; Jain, R.; Rawat, A.; McNair, S.; Caseltine, S.; Mojsilovic, A.; Gupta, P.; Neethling, F.; et al. An hla-presented fragment of macrophage migration inhibitory factor is a therapeutic target for invasive breast cancer. J. Immunol. 2011, 186, 6607–6616. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Hoffmann, N.; Farago, M.; Hoogenboom, H.R.; Eisenbach, L.; Reiter, Y. Direct detection and quantitation of a distinct t-cell epitope derived from tumor-specific epithelial cell-associated mucin using human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of t cells. Cancer Res. 2002, 62, 5835–5844. [Google Scholar] [PubMed]
- Denkberg, G.; Cohen, C.J.; Lev, A.; Chames, P.; Hoogenboom, H.R.; Reiter, Y. Direct visualization of distinct t cell epitopes derived from a melanoma tumor-associated antigen by using human recombinant antibodies with mhc- restricted t cell receptor-like specificity. Proc. Natl. Acad. Sci. USA 2002, 99, 9421–9426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting nk-92 for anti-melanoma activity by a tcr-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Denkberg, G.; Lev, A.; Eisenbach, L.; Benhar, I.; Reiter, Y. Selective targeting of melanoma and apcs using a recombinant antibody with tcr-like specificity directed toward a melanoma differentiation antigen. J. Immunol. 2003, 171, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Denkberg, G.; Cohen, C.J.; Tzukerman, M.; Skorecki, K.L.; Chames, P.; Hoogenboom, H.R.; Reiter, Y. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of t cells directed toward the widely expressed tumor t-cell epitopes of the telomerase catalytic subunit. Cancer Res. 2002, 62, 3184–3194. [Google Scholar] [PubMed]
- Cohen, C.J.; Sarig, O.; Yamano, Y.; Tomaru, U.; Jacobson, S.; Reiter, Y. Direct phenotypic analysis of human mhc class i antigen presentation: Visualization, quantitation, and in situ detection of human viral epitopes using peptide-specific, mhc-restricted human recombinant antibodies. J. Immunol. 2003, 170, 4349–4361. [Google Scholar] [CrossRef] [PubMed]
- Biddison, W.E.; Turner, R.V.; Gagnon, S.J.; Lev, A.; Cohen, C.J.; Reiter, Y. Tax and m1 peptide/hla-a2-specific fabs and t cell receptors recognize nonidentical structural features on peptide/hla-a2 complexes. J. Immunol. 2003, 171, 3064–3074. [Google Scholar] [CrossRef] [PubMed]
- Held, G.; Matsuo, M.; Epel, M.; Gnjatic, S.; Ritter, G.; Lee, S.Y.; Tai, T.Y.; Cohen, C.J.; Old, L.J.; Pfreundschuh, M.; et al. Dissecting cytotoxic t cell responses towards the ny-eso-1 protein by peptide/mhc-specific antibody fragments. Eur. J. Immunol. 2004, 34, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Jones, G.; Wadle, A.; Hombach, A.; Shenderov, E.; Held, G.; Fischer, E.; Kleber, S.; Nuber, N.; Stenner-Liewen, F.; Bauer, S.; et al. Rational development of high-affinity t-cell receptor-like antibodies. Proc. Natl. Acad. Sci. USA 2009, 106, 5784–5788. [Google Scholar] [CrossRef] [PubMed]
- Held, G.; Wadle, A.; Dauth, N.; Stewart-Jones, G.; Sturm, C.; Thiel, M.; Zwick, C.; Dieckmann, D.; Schuler, G.; Hoogenboom, H.R.; et al. Mhc-peptide-specific antibodies reveal inefficient presentation of an hla-a*0201-restricted, melan-a-derived peptide after active intracellular processing. Eur. J. Immunol. 2007, 37, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Klechevsky, E.; Gallegos, M.; Denkberg, G.; Palucka, K.; Banchereau, J.; Cohen, C.; Reiter, Y. Antitumor activity of immunotoxins with t-cell receptor-like specificity against human melanoma xenografts. Cancer Res. 2008, 68, 6360–6367. [Google Scholar] [CrossRef] [PubMed]
- Wittman, V.P.; Woodburn, D.; Nguyen, T.; Neethling, F.A.; Wright, S.; Weidanz, J.A. Antibody targeting to a class i mhc-peptide epitope promotes tumor cell death. J. Immunol. 2006, 177, 4187–4195. [Google Scholar] [CrossRef] [PubMed]
- Neethling, F.A.; Ramakrishna, V.; Keler, T.; Buchli, R.; Woodburn, T.; Weidanz, J.A. Assessing vaccine potency using tcrmimic antibodies. Vaccine 2008, 26, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Bernardeau, K.; Gouard, S.; David, G.; Ruellan, A.L.; Devys, A.; Barbet, J.; Bonneville, M.; Cherel, M.; Davodeau, F. Assessment of cd8 involvement in t cell clone avidity by direct measurement of hla-a2/mage3 complex density using a high-affinity tcr like monoclonal antibody. Eur. J. Immunol. 2005, 35, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.; Alatrash, G.; He, H.; Ruisaard, K.; Lu, S.; Wygant, J.; McIntyre, B.W.; Ma, Q.; Li, D.; St John, L.; et al. An anti-pr1/hla-a2 t-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 2011, 117, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Verma, B.; Hawkins, O.E.; Neethling, F.A.; Caseltine, S.L.; Largo, S.R.; Hildebrand, W.H.; Weidanz, J.A. Direct discovery and validation of a peptide/mhc epitope expressed in primary human breast cancer cells using a tcrm monoclonal antibody with profound antitumor properties. Cancer Immunol. Immunother. CII 2010, 59, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Weidanz, J.A.; Nguyen, T.; Woodburn, T.; Neethling, F.A.; Chiriva-Internati, M.; Hildebrand, W.H.; Lustgarten, J. Levels of specific peptide-hla class i complex predicts tumor cell susceptibility to ctl killing. J. Immunol. 2006, 177, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.R.; Koide, A.; Leung, B.; Fitzsimmons, J.; Yoder, B.; Yuan, H.; Jay, M.; Sidhu, S.S.; Koide, S.; Collins, E.J. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS ONE 2012, 7, e43746. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rawat, A.; Verma, B.; Markiewski, M.M.; Weidanz, J.A. Antitumor activity of a monoclonal antibody targeting major histocompatibility complex class I–Her2 peptide complexes. J. Nat. Cancer Inst. 2013, 105, 202–218. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, A.Y.; Dao, T.; Gejman, R.S.; Jarvis, C.A.; Scott, A.; Dubrovsky, L.; Mathias, M.D.; Korontsvit, T.; Zakhaleva, V.; Curcio, M.; et al. A therapeutic t cell receptor mimic antibody targets tumor-associated prame peptide/hla-i antigens. J. Clin. Investig. 2017, 127, 2705–2718. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting alpha-fetoprotein (afp)-mhc complex with car t-cell therapy for liver cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Dao, T.; Yan, S.; Veomett, N.; Pankov, D.; Zhou, L.; Korontsvit, T.; Scott, A.; Whitten, J.; Maslak, P.; Casey, E.; et al. Targeting the intracellular wt1 oncogene product with a therapeutic human antibody. Sci. Transl. Med. 2013, 5, 176ra133. [Google Scholar] [CrossRef]
- Ataie, N.; Xiang, J.; Cheng, N.; Brea, E.J.; Lu, W.; Scheinberg, D.A.; Liu, C.; Ng, H.L. Structure of a tcr-mimic antibody with target predicts pharmacogenetics. J. Mol. Biol. 2016, 428, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Oren, R.; Hod-Marco, M.; Haus-Cohen, M.; Thomas, S.; Blat, D.; Duvshani, N.; Denkberg, G.; Elbaz, Y.; Benchetrit, F.; Eshhar, Z.; et al. Functional comparison of engineered t cells carrying a native tcr versus tcr-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J. Immunol. 2014, 193, 5733–5743. [Google Scholar] [CrossRef]
- Zhao, Q.; Ahmed, M.; Tassev, D.V.; Hasan, A.; Kuo, T.Y.; Guo, H.F.; O’Reilly, R.J.; Cheung, N.K. Affinity maturation of t-cell receptor-like antibodies for wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015, 29, 2238–2247. [Google Scholar] [CrossRef]
- Sim, A.C.N.; Too, C.T.; Oo, M.Z.; Lai, J.; Eio, M.Y.; Song, Z.; Srinivasan, N.; Tan, D.A.L.; Pang, S.W.; Gan, S.U.; et al. Defining the expression hierarchy of latent t-cell epitopes in epstein-barr virus infection with tcr-like antibodies. Sci. Rep. 2013, 3, 3232. [Google Scholar] [CrossRef]
- Ahmed, M.; Lopez-Albaitero, A.; Pankov, D.; Santich, B.H.; Liu, H.; Yan, S.; Xiang, J.; Wang, P.; Hasan, A.N.; Selvakumar, A.; et al. Tcr-mimic bispecific antibodies targeting lmp2a show potent activity against ebv malignancies. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.B.; Cao, J.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of manabodies specific to hla-restricted epitopes encoded by somatically mutated genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [PubMed]
- Epel, M.; Carmi, I.; Soueid-Baumgarten, S.; Oh, S.K.; Bera, T.; Pastan, I.; Berzofsky, J.; Reiter, Y. Targeting tarp, a novel breast and prostate tumor-associated antigen, with t cell receptor-like human recombinant antibodies. Eur. J. Immunol. 2008, 38, 1706–1720. [Google Scholar] [CrossRef] [PubMed]
- Dass, S.A.; Norazmi, M.N.; Dominguez, A.A.; Miguel, M.; Tye, G.J. Generation of a t cell receptor (tcr)-like single domain antibody (sdab) against a mycobacterium tuberculosis (mtb) heat shock protein (hsp) 16kda antigen presented by human leukocyte antigen (hla)-a*02. Mol. Immunol. 2018, 101, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Weidanz, J.A.; Piazza, P.; Hickman-Miller, H.; Woodburn, D.; Nguyen, T.; Wahl, A.; Neethling, F.; Chiriva-Internati, M.; Rinaldo, C.R.; Hildebrand, W.H. Development and implementation of a direct detection, quantitation and validation system for class i mhc self-peptide epitopes. J. Immunol. Methods 2007, 318, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, Y.; Akahori, Y.; Murayama, Y.; Shiraishi, K.; Tsuzuki-Iba, S.; Endoh, A.; Tsujikawa, J.; Demachi-Okamura, A.; Hiramatsu, K.; Saji, H.; et al. Construction and molecular characterization of a t-cell receptor-like antibody and car-t cells specific for minor histocompatibility antigen ha-1h. Gene Ther. 2014, 21, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Michaeli, Y.; Denkberg, G.; Sinik, K.; Lantzy, L.; Chih-Sheng, C.; Beauverd, C.; Ziv, T.; Romero, P.; Reiter, Y. Expression hierarchy of t cell epitopes from melanoma differentiation antigens: Unexpected high level presentation of tyrosinase-hla-a2 complexes revealed by peptide-specific, mhc-restricted, tcr-like antibodies. J. Immunol. 2009, 182, 6328–6341. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bentley, C.; Yates, J.; Salimi, M.; Greig, J.; Wiblin, S.; Hassanali, T.; Banham, A.H. Engineering chimeric human and mouse major histocompatibility complex (mhc) class i tetramers for the production of t-cell receptor (tcr) mimic antibodies. PLoS ONE 2017, 12, e0176642. [Google Scholar] [CrossRef] [PubMed]
- Spanier, J.A.; Frederick, D.R.; Taylor, J.J.; Heffernan, J.R.; Kotov, D.I.; Martinov, T.; Osum, K.C.; Ruggiero, J.L.; Rust, B.J.; Landry, S.J.; et al. Efficient generation of monoclonal antibodies against peptide in the context of mhcii using magnetic enrichment. Nat. Commun. 2016, 7, 11804. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.B.; Lo, D.; Rath, S.; Brinster, R.L.; Flavell, R.A.; Slanetz, A.; Janeway, C.A., Jr. A novel mhc class ii epitope expressed in thymic medulla but not cortex. Nature 1989, 338, 765–768. [Google Scholar] [CrossRef]
- Muraille, E.; Gounon, P.; Cazareth, J.; Hoebeke, J.; Lippuner, C.; Davalos-Misslitz, A.; Aebischer, T.; Muller, S.; Glaichenhaus, N.; Mougneau, E. Direct visualization of peptide/mhc complexes at the surface and in the intracellular compartments of cells infected in vivo by leishmania major. PLoS Pathog. 2010, 6, e1001154. [Google Scholar] [CrossRef]
- Zhang, L.; Crawford, F.; Yu, L.; Michels, A.; Nakayama, M.; Davidson, H.W.; Kappler, J.W.; Eisenbarth, G.S. Monoclonal antibody blocking the recognition of an insulin peptide-mhc complex modulates type 1 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Reis e Sousa, C.; Germain, R.N. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class ii complexes formed by processing of exogenous protein. Proc. Natl. Acad. Sci. USA 1997, 94, 13856–13861. [Google Scholar] [CrossRef] [PubMed]
- Dadaglio, G.; Nelson, C.A.; Deck, M.B.; Petzold, S.J.; Unanue, E.R. Characterization and quantitation of peptide-mhc complexes produced from hen egg lysozyme using a monoclonal antibody. Immunity 1997, 6, 727–738. [Google Scholar] [CrossRef]
- Aharoni, R.; Teitelbaum, D.; Arnon, R.; Puri, J. Immunomodulation of experimental allergic encephalomyelitis by antibodies to the antigen-ia complex. Nature 1991, 351, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Reay, P.A.; Matsui, K.; Haase, K.; Wulfing, C.; Chien, Y.H.; Davis, M.M. Determination of the relationship between t cell responsiveness and the number of mhc-peptide complexes using specific monoclonal antibodies. J. Immunol. 2000, 164, 5626–5634. [Google Scholar] [CrossRef]
- Wolpl, A.; Halder, T.; Kalbacher, H.; Neumeyer, H.; Siemoneit, K.; Goldmann, S.F.; Eiermann, T.H. Human monoclonal antibody with t-cell-like specificity recognizes mhc class i self-peptide presented by hla-dr1 on activated cells. Tissue Antigens 1998, 51, 258–269. [Google Scholar] [CrossRef]
- Krogsgaard, M.; Wucherpfennig, K.W.; Cannella, B.; Hansen, B.E.; Svejgaard, A.; Pyrdol, J.; Ditzel, H.; Raine, C.; Engberg, J.; Fugger, L. Visualization of myelin basic protein (mbp) t cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (hla)-dr2-mbp 85-99 complex. J. Exp. Med. 2000, 191, 1395–1412. [Google Scholar] [CrossRef]
- Dahan, R.; Tabul, M.; Chou, Y.K.; Meza-Romero, R.; Andrew, S.; Ferro, A.J.; Burrows, G.G.; Offner, H.; Vandenbark, A.A.; Reiter, Y. Tcr-like antibodies distinguish conformational and functional differences in two- versus four-domain auto reactive mhc class ii-peptide complexes. Eur. J. Immunol. 2011, 41, 1465–1479. [Google Scholar] [CrossRef]
- Dahan, R.; Gebe, J.A.; Preisinger, A.; James, E.A.; Tendler, M.; Nepom, G.T.; Reiter, Y. Antigen-specific immunomodulation for type 1 diabetes by novel recombinant antibodies directed against diabetes-associates auto-reactive t cell epitope. J. Autoimmun. 2013, 47, 83–93. [Google Scholar] [CrossRef]
- Steenbakkers, P.G.; Baeten, D.; Rovers, E.; Veys, E.M.; Rijnders, A.W.; Meijerink, J.; De Keyser, F.; Boots, A.M. Localization of mhc class ii/human cartilage glycoprotein-39 complexes in synovia of rheumatoid arthritis patients using complex-specific monoclonal antibodies. J. Immunol. 2003, 170, 5719–5727. [Google Scholar] [CrossRef]
- Høydahl, L.S.; Richter, L.; Frick, R.; Snir, O.; Gunnarsen, K.S.; Landsverk, O.J.; Iversen, R.; Jeliazkov, J.R.; Gray, J.J.; Bergseng, E.; et al. Plasma cells are the most abundant gluten peptide mhc-expressing cells in inflamed intestinal tissues from patients with celiac disease. Gastroenterology 2018, 156, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Ponsel, D.; Neugebauer, J.; Ladetzki-Baehs, K.; Tissot, K. High affinity, developability and functional size: The holy grail of combinatorial antibody library generation. Molecules 2011, 16, 3675–3700. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A. Antibodies from phage antibody libraries. J. Immunol. Methods 2004, 290, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Høydahl, L.S.; Nilssen, N.R.; Gunnarsen, K.S.; Pre, M.F.; Iversen, R.; Roos, N.; Chen, X.; Michaelsen, T.E.; Sollid, L.M.; Sandlie, I.; et al. Multivalent pix phage display selects for distinct and improved antibody properties. Sci. Rep. 2016, 6, 39066. [Google Scholar] [CrossRef] [PubMed]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Noy, R.; Eppel, M.; Haus-Cohen, M.; Klechevsky, E.; Mekler, O.; Michaeli, Y.; Denkberg, G.; Reiter, Y. T-cell receptor-like antibodies: Novel reagents for clinical cancer immunology and immunotherapy. Expert Rev. Anticancer Ther. 2005, 5, 523–536. [Google Scholar] [CrossRef]
- Bradbury, A.R.; Sidhu, S.; Dubel, S.; McCafferty, J. Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol. 2011, 29, 245–254. [Google Scholar] [CrossRef]
- Willemsen, R.A.; Ronteltap, C.; Chames, P.; Debets, R.; Bolhuis, R.L. T cell retargeting with mhc class i-restricted antibodies: The cd28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J. Immunol. 2005, 174, 7853–7858. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef] [PubMed]
- Sethi, D.K.; Schubert, D.A.; Anders, A.K.; Heroux, A.; Bonsor, D.A.; Thomas, C.P.; Sundberg, E.J.; Pyrdol, J.; Wucherpfennig, K.W. A highly tilted binding mode by a self-reactive t cell receptor results in altered engagement of peptide and mhc. J. Exp. Med. 2011, 208, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Tynan, F.E.; Burrows, S.R.; Buckle, A.M.; Clements, C.S.; Borg, N.A.; Miles, J.J.; Beddoe, T.; Whisstock, J.C.; Wilce, M.C.; Silins, S.L.; et al. T cell receptor recognition of a ‘super-bulged’ major histocompatibility complex class i-bound peptide. Nat. Biotechnol. 2005, 6, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.J.; Narayanan, S.; Liu, B.; Birnbaum, M.E.; Kruse, A.C.; Bowerman, N.A.; Chen, W.; Levin, A.M.; Connolly, J.M.; Zhu, C.; et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 2011, 35, 681–693. [Google Scholar] [CrossRef]
- Beringer, D.X.; Kleijwegt, F.S.; Wiede, F.; van der Slik, A.R.; Loh, K.L.; Petersen, J.; Dudek, N.L.; Duinkerken, G.; Laban, S.; Joosten, A.; et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat. Biotechnol. 2015, 16, 1153–1161. [Google Scholar] [CrossRef]
- Leem, J.; de Oliveira, S.H.P.; Krawczyk, K.; Deane, C.M. Stcrdab: The structural t-cell receptor database. Nucleic Acids Res. 2018, 46, D406–D412. [Google Scholar] [CrossRef]
- Liu, Y.C.; Miles, J.J.; Neller, M.A.; Gostick, E.; Price, D.A.; Purcell, A.W.; McCluskey, J.; Burrows, S.R.; Rossjohn, J.; Gras, S. Highly divergent t-cell receptor binding modes underlie specific recognition of a bulged viral peptide bound to a human leukocyte antigen class i molecule. J. Biol. Chem. 2013, 288, 15442–15454. [Google Scholar] [CrossRef]
- Hahn, M.; Nicholson, M.J.; Pyrdol, J.; Wucherpfennig, K.W. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune t cell receptor. Nat. Biotechnol. 2005, 6, 490–496. [Google Scholar] [CrossRef]
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity 2009, 30, 348–357. [Google Scholar] [CrossRef]
- Collins, E.J.; Riddle, D.S. Tcr-mhc docking orientation: Natural selection, or thymic selection? Immunol. Res. 2008, 41, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.C.; Adams, E.J. How the t cell receptor sees antigen—A structural view. Cell 2005, 122, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, F.; Sarafova, S.D.; Park, J.-H.; Tai, X.; Pobezinsky, L.; Guinter, T.I.; Adoro, S.; Adams, A.; Sharrow, S.O.; Feigenbaum, L.; et al. Deletion of cd4 and cd8 coreceptors permits generation of αβt cells that recognize antigens independently of the mhc. Immunity 2007, 27, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, F.; Tikhonova, A.N.; Pobezinsky, L.A.; Tai, X.; Kimura, M.Y.; Le Saout, C.; Guinter, T.I.; Adams, A.; Sharrow, S.O.; Bernhardt, G.; et al. Lck availability during thymic selection determines the recognition specificity of the t cell repertoire. Cell 2013, 154, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Scott-Browne, J.P.; White, J.; Kappler, J.W.; Gapin, L.; Marrack, P. Germline-encoded amino acids in the alphabeta t-cell receptor control thymic selection. Nature 2009, 458, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Garcia, K.C.; Adams, J.J.; Feng, D.; Ely, L.K. The molecular basis of tcr germline bias for mhc is surprisingly simple. Nat. Biotechnol. 2009, 10, 143–147. [Google Scholar]
- Sharon, E.; Sibener, L.V.; Battle, A.; Fraser, H.B.; Garcia, K.C.; Pritchard, J.K. Genetic variation in mhc proteins is associated with t cell receptor expression biases. Nat. Genet. 2016, 48, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Stanfield, R.L. Antibody-antigen interactions: New structures and new conformational changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867. [Google Scholar] [CrossRef]
- Mareeva, T.; Martinez-Hackert, E.; Sykulev, Y. How a t cell receptor-like antibody recognizes major histocompatibility complex-bound peptide. J. Biol. Chem. 2008, 283, 29053–29059. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; LeMaoult, J.; Nikolic-Zugic, J. The mode of ligand recognition by two peptide:Mhc class i-specific monoclonal antibodies. J. Immunol. 1999, 163, 3286–3294. [Google Scholar] [PubMed]
- Stryhn, A.; Andersen, P.S.; Pedersen, L.O.; Svejgaard, A.; Holm, A.; Thorpe, C.J.; Fugger, L.; Buus, S.; Engberg, J. Shared fine specificity between t-cell receptors and an antibody recognizing a peptide/major histocompatibility class i complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10338–10342. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.V.; Unanue, E.R. Quantitation of antigen-presenting cell mhc class ii/peptide complexes necessary for t-cell stimulation. Nature 1990, 346, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Biotechnol. 2004, 5, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Tan, W.J.; Too, C.T.; Choo, J.A.; Wong, L.H.; Mustafa, F.B.; Srinivasan, N.; Lim, A.P.; Zhong, Y.; Gascoigne, N.R.; et al. Targeting epstein-barr virus-transformed b lymphoblastoid cells using antibodies with t-cell receptor-like specificities. Blood 2016, 128, 1396–1407. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sosa, R.A.; Murphey, C.; Ji, N.; Cardona, A.E.; Forsthuber, T.G. The kinetics of myelin antigen uptake by myeloid cells in the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 5848–5857. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Steenbakkers, P.G.; Rijnders, A.M.; Boots, A.M.; Veys, E.M.; De Keyser, F. Detection of major histocompatibility complex/human cartilage gp-39 complexes in rheumatoid arthritis synovitis as a specific and independent histologic marker. Arthritis Rheum. 2004, 50, 444–451. [Google Scholar] [CrossRef]
- Reiter, Y.; Di Carlo, A.; Fugger, L.; Engberg, J.; Pastan, I. Peptide-specific killing of antigen-presenting cells by a recombinant antibody-toxin fusion protein targeted to major histocompatibility complex/peptide class i complexes with t cell receptor-like specificity. Proc. Natl. Acad. Sci. USA 1997, 94, 4631–4636. [Google Scholar] [CrossRef]
- Veomett, N.; Dao, T.; Liu, H.; Xiang, J.; Pankov, D.; Dubrovsky, L.; Whitten, J.A.; Park, S.M.; Korontsvit, T.; Zakhaleva, V.; et al. Therapeutic efficacy of an fc-enhanced tcr-like antibody to the intracellular wt1 oncoprotein. Clin. Cancer Res. 2014, 20, 4036–4046. [Google Scholar] [CrossRef]
- Dao, T.; Pankov, D.; Scott, A.; Korontsvit, T.; Zakhaleva, V.; Xu, Y.; Xiang, J.; Yan, S.; de Morais Guerreiro, M.D.; Veomett, N.; et al. Therapeutic bispecific t-cell engager antibody targeting the intracellular oncoprotein wt1. Nat. Biotechnol. 2015, 33, 1079–1086. [Google Scholar] [CrossRef]
- Rafiq, S.; Purdon, T.J.; Daniyan, A.F.; Koneru, M.; Dao, T.; Liu, C.; Scheinberg, D.A.; Brentjens, R.J. Optimized t-cell receptor-mimic chimeric antigen receptor t cells directed toward the intracellular wilms tumor 1 antigen. Leukemia 2017, 31, 1788–1797. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a t cell–engaging antibody. Science (New York, N.Y.) 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. 2010, 10, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sosinowski, T.; Aaron, R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (car) t cells targeting a pathogenic mhc class ii:Peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2018, 96, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Boitard, C.; Bendelac, A.; Richard, M.F.; Carnaud, C.; Bach, J.F. Prevention of diabetes in nonobese diabetic mice by anti-i-a monoclonal antibodies: Transfer of protection by splenic t cells. Proc. Natl. Acad. Sci. USA 1988, 85, 9719–9723. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Adams, K.J.; Bossi, G.; Wright, D.E.; Stacey, A.R.; Bedke, N.; Martinez-Hague, R.; Blat, D.; Humbert, L.; Buchanan, H.; et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered t cell receptor anti-cd3 bispecific (immtac) molecules. PLoS ONE 2018, 13, e0205491. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, R.S.; Dittel, B.; Hafler, D.; von Herrath, M.; Nestle, F.O. Unraveling the autoimmune translational research process layer by layer. Nat. Med. 2012, 18, 35–41. [Google Scholar] [CrossRef] [PubMed]
- The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [CrossRef]
- Bassani-Sternberg, M.; Chong, C.; Guillaume, P.; Solleder, M.; Pak, H.; Gannon, P.O.; Kandalaft, L.E.; Coukos, G.; Gfeller, D. Deciphering hla-i motifs across hla peptidomes improves neo-antigen predictions and identifies allostery regulating hla specificity. PLoS Comput. Biol. 2017, 13, e1005725. [Google Scholar] [CrossRef]
- Kisielow, J.; Obermair, F.J.; Kopf, M. Deciphering cd4+ t cell specificity using novel mhc-tcr chimeric receptors. Nat. Immunol. 2019, 20, 652–662. [Google Scholar] [CrossRef]
- Gee, M.H.; Han, A.; Lofgren, S.M.; Beausang, J.F.; Mendoza, J.L.; Birnbaum, M.E.; Bethune, M.T.; Fischer, S.; Yang, X.; Gomez-Eerland, R.; et al. Antigen identification for orphan t cell receptors expressed on tumor-infiltrating lymphocytes. Cell 2018, 172, 549–563.e16. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Antigen-specific therapeutic approaches for autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.; Santamaria, P. Nanoparticle-based approaches to immune tolerance for the treatment of autoimmune diseases. Eur. J. Immunol. 2018, 48, 751–756. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antigen | Epitope | MHC | Clone | Indication | Affinity 1 | Selection Method | References |
|---|---|---|---|---|---|---|---|
| PR8 | NA | H-2Dk/H-2Kb | NA | Infection | ND | Hybridoma | [35] |
| SV40 | NA | H-2Kb | Infection | ND | Hybridoma | [36] | |
| NP | NA | Kd | X5.3.7 | Infection | ND | Hybridoma | [37] |
| HA | FESTGNLI | Kk | Fab13.4.1 | Infection | 50 nM | Immunization/phage | [38] |
| pOV8 | SIINFEKL | Kk | 25-D1.16 2 | Model antigen | ND | Hybridoma | [39] |
| MAGE-A1 | EADPTGHSY | HLA-A*0101 | G8 | Cancer | 250 nM | Phage | [40] |
| MAGE-A1 | EADPTGHSY | HLA-A*0101 | Hyb3 2,3 | Cancer | 14 nM | Phage | [41,42] |
| MIF | FLSELTQQL | HLA-A*0201 | RL21A | Cancer | 24.4 nM | Hybridoma | [43] |
| MUC1 | LLLTVLTVV | HLA-A*0201 | M3A1, M3B8 | Cancer | ND | Phage | [44] |
| gp100 | KTWGQYWQV | HLA-A*0201 | G2D12, | Cancer | ND | Phage | [45] |
| gp100 | ITDQVPFSV | HLA-A*0201 | 1A7 | Cancer | ND | Phage | [45] |
| gp100 | YLEPGPVTA | HLA-A*0201 | 2F1 | Cancer | ND | Phage | [45] |
| gp100 | ITDQVPFSV | HLA-A*0201 | GPA7 4 | Cancer | 180 nM | Phage | [46] |
| gp100 | IMDQVPFSV | HLA-A*0201 | G1 | Cancer | ND | Phage | [47] |
| hTERT | ILAKFLHWL | HLA-A*0201 | 4A9, 4G9 | Cancer | ND | Phage | [48] |
| hTERT | RLVDDFLLV | HLA-A*0201 | 3G3, 3H2 | Cancer | ND | Phage | [48] |
| HTLV-1 | LLFGYPVYV | HLA-A*0201 | T3E3, T3F2 | Infection | ND | Phage | [49] |
| M1 | GILGFVFTL | HLA-A*0201 | M1-A2, M1-D1, M1-D12, M1-G8 | Infection | ND | Phage | [50] |
| NY-ESO-1 | SLLMWITQC | HLA-A*0201 | 3M4E5, 3M4F4 2 | Cancer | 46–95 nM | Phage | [51,52] |
| NY-ESO-1 | SLLMWITQC | HLA-A*0201 | T1 3 | Cancer | 2–4 nM | Phage | [52] |
| MelanA/MART-1 | EAAGIGILTV | HLA-A*0201 | E5, H4 | Cancer | ND | Phage | [53] |
| MelanA/MART-1 | ELAGIGILTV | HLA-A*0201 | 2M3F11, 3N4E9, 2N4B4, 3N4B5 | Cancer | ND | Phage | [53] |
| MelanA/MART-1 | EAAGIGILTV | HLA-A*0201 | CAG10, CLA12 | Cancer | ND | Phage | [54] |
| hCGβ | GVLPALPQV | HLA-A*0201 | RL4B/3.2G1 | Cancer | ND | Hybridoma | [55] |
| hCGβ | GVLPALPQV | HLA-A*0201 | 1B10 | Cancer | ND | Hybridoma | [56] |
| hCGβ | TMTRVLQGV | HLA-A*0201 | 3F9 | Cancer | ND | Hybridoma | [56] |
| MAGE3 | FLWGPRALV | HLA-A*0201 | 7D4 | Cancer | ND | Hybridoma | [57] |
| PR1 | VLQELNVTV | HLA-A*0201 | 8F4 | Cancer | 9.9 nM | Hybridoma | [58] |
| P68 RNA Helicase | YLLPAIVHI | HLA-A*0201 | RL6A | Cancer | 0.42 nM | Hybridoma | [59] |
| HER2/Neu | KIFGSLAFL | HLA-A*0201 | 1B8 | Cancer | ND | Hybridoma | [60] |
| HER2/Neu | KIFGSLAFL | HLA-A*0201 | fE75 | Cancer | 59 nM | Phage | [61] |
| HER2/Neu | KIFGSLAFL | HLA-A*0201 | RL1B | Cancer | 2.69 nM | Hybridoma | [62] |
| Calreticulin | MLSVPLLL | HLA-A*0201 | fML | Cancer | 79 nM | Phage | [61] |
| PRAME | ALYVDSLFFL | HLA-A*0201 | Pr20 | Cancer | ND | Phage | [63] |
| AFP | FMNKFIYEI | HLA-A*0201 | ET1402L1 | Cancer | ND | Phage | [64] |
| WT1 | RMFPNAPYL | HLA-A*0201 | ESK1 2 | Cancer | ND | Phage | [65,66] |
| WT1 | RMFPNAPYL | HLA-A*0201 | F2, F3 | Cancer | 400, 30 nM | Phage | [67] |
| WT1 | RMFPNAPYL | HLA-A*0201 | Clone45 | Cancer | 263 nM | Phage | [68] |
| WT1 | RMFPNAPYL | HLA-A*0201 | Q2L 3,5 | Cancer | 3 nM | Yeast | [68] |
| LMP1 | YLLEMLWRL | HLA-A*0201 | L1 | EBV-cancer | 1.85 nM | Hybridoma | [69] |
| LMP2A | CLGGLLTMV | HLA-A*0201 | L2 | EBV-cancer | 6.98 nM | Hybridoma | [69] |
| EBNA1 | FMVFLQTHI | HLA-A*0201 | E1 | EBV-cancer | 6.02 nM | Hybridoma | [69] |
| LMP2A | CLGGLLTMV | HLA-A*0201 | 38 | EBV-cancer | ND | Phage | [70] |
| LMP2A | CLGGLLTMV | HLA-A*0201 | 38-2 3 | EBV-cancer | ND | Phage | [70] |
| KRAS | KLVVVGAVGV | HLA-A*0201 | D10 | Cancer | ND | Phage | [71] |
| KRAS | KLVVVGAVGV | HLA-A*0201 | D10-7 3 | Cancer | ND | Phage | [71] |
| EGFR | KITDFGRAK | HLA-A3 | C9 | Cancer | ND | Phage | [71] |
| TARP | FLRNFSLML | HLA-A*0201 | D2 | Cancer | ND | Phage | [72] |
| HSP16 | GILTVSVAV | HLA-A*0201 | A2/Ab(clone3) 4 | Infection | ND | Phage | [73] |
| eIF4G | VLMTEDIKL | HLA-A*0201 | 4F7 | Infection | ND | Hybridoma | [74] |
| HA-1H | VLHDDLLEA | HLA-A*0201 | #131 | Cancer | 19.9 nM | Phage | [75] |
| Tyrosinase | YMDGTMSQV | HLA-A*0201 | TA2 | Cancer | ND | Phage | [76] |
| p53 | RMPEAAPPV | HLA-A*0201 | T1-116C | Cancer | ND | Hybridoma | |
| p53 | RMPEAAPPV | HLA-A*0201 | T1-29D and T1-84C | Cancer | ND | Hybridoma | [77] |
| p53 | GLAPPQHLIRV | HLA-A*0201 | T2-108A, T2-2A, T2-116A | Cancer | ND | Hybridoma | [77] |
| Antigen | Epitope | MHC | Clone | Indication | Affinity 1 | Selection method | References |
|---|---|---|---|---|---|---|---|
| 2W | EAWGALANWAVDSA | I-Ab | W6 | Infection | 3.4 nM | Hybridoma | [78] |
| Eα | ASFEAQGALANIAVDKA | I-Ab | Y-Ae | Self-peptide | 0.48 nM | Hybridoma | [79] |
| LACK | ICFSPSLEHPIVVSGSWD | I-Ad | 2C44 | Infection | ND | Hybridoma | [80] |
| insulin | HLVERLYLVCGEEG | I-Ag7 | mAb287 | Autoimmunity | 130 nM | Hybridoma | [81] |
| p63 | RTRPLWVRME | I-Ag7 | FS1 | Autoimmunity | 0.02 nM | Hybridoma | [78] |
| HEL | NTDGSTDYGILQINSR | I-Ak | B6Ge1 | Model antigen | ND | Hybridoma | [82] |
| HEL | KGTDVQAWIRGCRL | I-Ak | D8H21 | Model antigen | ND | Hybridoma | [82] |
| HEL | DGSTDYGILQINSRW | I-Ak | Aw3.18 | Model antigen | 12.4 nM | Hybridoma | [83] |
| MBP | VHFFKNIVTPRTP | I-As | B-7-1, B-18-7, C34-72 | Autoimmunity | ND | Hybridoma | [84] |
| MCC | IAYLKQATK | I-Ek | D4,G32,G35 | Model antigen | 700 nM | Hybridoma | [85] |
| HLA-A2 | SDWRFLRGYHQYA | HLA-DR1 | UL-5A1 | Self-peptide | ND | Hybridoma | [86] |
| MBP | ENPVVHFFKNIVTPR | HLA-DR2b | MK16 | Autoimmunity | ND | Immunization/phage | [87] |
| MOG | MEVGWYRPPFSRVVHLYRNGK | HLA-DR2b | 2E4, 1F11, 3A3, 3H5, 2C3 | Autoimmunity | 30–150 nM | Phage | [88] |
| GAD65 | NFFRMVISNPAAT | HLA-DR4.1 | G1H12, G3H8, D2 | Autoimmunity | 64 nM, 104 nM | Phage | [89] |
| HC gp-39 | RSFTLASSETGVG | HLA-DR4.1 | 12A | Autoimmunity | ND | Hybridoma | [90] |
| Gluten | QLQPFPQPELPY | HLA-DQ2.5 | 106, 107 2 | Autoimmunity | 70 nM, 100 nM | Phage | [91] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Høydahl, L.S.; Frick, R.; Sandlie, I.; Løset, G.Å. Targeting the MHC Ligandome by Use of TCR-Like Antibodies. Antibodies 2019, 8, 32. https://doi.org/10.3390/antib8020032
Høydahl LS, Frick R, Sandlie I, Løset GÅ. Targeting the MHC Ligandome by Use of TCR-Like Antibodies. Antibodies. 2019; 8(2):32. https://doi.org/10.3390/antib8020032
Chicago/Turabian StyleHøydahl, Lene Støkken, Rahel Frick, Inger Sandlie, and Geir Åge Løset. 2019. "Targeting the MHC Ligandome by Use of TCR-Like Antibodies" Antibodies 8, no. 2: 32. https://doi.org/10.3390/antib8020032
APA StyleHøydahl, L. S., Frick, R., Sandlie, I., & Løset, G. Å. (2019). Targeting the MHC Ligandome by Use of TCR-Like Antibodies. Antibodies, 8(2), 32. https://doi.org/10.3390/antib8020032