
Antiphospholipid Antibodies: Their Origin and Development

Abstract
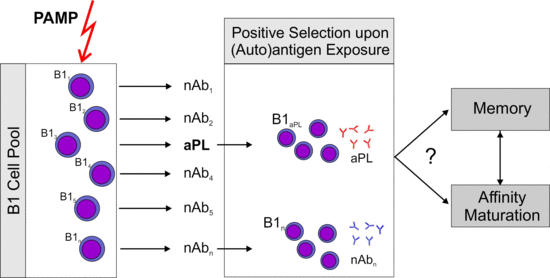
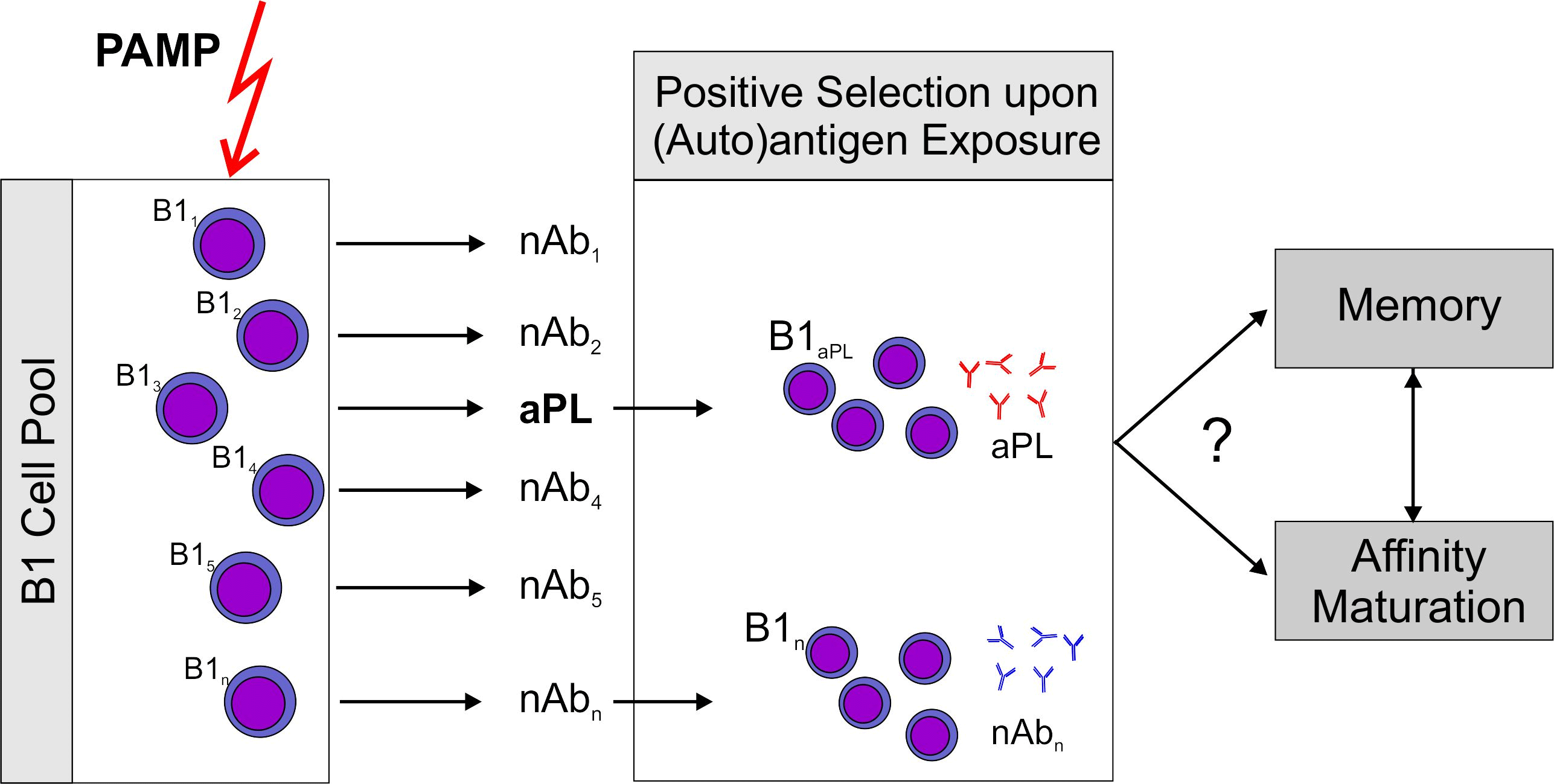
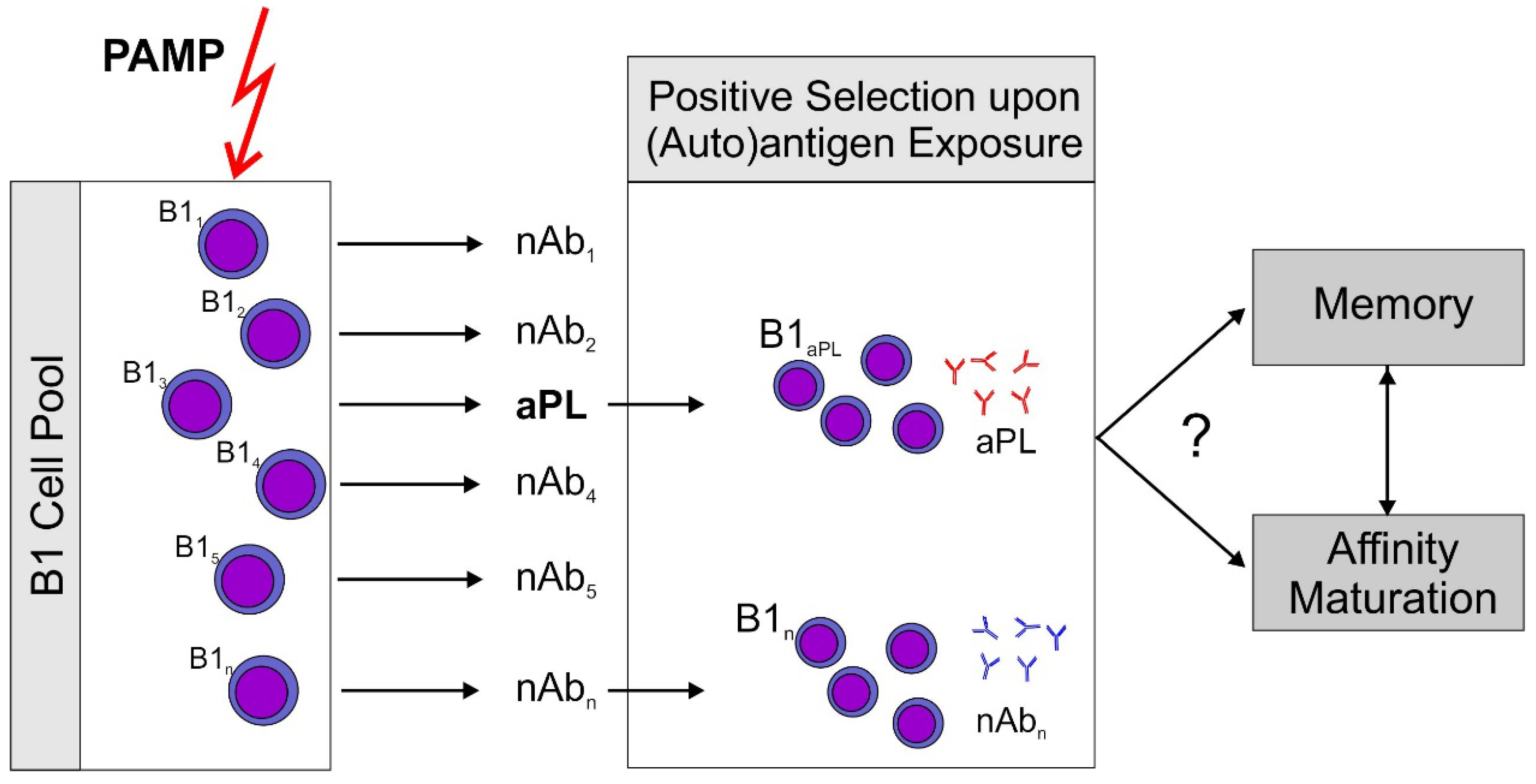
:
{kind=link}
{kind=link}
1. Introduction
2. Are aPL Part of the Natural Antibody Repertoire?
2.1. Animal Models
2.2. Infections and aPL
2.3. Analysis of Human Monoclonal aPL
2.4. What Is the Role of Antigen Driven Maturation?
2.5. Memory B-cells
3. Genetic Aspects of aPL
4. Conclusions and Outlook
Conflicts of Interest
Glossary
| Germline sequence | Antibodies are encoded in the genome as every other protein. However, for certain segments (V, D, and J) of the variable chains of antibodies there are several coding gene segments. The term germline sequence refers to an antibody sequence encoded in the genome. Germline sequences can be modified by→antigen driven maturation. If an antibody has a germline encoded sequence this suggests that no antigen driven maturation has occurred, yet. |
| V,(D), J (or somatic) -recombination | The process of combining the gene segments for the desired V, D, and J-segments of the variable chains and of removal of surplus gene segments from the B cell genome is referred to as somatic recombination or V, (D), J-recombination. It is mediated by VDJ-recombinase, a multi-enzyme complex. Somatic recombination is the first step in antibody production that generates a huge potential diversity with more than 1011 theoretical combinations. |
| Somatic (hyper)mutation | During B-cell proliferation which occurs after antigen contact, the B-cell receptor locus can undergo an extremely high rate of somatic mutations which is several orders of magnitude greater than the spontaneous mutation rate. Most of the somatic mutations are found in specific regions of the antibody molecule, the so called hypervariable or complementarity determining regions (CDR). Somatic mutation generates B-cell clones which produce antibodies with different affinity to their antigen. The clones producing higher affinity antibodies are positively selected. Thus, somatic hypermutation is key to→antigen driven maturation of B-cell clones. |
| Antigen driven maturation | Also called affinity maturation, antigen driven maturation is the central process of adaptive immunity. By selecting higher affinity clones and deleting lower affinity clones, there is continuous improvement of antibody affinity to the relevant target antigen. A significant deviation of the sequence of an antibody from the known germline genes indicates antigen driven maturation. |
| Anti-idiotype | An idiotype describes the sum of the variable parts of a specific antibody. By this, it also includes the antigen binding site of the antibody. An anti-idiotype is an antibody that binds to a specific idiotype. In theory anti-idiotypes may mimick the antigen/epitope of the original antibody. |
Abbreviations
| aPL | antiphospholipid antibody |
| APS | antiphospholipid syndrome |
| β2GPI | β2 glycoprotein I |
| SLE | systemic lupus erythematosus |
References
- Wassermann, A.; Neisser, A.; Bruck, C. Eine serodiagnostische Reaktion bei Syphilis. Dtsch. Med. Wochenschr. 1906, 31, 745–746. (In German) [Google Scholar] [CrossRef]
- Landsteiner, K.; Müller, R.; Poetzl, O. Zur Frage der Komplementbindungsreaktionen bei Syphilis. Wien. Klin. Wochenschr. 1907, 20, 1565–1567. (In German) [Google Scholar]
- Pangborn, M. Isolation and purification of a serologically active phospholipid from beef heart. J. Biol. Chem. 1942, 143, 247–256. [Google Scholar]
- Harris, E.N.; Gharavi, A.E.; Boey, M.L.; Patel, B.M.; Mackworth-Young, C.G.; Loizou, S.; Hughes, G.R. Anticardio-lipin antibodies: Detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 1983, 8361, 1211–1214. [Google Scholar] [CrossRef]
- Hughes, G.R. The anticardiolipin syndrome. Clin. Exp. Rheumatol. 1985, 3, 285–286. [Google Scholar] [PubMed]
- Bertolaccini, M.L.; Amengual, O.; Andreoli, L.; Atsumi, T.; Chighizola, C.B.; Forastiero, R.; de Groot, P.; Lakos, G.; Lambert, M.; Meroni, P.; et al. 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun. Rev. 2014, 13, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of the antiphospholipid syndrome: understand-ding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, Y. The Michael Mason prize: Pathogenic antiphospholipid antibodies, stressed out antigens and the deployment of decoys. Rheumatology 2012, 51, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Poulton, K.; Rahman, A.; Giles, I. Examining how antiphospholipid antibodies activate intracellular signaling pathways: A systematic review. Sem. Arthritis. Rheum. 2012, 41, 720–736. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. New Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Du, V.X.; Kelchtermans, H.; de Groot, P.G.; de Laat, B. From antibody to clinical phenotype, the black box of the antiphospholipid syndrome: Pathogenic mechanisms of the antiphospholipid syndrome. Thromb. Res. 2013, 132, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Merashli, M.; Noureldine, J.H.A.; Uthman, I.; Khamashta, M. Antiphospholipid syndrome: An update. Eur. J. Clin. Investig. 2015, 45, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Ding, J.K. Natural antibodies bridge innate and adaptive immunity. J. Immunol. 2015, 194, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Griffin, D.O.; Holodick, N.E.; Quach, T.D.; Kaku, H. Human B-1 cells take the stage. Ann. New York Acad. Sci. 2013, 1285, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Youinou, P.; Renaudineau, Y. The antiphospholipid syndrome as a model for B cell-induced autoimmune diseases. Thromb. Res. 2004, 114, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T. Do antiphospholipid antibodies develop for a purpose? Curr. Rheumatol. Rep. 2006, 8, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Bakimer, R.; Fishman, P.; Blank, M.; Sredni, B.; Djaldetti, M.; Shoenfeld, Y. Induction of primary antiphospholipid syndrome in mice by immunization with a human monoclonal anticardiolipin antibody (H-3). J. Clin. Investig. 1992, 89, 1558–1563. [Google Scholar] [CrossRef] [PubMed]
- Shoenfeld, Y. Idiotypic induction of autoimmunity: A new aspect of the idiotypic network. FASEB J. 1994, 8, 1296–1301. [Google Scholar] [PubMed]
- Pierangeli, S.S.; Harris, E.N. Induction of phospholipid-binding antibodies in mice and rabbits by immunization with human β2 glycoprotein 1 or anticardiolipin antibodies alone. Clin. Exp. Immunol. 1993, 93, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, S.S.; Liu, S.W.; Anderson, G.; Barker, J.H.; Harris, E.N. Thrombogenic properties of murine anti-cardiolipin antibodies induced by beta 2 glycoprotein 1 and human immunoglobulin G antiphospholipid antibodies. Circulation 1996, 94, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Prinz, N.; Clemens, N.; Strand, D.; Pütz, I.; Lorenz, M.; Daiber, A.; Stein, P.; Degreif, A.; Radsak, M.; Schild, H.; et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 2011, 118, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Saitoh, S.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Yokogawa, M.; Takaishi, M.; Nakajima, K.; Kamijima, R.; Fujimoto, C.; Kataoka, S.; Terada, Y.; Sano, S. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: A new model of systemic Lupus erythematosus. Arthritis Rheumatol. 2014, 66, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Arimura, K.; Uto, T.; Fukaya, T.; Nakamura, T.; Choijookhuu, N.; Hishikawa, Y.; Sato, K. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, M.; Matsuda, J. Induction of anticardiolipin antibody and/or lupus anticoagulant in rabbits by immunization with lipoteichoic acid, lipopolysaccharide and lipid, A. Lupus 1996, 5, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Lopez-Olivo, M.A.; Pinto-Patarroyo, G.P.; Suarez-Almazor, M.E. Systematic review of case reports of antiphospholipid syndrome following infection. Lupus 2016, in press. [Google Scholar] [CrossRef]
- Gharavi, A.E.; Pierangeli, S.S.; Espinola, R.G.; Liu, X.; Colden-Stanfield, M.; Harris, E.N. Antiphospholipid antibodies induced in mice by immunization with a cytomegalovirus-derived peptide cause thrombosis and activation of endothelial cells in vivo. Arthritis Rheum. 2002, 46, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Krause, I.; Fridkin, M.; Keller, N.; Kopolovic, J.; Goldberg, I.; Tobar, A.; Shoenfeld, Y. Bacterial induction of autoantibodies to beta2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J. Clin. Investig. 2002, 109, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.E.; Pierangeli, S.S.; Harris, E.N. Viral origin of antiphospholipid antibodies: Endothelial cell activa-tion and thrombus enhancement by CMV peptide-induced APL antibodies. Immunobiology 2003, 207, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Shoenfeld, Y.; Blank, M.; Cervera, R.; Font, J.; Raschi, E.; Meroni, P.L. Infectious origin of the antiphospholipid syndrome. Ann. Rheum. Dis. 2006, 65, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Winn, R.; Nugent, K. Catastrophic antiphospholipid syndrome in a community-acquired methicillin-resistant Staphylococcus aureus infection: A review of pathogenesis with a case for molecular mimicry. Autoimmun. Rev. 2011, 10, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Justo, D.; Finn, T.; Atzmony, L.; Guy, N.; Steinvil, A. Thrombosis associated with acute cytomegalo-virus infection: A meta-analysis. Eur. J. Intern. Med. 2011, 22, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Uthman, I.; Tabbarah, Z.; Gharavi, A.E. Hughes syndrome associated with cytomegalovirus infection. Lupus 1999, 8, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Akahoshi, M.; Irino, K.; Kimoto, Y.; Arinobu, Y.; Niiro, H.; Tsukamoto, H.; Horiuchi, T.; Akashi, K. Transient antiphospholipid syndrome associated with primary cytomegalovirus infection: A case report and literature review. Case Rep. Rheumatol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Giles, I.P.; Haley, J.D.; Nagl, S.; Isenberg, D.A.; Latchman, D.S.; Rahman, A. A systematic analysis of sequences of human antiphospholipid and anti-b2-glycoprotein I antibodies: The importance of somatic mutations and certain sequence motifs. Semin. Arthritis Rheum. 2003, 32, 246–265. [Google Scholar] [CrossRef] [PubMed]
- Elkon, K.; Casali, P. Nature and functions of autoantibodies. Nat. Clin. Pract. Rheumatol. 2008, 4, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Lieby, P.; Soley, A.; Levallois, H.; Hugel, B.; Freyssinet, J.-M.; Cerutti, M.; Pasquali, J.-L.; Martin, T. The clonal analysis of anticardiolipin antibodies in a single patient with primary antiphospholipid syndrome reveals extreme antibody heterogeneity. Blood 2001, 97, 3820–3828. [Google Scholar] [CrossRef] [PubMed]
- Lieby, P.; Soley, A.; Knapp, A.-M.; Cerutti, M.; Freyssinet, J.-M.; Pasqualit, J.-L.; Martin, T. Memory B cells producing somatically mutated antiphospholipid antibodies are present in healthy individuals. Blood 2003, 102, 2459–2465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pasquali, J.-L.; Nehme, H.; Korganow, A.-S.; Martin, T. Antiphospholipid antibodies: Recent progresses on their origin and pathogenicity. Joint Bone Spine 2004, 71, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Lieby, P.; Poindron, V.; Roussi, S.; Klein, C.; Knapp, A.M.; Garaud, J.C.; Cerutti, M.; Martin, T.; Pasquali, J.L. Patho-genic antiphospholipid antibody: An antigen-selected needle in a haystack. Blood 2004, 104, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- von Landenberg, C.; Lackner, K.J.; von Landenberg, P.; Lang, B.; Schmitz, G. Isolation and character-rization of two human monoclonal antiphospholipid IgG from patients with autoimmune disease. J. Autoimmun. 1999, 13, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, C.; Fischer, C.; Ochsenhirt, V.; Neukirch, C.; Lackner, K.J.; von Landenberg, P. Generation and characterization of three monoclonal IgM antiphospholipid antibodies recognizing different phospholipid antigens. Ann. N. Y. Acad. Sci. 2005, 1051, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Prinz, N.; Häuser, F.; Lorenz, M.; Lackner, K.J.; von Landenberg, P. Structural and functional characterization of a human IgG monoclonal antiphospholipid antibody. Immunobiology 2011, 216, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Prinz, N.; Clemens, N.; Canisius, A.; Lackner, K.J. Endosomal NADPH-oxidase is critical for induction of the tissue factor gene in monocytes and endothelial cells. Lessons from the antiphospholipid syndrome. Thromb. Haemost. 2013, 109, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Müller-Calleja, N.; Köhler, A.; Siebald, B.; Canisius, A.; Orning, C.; Radsak, M.; Stein, P.; Mönnikes, R.; Lackner, K.J. Cofactor-independent antiphospholipid antibodies activate the NLRP3-inflammasome via endosomal NADPH-oxidase: Implications for the antiphospholipid syndrome. Thromb. Haemost. 2015, 113, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, D.; Müller-Calleja, N.; Jäckel, S.; Luchmann, K.; Mönnikes, R.; Kiouptsi, K.; Reinhardt, C.; Jurk, K.; Walter, U.; Lackner, K.J. Cofactor Independent Human Antiphospholipid Antibodies Induce Venous Thrombosis in Mice. J. Thromb. Haemost. 2016, 14, 1011–1020. [Google Scholar] [PubMed]
- Girardi, G.; Berman, J.; Redecha, P.; Spruce, L.; Thurman, J.M.; Kraus, D.; Hollmann, T.J.; Casali, P.; Caroll, M.C.; Wetsel, R.A.; et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Investig. 2003, 112, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Ikematsu, W.; Luan, F.L.; La Rosa, L.; Beltrami, B.; Nicoletti, F.; Buyon, J.P.; Meroni, P.L.; Balestrieri, G.; Casali, P. Human anticardiolipin monoclonal autoantibodies cause placental necrosis and fetal loss in BALB/c mice. Arthritis Rheum. 1998, 41, 1026–1039. [Google Scholar]
- Ritchie, D.S.; Sainani, A.; D'Souza, A.; Grigg, A.P. Passive donor-to-recipient transfer of antiphospholipid syndrome following allogeneic stem-cell transplantation. Am. J. Hematol. 2005, 79, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.O.; Holodick, N.E.; Rothstein, T.L. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70−. J. Exp. Med. 2011, 208, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G. To B1 or not to B1: That really is still the question! Blood 2013, 121, 5109–5110. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Hirota, S.; Hirano, K.; Fujii, H.; Sugahara-Tobinai, A.; Ishii, T.; Harigae, H.; Takai, T. Human CD43+ B cells are closely related not only to memory B cells phenotypically but also to plasmablasts developmentally in healthy individuals. Int. Immunol. 2015, 27, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Allugupalli, K.R.; Leong, J.M.; Woodland, R.T.; Muramatsu, M.; Honjo, T.; Gerstein, R.M. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity 2004, 21, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ghosn, E.E.; Cole, L.E.; Obukhanych, T.V.; Sadate-Ngatchou, P.; Vogel, S.N.; Herzenberg, L.A.; Herzenberg, L.A. Antigen-specific memory in B-1a and its relationship to natural immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 5388–5393. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ghosn, E.E.; Cole, L.E.; Obukhanych, T.V.; Sadate-Ngatchou, P.; Vogel, S.N.; Herzenberg, L.A.; Herzenberg, L.A. Antigen-specific antibody responses in B-1a and their relationship to natural immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 5382–5387. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Wang, X.; Kao, A.H.; Barmada, M.M.; Clarke, A.; Ramsey-Goldman, R.; Manzi, S.; Demirci, F.Y. Genome-wide association study of antiphospholipid antibodies. Autoimmun. Dis. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Müller-Calleja, N.; Rossmann, H.; Müller, C.; Wild, P.; Blankenberg, S.; Pfeiffer, N.; Binder, H.; Beutel, M.E.; Manukyan, D.; Zeller, T.; et al. Antiphospholipid antibodies in a large population-based cohort: genome-wide associations and effects on monocyte gene expression. Thromb. Haemost. 2016, 116. [Google Scholar] [CrossRef] [PubMed]
- Hirose, N.; Williams, R.; Alberts, A.R.; Furie, R.A.; Chartash, E.K.; Jain, R.I.; Sison, C.; Lahita, R.G.; Merrill, J.T.; Cucurull, E.; et al. A role for the polymorphism at position 247 of the beta2-glyco-protein I gene in the generation of anti-beta2-glycoprotein I antibodies in the antiphospholipid syndrome. Arthritis Rheum. 1999, 42, 1655–1661. [Google Scholar] [CrossRef]
- Chamorro, A.J.; Marcos, M.; Mirón-Canelo, J.A.; Cervera, R.; Eapinosa, G. Val247Leu β2-glycoprotein-I allelic variant is associated with antiphospholipid syndrome: Systematic review and meta-analysis. Autoimmun. Rev. 2012, 11, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.D.; Iuliano, A.; Cantarini, L.; Galeazzi, M. Genetic aspects of the antiphospholipid syndrome: An update. Autoimmun. Rev. 2016, 15, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, R.; Zeth, K.; Diederichs, K.; Gries, A.; Kostner, G.M.; Laggner, P.; Prassl, R. Crystal structure of human beta2-glycoprotein I: Implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. 1999, 18, 6228–6239. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Kriechbaum, M.; Gries, A.; Kostner, G.M.; Laggner, P.; Prassl, R. Solution structure of human and bovine beta(2)-glycoprotein I revealed by small-angle X-ray scattering. J. Mol. Biol. 2002, 321, 85–97. [Google Scholar] [CrossRef]
- Agar, C.; van Os, G.M.; Mörgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T.; Derksen, R.H.; Meijers, J.C.; de Groot, P.G. Beta2-glycoprotein I can exist in 2 conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Ninivaggi, M.; Kelchtermans, H.; Lindhout, T.; de Laat, B. Conformation of beta2glycoprotein I and its effect on coagulation. Thromb. Res. 2012, 130, S33–S36. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lackner, K.J.; Müller-Calleja, N. Antiphospholipid Antibodies: Their Origin and Development. Antibodies 2016, 5, 15. https://doi.org/10.3390/antib5020015
Lackner KJ, Müller-Calleja N. Antiphospholipid Antibodies: Their Origin and Development. Antibodies. 2016; 5(2):15. https://doi.org/10.3390/antib5020015
Chicago/Turabian StyleLackner, Karl J., and Nadine Müller-Calleja. 2016. "Antiphospholipid Antibodies: Their Origin and Development" Antibodies 5, no. 2: 15. https://doi.org/10.3390/antib5020015
APA StyleLackner, K. J., & Müller-Calleja, N. (2016). Antiphospholipid Antibodies: Their Origin and Development. Antibodies, 5(2), 15. https://doi.org/10.3390/antib5020015