
B Cell Epitope-Based Vaccination Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Peptide Vaccines Induce Specific Cytotoxic T Cells
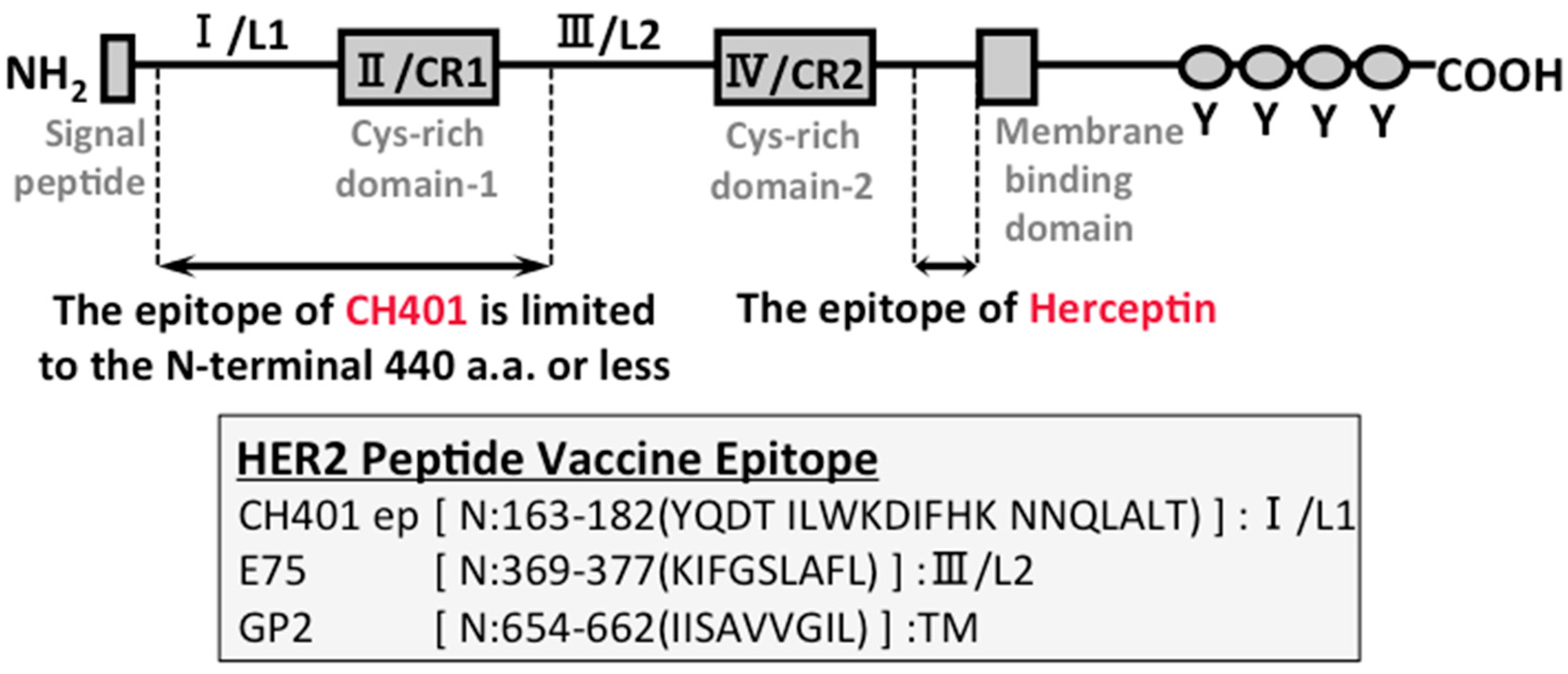
3. Why Can Peptide Vaccines Not Fully Induce Cellular Immunity to Reject Established Cancerous Conditions?
4. Immune Checkpoint Antibodies
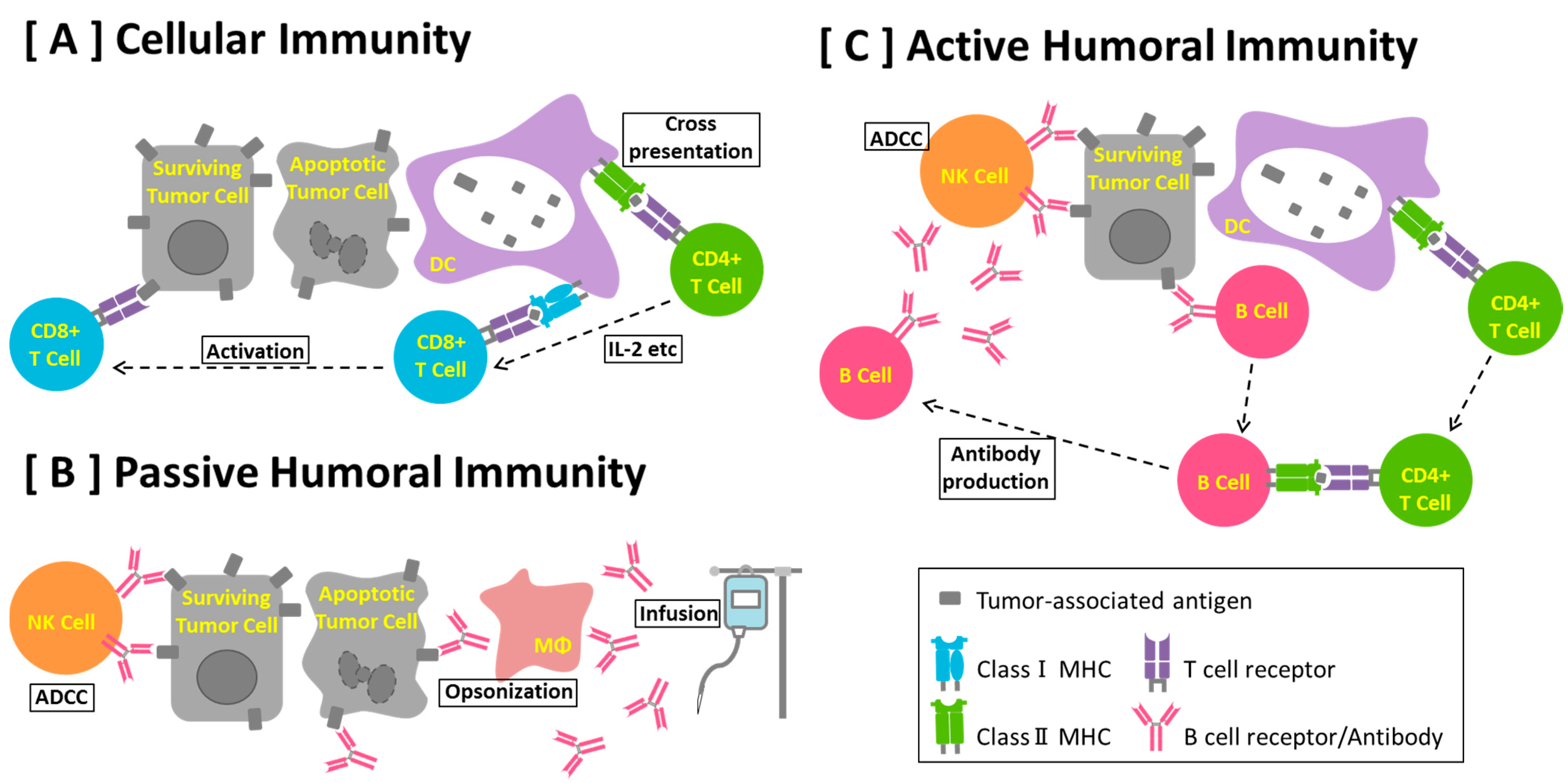
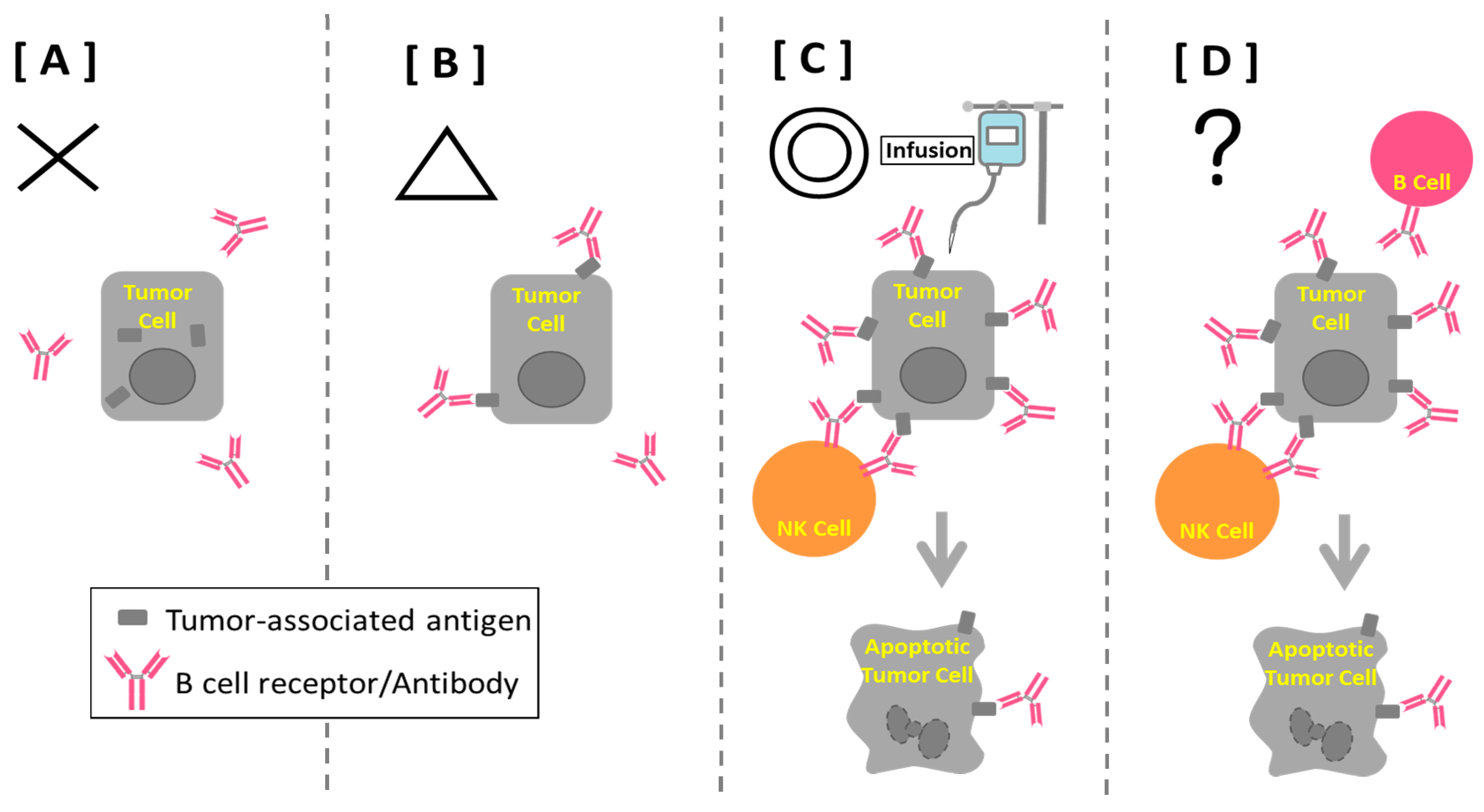
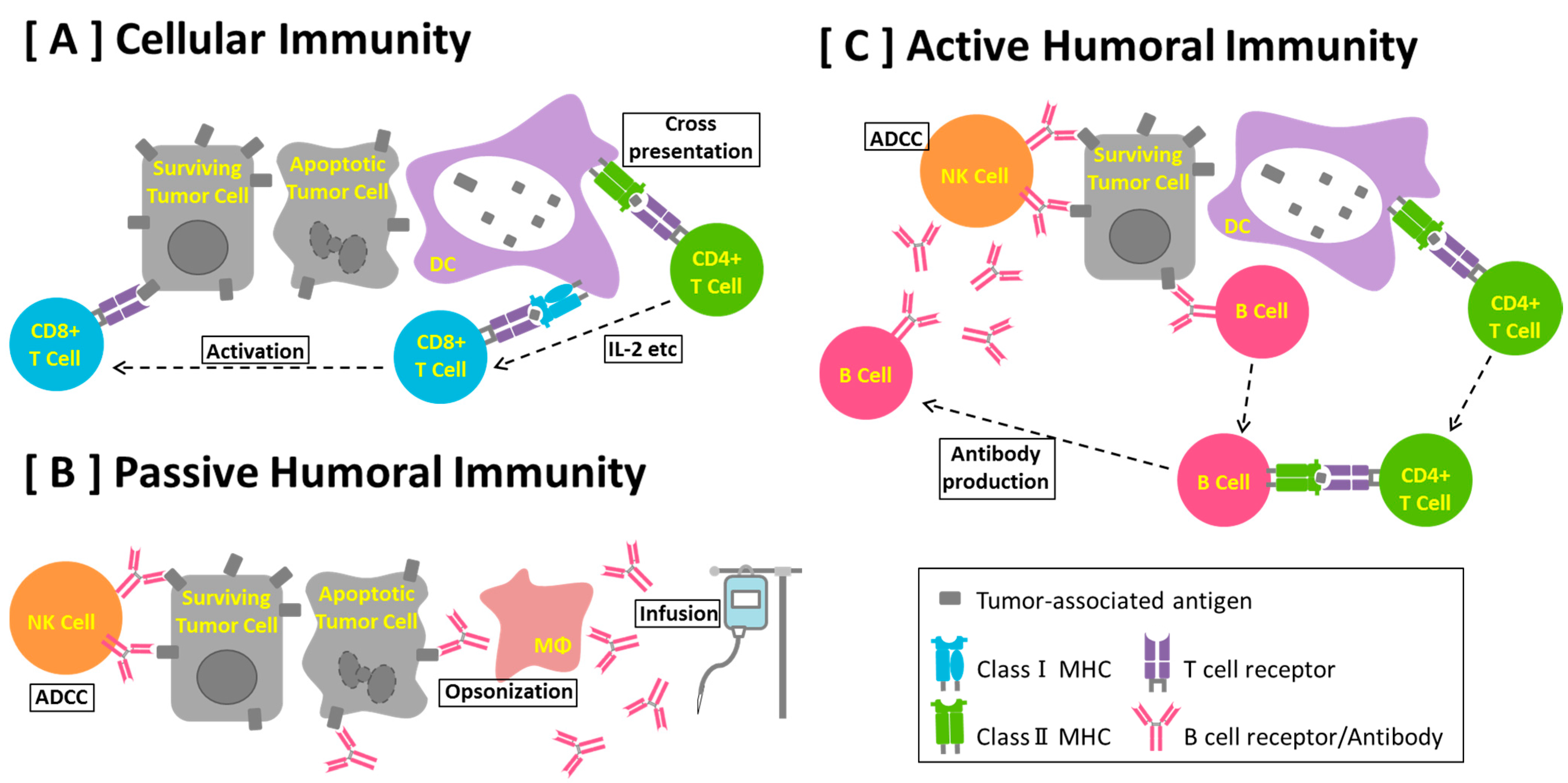
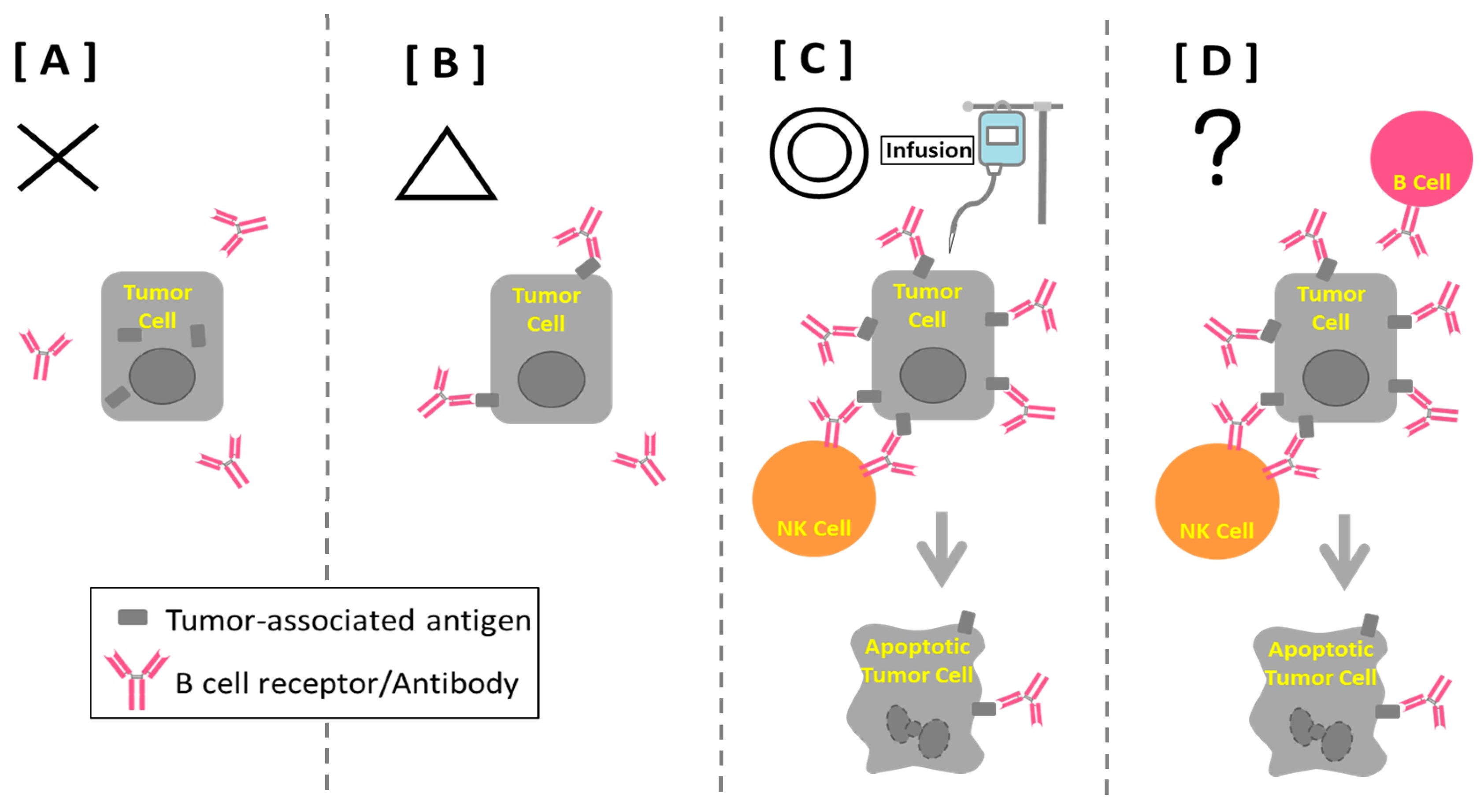
5. Anti-Cancer Antibody
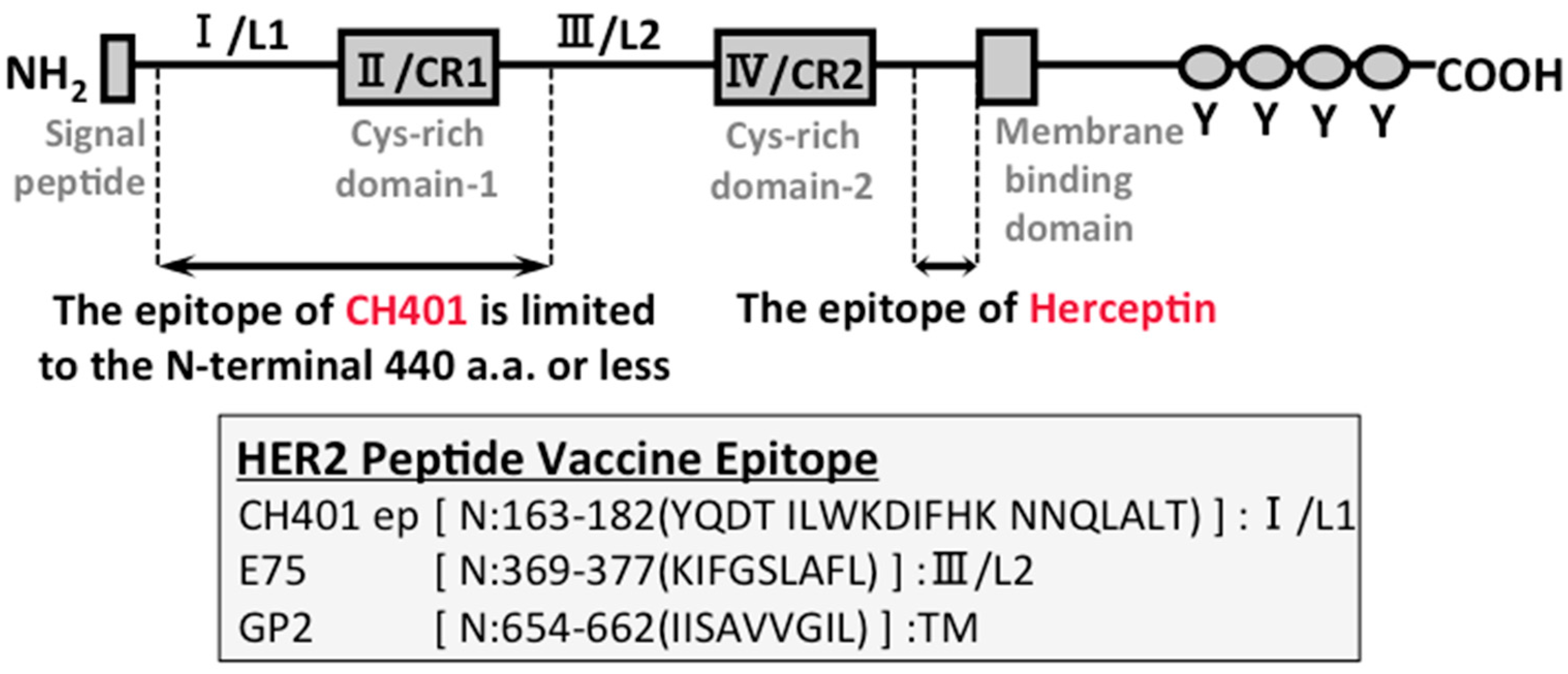
6. Why is Herceptin Successful?
7. B Cell-based Peptide Vaccine
8. B Cells and Cancer Immunity
9. Humanized Mouse Model for Peptide Vaccination
10. Future Views
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Stagg, J.; Andre, F.; Loi, S. Immunomodulation via chemotherapy and targeted therapy: A new paradigm in breast cancer therapy? Breast Care 2012, 7, 2670272. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Dieci, M.; Dubsky, P.; Sotiriou, C.; Curigliano, G.; Denkert, C.; Loi, S. Molecular pathways: Involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin. Cancer Res. 2013, 19, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Rammensee, H.; Bachmann, J.; Emmerich, N.P.; Bachor, O.A.; Stevanovic, S. Syfpeithi: Database for mhc ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.C.; Bednarek, M.A.; Coligan, J.E. Scheme for ranking potential hla-a2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 1994, 152, 163–175. [Google Scholar] [PubMed]
- Peters, B.; Sidney, J.; Bourne, P.; Bui, H.H.; Buus, S.; Doh, G.; Fleri, W.; Kronenberg, M.; Kubo, R.; Lund, O.; et al. The immune epitope database and analysis resource: From vision to blueprint. PLoS Biol. 2005, 3, e91. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-t immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against hpv-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Neelapu, S.S.; Gause, B.L.; Janik, J.E.; Muggia, F.M.; Gockerman, J.P.; Winter, J.N.; Flowers, C.R.; Nikcevich, D.A.; Sotomayor, E.M.; et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 2011, 29, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Lizee, G.; Overwijk, W.W.; Radvanyi, L.; Gao, J.; Sharma, P.; Hwu, P. Harnessing the power of the immune system to target cancer. Ann. Rev. Med. 2013, 64, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Harao, M.; Mittendorf, E.A.; Radvanyi, L.G. Peptide-based vaccination and induction of cd8+ t-cell responses against tumor antigens in breast cancer. BioDrugs 2015, 29, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Baxter, D. Active and passive immunization for cancer. Hum. Vaccines Immunother. 2014, 10, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Schneble, E.J.; Berry, J.S.; Trappey, F.A.; Clifton, G.T.; Ponniah, S.; Mittendorf, E.; Peoples, G.E. The her2 peptide nelipepimut-s (e75) vaccine (neuvax) in breast cancer patients at risk for recurrence: Correlation of immunologic data with clinical response. Immunotherapy 2014, 6, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Sasada, T.; Noguchi, M.; Itoh, K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013, 104, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.; Singhania, S.; Ramsey, K.; Murthy, S. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Mittendorf, E.; Clifton, G.T.; Holmes, J.; Clive, K.; Patil, R.; Benavides, L.; Gates, J.; Sears, A.; Stojadinovic, A.; Ponniah, S.; et al. Clinical trial results of the her-2/neu (e75) vaccine to prevent breast cancer recurrence in high-risk patients: From us millitary cancer institute clinical trials group study i-01 and i-02. Cancer 2012, 118, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Clive, K.; Tyler, J.; Travis Clifton, G.; Holmes, J.; Ponniah, S.; Peoples, G.; Mittendorf, E. The gp2 peptide: A her2/neu-based breast cancer vaccine. J. Surg. Oncol. 2012, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The erbb/her family of protein-tyrosine kinases and cancer. Pharm. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific cd8(+) t cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, D.; Pena, M.J.; Mijares, M.; Martinez, G.; Blanca, I.; De Sanctis, J.B. Peptide vaccines for cancer therapy. Recent Patents Inflam. Allergy Drug Discov. 2015, 9, 38–45. [Google Scholar] [CrossRef]
- Chang, D.Z.; Lomazow, W.; Joy Somberg, C.; Stan, R.; Perales, M.A. Granulocyte-macrophage colony stimulating factor: An adjuvant for cancer vaccines. Hematology 2004, 9, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Immunological adjuvants and their modes of action. Arch. Immunol. Ther. Exp. 1997, 45, 141–147. [Google Scholar]
- Roychoudhuri, R.; Eil, R.L.; Restifo, N.P. The interplay of effector and regulatory T cells in cancer. Curr. Opin. Immunol. 2015, 33c, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The role of death receptor ligands in shaping tumor microenvironment. Immunol. Invest. 2007, 36, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Ledbetter, J.A. The role of the cd28 receptor during t cell responses to antigen. Ann. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.F.; Denizot, F.; Golstein, P. A differential molecular biology search for genes preferentially expressed in functional t lymphocytes: The ctla genes. Immunol. Rev. 1988, 103, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Iwai, Y.; Honjo, T. New regulatory co-receptors: Inducible co-stimulator and pd-1. Curr. Opin. Immunol. 2002, 14, 779–782. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of pd-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Collins, M. The b7 family of ligands and its receptors: New pathways for costimulation and inhibition of immune responses. Ann. Rev. Immunol. 2002, 20, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of pd-l1 on tumor cells with pd-1 on tumor-specific t cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immun. 2005, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; Macrae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and clinical effects of antibody blockade of cytotoxic t lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Clinical development of the anti-ctla-4 antibody tremelimumab. Semin. Oncol. 2010, 37, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Wolchok, J.D. Immune modulation for cancer therapy. Br. J. Cancer 2014, 111, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Horn, L.; Haile, S. The programmed death-1 immune-suppressive pathway: Barrier to antitumor immunity. J. Immunol. 2014, 193, 3835–3841. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.; Callahan, M.; Wolchok, J. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.; Harview, C.; Yearley, J.; Shintaku, I.; Taylor, E.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Gunturi, A.; McDermott, D.F. Nivolumab for the treatment of cancer. Expert Opin. Invest. Drug. 2015, 24, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, P.; Postow, M.A. Immunologic checkpoints in cancer therapy: Focus on the programmed death-1 (pd-1) receptor pathway. Pharmacogenomics Pers. Med. 2014, 7, 357–365. [Google Scholar]
- Wolchok, J.; Hodi, F.; Weber, J.; Allison, J.; Urba, W.; Robert, C.; O'Day, S.; Hoos, A.; Humphrey, R.; Berman, D.; et al. Development of ipilimumab: A novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. NY Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.; Kluger, H.; Callahan, M.; Postow, M.; Rizvi, N.; Lesokhin, A.; Segal, N.; Ariyan, C.; Gordon, R.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.; Lesokhin, A.; Borrello, I.; Halwani, A.; Scott, E.; Gutierrez, M.; Schuster, S.; Millenson, M.; Cattry, D.; Freeman, G.; et al. Pd-1 blockade with nivolumab in relapsed or refractory hodgkin's lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.; Gulley, J.; Tsang, K.; Schlom, J. Antibody-dependent cellular cytotoxicity activity of a novel anti-pd-l1 antibody avelumab (msb0010718c) on human tumor cells. Cancer Immunol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.A.; Schwartz, D.A.; Nordberg, J.E.; Weiner, D.B.; Greene, M.I.; Torney, L.; Robinson, R.A. P185neu expression in human lung adenocarcinomas predicts shortened survival. Cancer Res. 1990, 50, 5184–5187. [Google Scholar] [PubMed]
- Berchuck, A.; Rodriguez, G.; Kinney, R.B.; Soper, J.T.; Dodge, R.K.; Clarke-Pearson, D.L.; Bast, R.C., Jr. Overexpression of her-2/neu in endometrial cancer is associated with advanced stage disease. Am. J. Obstet. Gynecol. 1991, 164, 15–21. [Google Scholar] [CrossRef]
- Cirisano, F.D.; Karlan, B.Y. The role of the her-2/neu oncogene in gynecologic cancers. J. Soc. Gynecol. Invest. 1996, 3, 99–105. [Google Scholar] [CrossRef]
- Morrison, C.; Zanagnolo, V.; Ramirez, N.; Cohn, D.E.; Kelbick, N.; Copeland, L.; Maxwell, G.L.; Fowler, J.M. Her-2 is an independent prognostic factor in endometrial cancer: Association with outcome in a large cohort of surgically staged patients. J. Clin. Oncol. 2006, 24, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Doi, T.; Ohtsu, A.; Boku, N.; Hashizume, K.; Nakanishi, M.; Ochiai, A. Comparison of her2 gene amplification assessed by fluorescence in situ hybridization and her2 protein expression assessed by immunohistochemistry in gastric cancer. Oncol. Rep. 2006, 15, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, M.; Glodkowska-Mrowka, E.; Bil, J.; Golab, J. Molecular mechanisms of the antitumor effects of anti-cd20 antibodies. Front. Biosci. 2011, 16, 277–306. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Hasmann, M.; Leyland-Jones, B. Molecular determinants of trastuzumab efficacy: What is their clinical relevance? Cancer Treat. Rev. 2013, 39, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Marechal, R.; De Schutter, J.; Nagy, N.; Demetter, P.; Lemmers, A.; Deviere, J.; Salmon, I.; Tejpar, S.; Van Laethem, J.L. Putative contribution of cd56 positive cells in cetuximab treatment efficacy in first-line metastatic colorectal cancer patients. BMC Cancer 2010, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Mach, J.P. Introduction to monoclonal antibodies. Cancer Immun. 2012, 12, 11. [Google Scholar] [PubMed]
- Oldham, R.K.; Dillman, R.O. Monoclonal antibodies in cancer therapy: 25 years of progress. J. Clin. Oncol. 2008, 26, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in her2-positive breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Cortes, J.; Kim, S.B.; Im, S.A.; Hegg, R.; Im, Y.H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [PubMed]
- zum Buschenfelde, C.M.; Hermann, C.; Schmidt, B.; Peschel, C.; Bernhard, H. Antihuman epidermal growth factor receptor 2 (her2) monoclonal antibody trastuzumab enhances cytolytic activity of class i-restricted her2-specific T lymphocytes against her2-overexpressing tumor cells. Cancer Res. 2002, 62, 2244–2247. [Google Scholar]
- Banerjee, D.; Matthews, P.; Matayeva, E.; Kaufman, J.L.; Steinman, R.M.; Dhodapkar, K.M. Enhanced T-cell responses to glioma cells coated with the anti-egf receptor antibody and targeted to activating fcgammars on human dendritic cells. J. Immunother. 2008, 31, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-based treatment of her2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Lane, H.A.; Hynes, N.E. The role of overexpressed her2 in transformation. Ann. Oncol. 2001, 12 (Suppl. 1), S9–S13. [Google Scholar] [CrossRef] [PubMed]
- Paschen, A.; Eichmuller, S.; Schadendorf, D. Identification of tumor antigens and T-cell epitopes, and its clinical application. Cancer Immunol. Immun. 2004, 53, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Strome, S.E.; Sewell, D.A. Synthetic peptide-based cancer vaccines: Lessons learned and hurdles to overcome. Curr. Mol. Med. 2009, 9, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Pilla, L.; Rivoltini, L.; Patuzzo, R.; Marrari, A.; Valdagni, R.; Parmiani, G. Multipeptide vaccination in cancer patients. Exp. Opin. Biol. Ther. 2009, 9, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Simoes, E.; Tan, D.; Ohlsson, A.; Sales, V.; Wang, E. Respiratory syncytial virus vaccine: A systematic overview with emphasis on respiratory syncytial virus subunit vaccines. Vaccine 2002, 20, 954–960. [Google Scholar] [CrossRef]
- Beck, A.; Klinguer-Hamour, C.; Bussat, M.-C.; Champion, T.; Haeuw, J.-F.; Goetsch, L.; Wurch, T.; Sugawara, M.; Milon, A.; van Dorsselaer, A.; et al. Peptides as tools and drugs for immunotherapies. J. Peptide Sci. 2007, 13, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Foy, K.C.; Wygle, R.M.; Miller, M.J.; Overholser, J.P.; Bekaii-Saab, T.; Kaumaya, P.T. Peptide vaccines and peptidomimetics of egfr (her-1) ligand binding domain inhibit cancer cell growth in vitro and in vivo. J. Imunol. 2013, 191, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Foy, K.C.; Kaumaya, P.T. Cancer immunotherapy: Present status, future perspective, and a new paradigm of peptide immunotherapeutics. Discov. Med. 2013, 15, 166–176. [Google Scholar] [PubMed]
- Dakappagari, N.K.; Douglas, D.B.; Triozzi, P.L.; Stevens, V.C.; Kaumaya, P.T. Prevention of mammary tumors with a chimeric her-2 b-cell epitope peptide vaccine. Cancer Res. 2000, 60, 3782–3789. [Google Scholar] [PubMed]
- Dakappagari, N.K.; Pyles, J.; Parihar, R.; Carson, W.E.; Young, D.C.; Kaumaya, P.T. A chimeric multi-human epidermal growth factor receptor-2 b cell epitope peptide vaccine mediates superior antitumor responses. J. Immunol. 2003, 170, 4242–4253. [Google Scholar] [CrossRef] [PubMed]
- Dakappagari, N.K.; Lute, K.D.; Rawale, S.; Steele, J.T.; Allen, S.D.; Phillips, G.; Reilly, R.T.; Kaumaya, P.T. Conformational her-2/neu b-cell epitope peptide vaccine designed to incorporate two native disulfide bonds enhances tumor cell binding and antitumor activities. J. Biol. Chem. 2005, 280, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Dakappagari, N.K.; Sundaram, R.; Rawale, S.; Liner, A.; Galloway, D.R.; Kaumaya, P.T. Intracellular delivery of a novel multiepitope peptide vaccine by an amphipathic peptide carrier enhances cytotoxic t-cell responses in hla-a*201 mice. J. Peptide Res. 2005, 65, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kaumaya, P.T.; Foy, K.C.; Garrett, J.; Rawale, S.V.; Vicari, D.; Thurmond, J.M.; Lamb, T.; Mani, A.; Kane, Y.; Balint, C.R.; et al. Phase i active immunotherapy with combination of two chimeric, human epidermal growth factor receptor 2, b-cell epitopes fused to a promiscuous t-cell epitope in patients with metastatic and/or recurrent solid tumors. J. Clin. Oncol. 2009, 27, 5270–5277. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, R.; Lynch, M.; Rawale, S.; Sun, Y.; Kazanji, M.; Kaumaya, P. De novo design of peptide immunogens that mimic the coiled coil region of human t-cell leukemia virus type-1 glycoprotein 21 transmembrane subunit for induction of native protein reactive neutralizing antibodies. J. Biol. Chem. 2004, 279, 24141–24151. [Google Scholar] [CrossRef] [PubMed]
- Hinoda, Y.; Sasaki, S.; Ishida, T.; Imai, K. Monoclonal antibodies as effective therapeutic agents for solid tumors. Cancer Sci. 2004, 95, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Miyako, H.; Kametani, Y.; Katano, I.; Ito, R.; Tsuda, B.; Furukawa, A.; Saito, Y.; Ishikawa, D.; Ogino, K.; Sasaki, S.; et al. Antitumor effect of new her2 peptide vaccination based on b cell epitope. Anticancer Res. 2011, 31, 3361–3368. [Google Scholar] [PubMed]
- Ishida, T.; Tsujisaki, M.; Hinoda, Y.; Imai, K.; Yachi, A. Establishment and characterization of mouse-human chimeric monoclonal antibody to erbb-2 product. Jpn. J. Cancer Res. 1994, 85, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kametani, Y.; Shiina, M.; Katano, I.; Ito, R.; Ando, K.; Toyama, K.; Tsukamoto, H.; Matsumura, T.; Saito, Y.; Ishikawa, D.; et al. Development of human-human hybridoma from anti-her-2 peptide-producing b cells in immunized nog mouse. Exp. Hematol. 2006, 34, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, B.; Kametani, Y.; Goto, Y.; Saito, Y.; Suzuki, Y.; Habu, S.; Inoko, H.; Tokuda, Y. A human b cell receptor epitope-based erbb-2 peptide (n:163-182) with pan-reactivity to the t cells of japanese breast cancer patients. J. Vaccines Vaccination 2012, 3, 159. [Google Scholar] [CrossRef]
- Tsuda, B.; Kametani, Y.; Miyamoto, A.; Miyako, H.; Kumaki, N.; Ogiy, R.; Oshitanai, R.; Terao, M.; Morioka, T.; Niikura, N.; et al. The effect of peptide treatment on the hla-binding and antibody production in peripheral blood mononuclear cells obtained from japanese breast cancer patients. J. Vaccines Vaccination 2015, in press. [Google Scholar]
- Babusíková, O.; Novotná, L.; Korec, S.; Schneková, K.; Turková, D.; Havránková, M.; Plentová, K. Change in the proportion of t and b lymphocytes in human malignant neoplasia in relation to the clinical stage. Neoplasma 1975, 22, 413–421. [Google Scholar] [PubMed]
- Hunig, T. The storm has cleared: Lessons from the cd28 superagonist tgn1412 trial. Nat. Rev. Immunol. 2012, 12, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kobayashi, K.; Nakahata, T. Nod/shi-scid il2rgamma(null) (nog) mice more appropriate for humanized mouse models. Curr. Top. Microbiol. Immunol. 2008, 324, 53–76. [Google Scholar] [PubMed]
- Brehm, M.A.; Wiles, M.V.; Greiner, D.L.; Shultz, L.D. Generation of improved humanized mouse models for human infectious diseases. J. Immunol. Methods 2014, 410, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Shiina, M.; Saito, Y.; Tokuda, Y.; Kametani, Y.; Habu, S. Antigen-specific antibody production of human b cells in nog mice reconstituted with the human immune system. Curr. Top. Microbiol. Immunol. 2008, 324, 95–107. [Google Scholar] [PubMed]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell. Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Shultz, L.D.; Luban, J.; Greiner, D.L. Overcoming current limitations in humanized mouse research. J. Infect. Dis. 2013, 208 (Suppl. 2), S125–S130. [Google Scholar] [CrossRef] [PubMed]
- Aryee, K.E.; Shultz, L.D.; Brehm, M.A. Immunodeficient mouse model for human hematopoietic stem cell engraftment and immune system development. Method. Mol. Biol. 2014, 1185, 267–278. [Google Scholar]
- Matsumura, T.; Kametani, Y.; Ando, K.; Hirano, Y.; Katano, I.; Ito, R.; Shiina, M.; Tsukamoto, H.; Saito, Y.; Tokuda, Y.; et al. Functional cd5+ b cells develop predominantly in the spleen of nod/scid/gammac(null) (nog) mice transplanted either with human umbilical cord blood, bone marrow, or mobilized peripheral blood cd34+ cells. Exp. Hematol. 2003, 31, 789–797. [Google Scholar] [CrossRef]
- Wege, A.K.; Melkus, M.W.; Denton, P.W.; Estes, J.D.; Garcia, J.V. Functional and phenotypic characterization of the humanized blt mouse model. Curr. Top. Microbiol. Immunol. 2008, 324, 149–165. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kametani, Y.; Miyamoto, A.; Tsuda, B.; Tokuda, Y. B Cell Epitope-Based Vaccination Therapy. Antibodies 2015, 4, 225-239. https://doi.org/10.3390/antib4030225
Kametani Y, Miyamoto A, Tsuda B, Tokuda Y. B Cell Epitope-Based Vaccination Therapy. Antibodies. 2015; 4(3):225-239. https://doi.org/10.3390/antib4030225
Chicago/Turabian StyleKametani, Yoshie, Asuka Miyamoto, Banri Tsuda, and Yutaka Tokuda. 2015. "B Cell Epitope-Based Vaccination Therapy" Antibodies 4, no. 3: 225-239. https://doi.org/10.3390/antib4030225
APA StyleKametani, Y., Miyamoto, A., Tsuda, B., & Tokuda, Y. (2015). B Cell Epitope-Based Vaccination Therapy. Antibodies, 4(3), 225-239. https://doi.org/10.3390/antib4030225