Role and Redirection of IgE against Cancer

{kind=link}
Abstract
:1. Introduction
2. Players of the IgE System vs. Cancer
2.1. IgE and Its Receptors
2.2. IgE-Mediated Immune Response
2.3. Epidemiological Correlation on Allergy and Cancer
2.4. IgE-Related Effector Cells in Cancer
2.4.1. Mast Cells and Basophils
2.4.2. Eosinophils
2.4.3. Macrophages and Monocytes
2.4.4. Antigen-Presenting Cells
2.4.5. T Cells
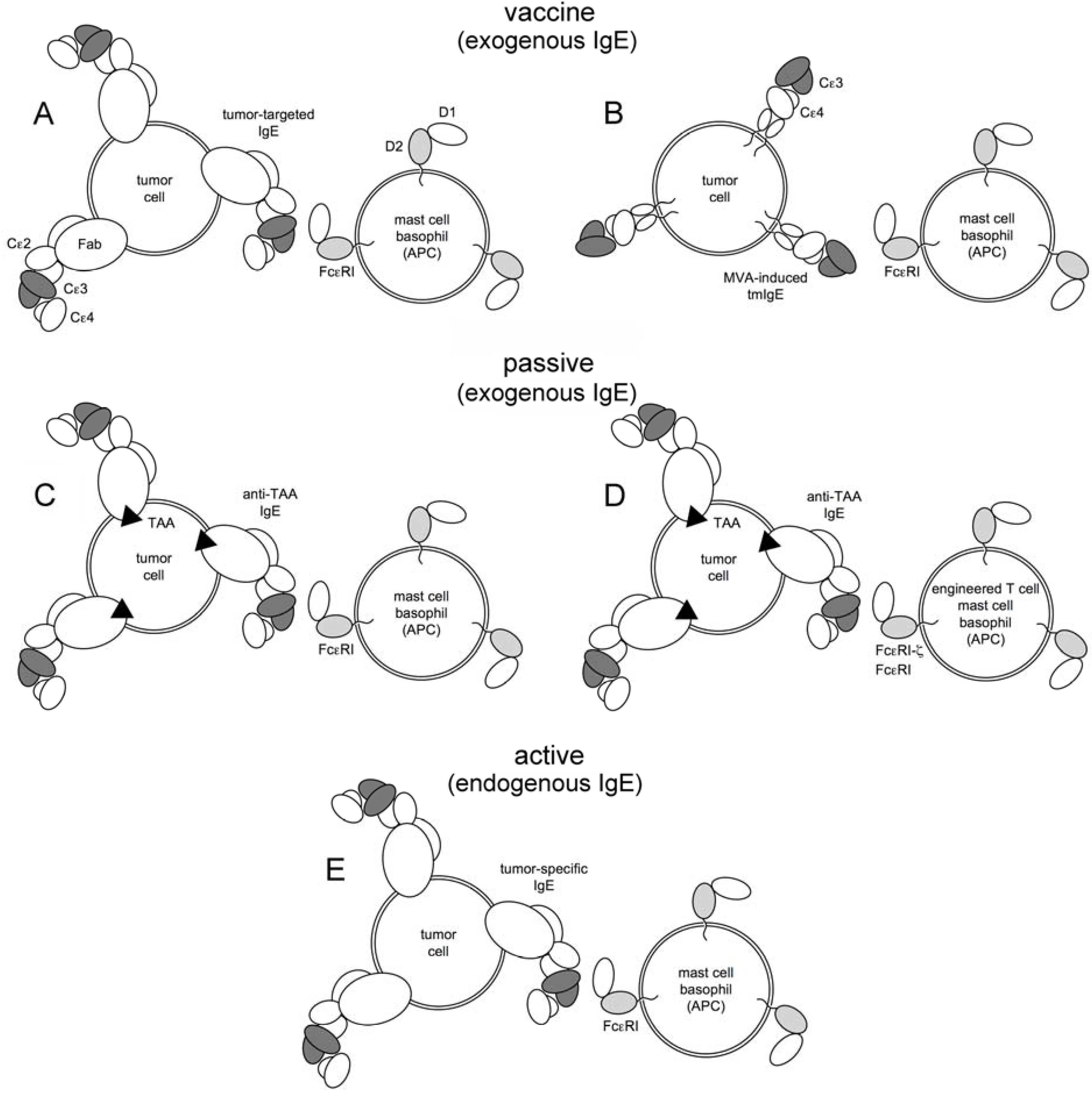
3. Experimental Approaches
3.1. IgE Antitumor Adjuvanticity
3.2. Induction of Endogenous IgE by Active Vaccination
3.3. IgE mAbs as Cancer Therapeutics
3.3.1. Murine Anti-gp36 IgE
3.3.2. Rat/Human Chimeric Anti-Murine Ly-2 IgE
3.3.3. Murine and Murine/Human Chimeric Anti-CCA IgE
3.3.4. Murine/Human Chimeric Anti-Human FRα IgE
3.3.5. Humanized and Human Anti-HER2/neu IgE
3.3.6. Murine/Human Chimeric Anti-Human MUC-1 IgE and Anti-Human CD20
3.3.7. Human Anti-EGFR IgE
3.3.8. Mouse/Human Chimeric Anti-PSA IgE
4. Conclusions
Conflict of Interest
References
- Johansson, S.G.O. The History of IgE: From discovery to 2010. Curr. Allergy Asthma Rep. 2011, 11, 173–177. [Google Scholar] [CrossRef]
- Dullaers, M.; De Bruyne, R.; Ramadani, F.; Gould, H.J.; Gevaert, P.; Lambrecht, B.N. The who, where, and when of IgE in allergic airway disease. J. Allergy Clin. Immunol. 2012, 129, 635–645. [Google Scholar] [CrossRef]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef]
- Erazo, A.; Kutchukhidze, N.; Leung, M.; Christ, A.P.G.; Urban, J.F.; Curotto de Lafaille, M.A.; Lafaille, J.J. Unique maturation program of the IgE response in vivo. Immunity 2007, 26, 191–203. [Google Scholar] [CrossRef]
- Talay, O.; Yan, D.; Brightbill, H.D.; Straney, E.E.M.; Zhou, M.; Ladi, E.; Lee, W.P.; Egen, J.G.; Austin, C.D.; Xu, M.; et al. IgE+ memory B cells and plasma cells generated through a germinal-center pathway. Nat. Immunol. 2012, 13, 396–404. [Google Scholar] [CrossRef]
- Yang, Z.; Sullivan, B.M.; Allen, C.D.C. Fluorescent in vivo detection reveals that IgE(+) B cells are restrained by an intrinsic cell fate predisposition. Immunity 2012, 36, 857–872. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Achatz, G.; Turner, M.C.; Karagiannis, S.; Legrand, F.; Capron, M.; Penichet, M.L.; Rodríguez, J.A.; Siccardi, A.G.; Vangelista, L.; et al. AllergoOncology: The role of IgE-mediated allergy in cancer. Allergy 2008, 63, 1255–1266. [Google Scholar] [CrossRef]
- Penichet, M.L.; Jensen-Jarolim, E. (Eds.) Cancer and IgE: Introducing the Concept of AllergoOncology, 1st ed.; Springer: New York, NY, USA, 2010; p. 280.
- Gould, H.J.; Sutton, B.J.; Beavil, A.J.; Beavil, R.L.; McCloskey, N.; Coker, H.A.; Fear, D.; Smurthwaite, L. The biology of IgE and the basis of allergic disease. Annu. Rev. Immunol. 2003, 21, 579–628. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Parodi, B. In vitro stimulation of IgE production at a single precursor level by human alloreactive T helper clones. Clin. Exp. Immunol. 1984, 55, 197–203. [Google Scholar]
- Cheng, L.E.; Wang, Z.-E.; Locksley, R.M. Murine B cells regulate serum IgE levels in a CD23-dependent manner. J. Immunol. 2010, 185, 5040–5047. [Google Scholar] [CrossRef]
- Kraft, S.; Kinet, J.-P. New developments in FcepsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef]
- Kinet, J.P. The high-affinity IgE receptor (Fc epsilon RI): From physiology to pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef]
- Vangelista, L.; Cesco-Gaspere, M.; Lamba, D.; Burrone, O. Efficient folding of the FcepsilonRI alpha-chain membrane-proximal domain D2 depends on the presence of the N-terminal domain D1. J. Mol. Biol. 2002, 322. [Google Scholar]
- Batista, F.D.F.; Anand, S.S.; Presani, G.G.; Efremov, D.G.D.; Burrone, O.R.O. The two membrane isoforms of human IgE assemble into functionally distinct B cell antigen receptors. J. Exp. Med. 1996, 184, 2197–2205. [Google Scholar] [CrossRef]
- Vangelista, L.; Soprana, E.; Cesco-Gaspere, M.; Mandiola, P.; Di Lullo, G.; Fucci, R.N.; Codazzi, F.; Palini, A.; Paganelli, G.; Burrone, O.R.; et al. Membrane IgE binds and activates Fc epsilon RI in an antigen-independent manner. J. Immunol. 2005, 174, 5602–5611. [Google Scholar]
- Vouldoukis, I.; Mazier, D.; Moynet, D.; Thiolat, D.; Malvy, D.; Mossalayi, M.D. IgE mediates killing of intracellular Toxoplasma gondii by human macrophages through CD23-dependent, interleukin-10 sensitive pathway. PLoS One 2011, 6, e18289. [Google Scholar]
- Vouldoukis, I.; Riveros-Moreno, V.; Dugas, B.; Ouaaz, F.; Bécherel, P.; Debré, P.; Moncada, S.; Mossalayi, M.D. The killing of Leishmania major by human macrophages is mediated by nitric oxide induced after ligation of the Fc epsilon RII/CD23 surface antigen. Proc. Natl. Acad. Sci. USA 1995, 92, 7804–7808. [Google Scholar] [CrossRef]
- Walter, K.; Fulford, A.J.C.; McBeath, R.; Joseph, S.; Jones, F.M.; Kariuki, H.C.; Mwatha, J.K.; Kimani, G.; Kabatereine, N.B.; Vennervald, B.J.; et al. Increased human IgE induced by killing Schistosoma mansoni in vivo is associated with pretreatment Th2 cytokine responsiveness to worm antigens. J. Immunol. 2006, 177, 5490–5498. [Google Scholar]
- Cooper, P.J.; Ayre, G.; Martin, C.; Rizzo, J.A.; Ponte, E.V.; Cruz, A.A. Geohelminth infections: a review of the role of IgE and assessment of potential risks of anti-IgE treatment. Allergy 2008, 63, 409–417. [Google Scholar] [CrossRef]
- Reddy, A.; Fried, B. Atopic disorders and parasitic infections. Adv. Parasitol. 2008, 66, 149–191. [Google Scholar] [CrossRef]
- Watanabe, N.; Bruschi, F.; Korenaga, M. IgE: A question of protective immunity in Trichinella spiralis infection. Trends Parasitol. 2005, 21, 175–178. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef]
- Gergen, P.J.; Turkeltaub, P.C.; Sempos, C.T. Is allergen skin test reactivity a predictor of mortality? Findings from a national cohort. Clin. Exp. Allergy 2000, 30, 1717–1723. [Google Scholar] [CrossRef]
- Turner, M.C.; Chen, Y.; Krewski, D.; Ghadirian, P. An overview of the association between allergy and cancer. Int. J. Cancer 2006, 118, 3124–3132. [Google Scholar] [CrossRef]
- Kero, J.; Gissler, M.; Hemminki, E.; Isolauri, E. Could TH1 and TH2 diseases coexist? Evaluation of asthma incidence in children with coeliac disease, type 1 diabetes, or rheumatoid arthritis: a register study. J. Allergy Clin. Immunol. 2001, 108, 781–783. [Google Scholar] [CrossRef]
- Olson, S.H.; Chou, J.F.; Ludwig, E.; O'Reilly, E.; Allen, P.J.; Jarnagin, W.R.; Bayuga, S.; Simon, J.; Gonen, M.; Reisacher, W.R.; et al. Allergies, obesity, other risk factors and survival from pancreatic cancer. Int. J. Cancer 2010, 127, 2412–2419. [Google Scholar] [CrossRef]
- Santillan, A.A.; Camargo, C.A.; Colditz, G.A. A meta-analysis of asthma and risk of lung cancer (United States). Cancer Causes Control 2003, 14, 327–334. [Google Scholar] [CrossRef]
- Arana, A.; Wentworth, C.E.; Fernández-Vidaurre, C.; Schlienger, R.G.; Conde, E.; Arellano, F.M. Incidence of cancer in the general population and in patients with or without atopic dermatitis in the U.K. Br. J. Dermatol. 2010, 163, 1036–1043. [Google Scholar] [CrossRef]
- Turner, M.C. Epidemiology: Allergy history, IgE, and cancer. Cancer Immunol. Immunother. 2012, 61, 1493–1510. [Google Scholar] [CrossRef]
- Rittmeyer, D.; Lorentz, A. Relationship between allergy and cancer: an overview. Int. Arch. Allergy Immunol. 2012, 159, 216–225. [Google Scholar] [CrossRef]
- Van Hemelrijck, M.; Garmo, H.; Binda, E.; Hayday, A.; Karagiannis, S.N.; Hammar, N.; Walldius, G.; Lambe, M.; Jungner, I.; Holmberg, L. Immunoglobulin E and cancer: A meta-analysis and a large Swedish cohort study. Cancer Causes Control 2010, 21, 1657–1667. [Google Scholar] [CrossRef]
- Brigati, C.; Noonan, D.M.; Albini, A.; Benelli, R. Tumors and inflammatory infiltrates: Friends or foes? Clin. Exp. Metastasis 2002, 19, 247–258. [Google Scholar] [CrossRef]
- Crivellato, E.; Nico, B.; Ribatti, D. Mast cells and tumour angiogenesis: New insight from experimental carcinogenesis. Cancer Lett. 2008, 269, 1–6. [Google Scholar] [CrossRef]
- Dabiri, S.; Huntsman, D.; Makretsov, N.; Cheang, M.; Gilks, B.; Badjik, C.; Gelmon, K.; Chia, S.; Hayes, M. The presence of stromal mast cells identifies a subset of invasive breast cancers with a favorable prognosis. Mod. Pathol. 2004, 17, 690–695. [Google Scholar] [CrossRef]
- Sinnamon, M.J.; Carter, K.J.; Sims, L.P.; Lafleur, B.; Fingleton, B.; Matrisian, L.M. A protective role of mast cells in intestinal tumorigenesis. Carcinogenesis 2008, 29, 880–886. [Google Scholar]
- Ribatti, D.; Molica, S.; Vacca, A.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Dammacco, F. Tryptase-positive mast cells correlate positively with bone marrow angiogenesis in B-cell chronic lymphocytic leukemia. Leukemia 2003, 17, 1428–1430. [Google Scholar] [CrossRef]
- Molin, D.; Edström, A.; Glimelius, I.; Glimelius, B.; Nilsson, G.; Sundström, C.; Enblad, G. Mast cell infiltration correlates with poor prognosis in Hodgkin’s lymphoma. Br. J. Haematol. 2002, 119, 122–124. [Google Scholar] [CrossRef]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikström, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin.Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef]
- Dalton, D.K.; Noelle, R.J. The roles of mast cells in anticancer immunity. Cancer Immunol. Immunother. 2012, 61, 1511–1520. [Google Scholar] [CrossRef]
- Di Carlo, E.; Modesti, A.; Coletti, A.; Colombo, M.P.; Giovarelli, M.; Forni, G.; Diodoro, M.G.; Musiani, P. Interaction between endothelial cells and the secreted cytokine drives the fate of an IL4- or an IL5-transduced tumour. J. Pathol. 1998, 186, 390–397. [Google Scholar] [CrossRef]
- Valent, P.; Agis, H.; Sperr, W.; Sillaber, C.; Horny, H.-P. Diagnostic and prognostic value of new biochemical and immunohistochemical parameters in chronic myeloid leukemia. Leuk. Lymphoma 2008, 49, 635–638. [Google Scholar] [CrossRef]
- Munitz, A.; Levi-Schaffer, F. Eosinophils: ‘new’ roles for ‘old’ cells. Allergy 2004, 59, 268–275. [Google Scholar] [CrossRef]
- Fernández-Aceñero, M.J.; Galindo-Gallego, M.; Sanz, J.; Aljama, A. Prognostic influence of tumor-associated eosinophilic infiltrate in colorectal carcinoma. Cancer 2000, 88, 1544–1548. [Google Scholar] [CrossRef]
- Dorta, R.G.; Landman, G.; Kowalski, L.P.; Lauris, J.R.P.; Latorre, M.R.D.O.; Oliveira, D.T. Tumour-associated tissue eosinophilia as a prognostic factor in oral squamous cell carcinomas. Histopathology 2002, 41, 152–157. [Google Scholar]
- Ishibashi, S.; Ohashi, Y.; Suzuki, T.; Miyazaki, S.; Moriya, T.; Satomi, S.; Sasano, H. Tumor-associated tissue eosinophilia in human esophageal squamous cell carcinoma. Anticancer Res. 2006, 26, 1419–1424. [Google Scholar]
- von Wasielewski, R.; Seth, S.; Franklin, J.; Fischer, R.; Hübner, K.; Hansmann, M.L.; Diehl, V.; Georgii, A. Tissue eosinophilia correlates strongly with poor prognosis in nodular sclerosing Hodgkin’s disease, allowing for known prognostic factors. Blood 2000, 95, 1207–1213. [Google Scholar]
- Gleich, G.J.; Adolphson, C.R. The eosinophilic leukocyte: Structure and function. Adv. Immunol. 1986, 39, 177–253. [Google Scholar] [CrossRef]
- Newton, D.L.; Rybak, S.M. Unique recombinant human ribonuclease and inhibition of Kaposi’s sarcoma cell growth. J. Natl. Cancer Inst. 1998, 90, 1787–1791. [Google Scholar] [CrossRef]
- Huland, E.; Huland, H. Tumor-associated eosinophilia in interleukin-2-treated patients: evidence of toxic eosinophil degranulation on bladder cancer cells. J. Cancer Res. Clin. Oncol. 1992, 118, 463–467. [Google Scholar] [CrossRef]
- Cormier, S.A.; Taranova, A.G.; Bedient, C.; Nguyen, T.; Protheroe, C.; Pero, R.; Dimina, D.; Ochkur, S.I.; O’Neill, K.; Colbert, D.; et al. Pivotal advance: Eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J. Leukoc. Biol. 2006, 79, 1131–1139. [Google Scholar] [CrossRef]
- Simson, L.; Ellyard, J.I.; Dent, L.A.; Matthaei, K.I.; Rothenberg, M.E.; Foster, P.S.; Smyth, M.J.; Parish, C.R. Regulation of carcinogenesis by IL-5 and CCL11: A potential role for eosinophils in tumor immune surveillance. J. Immunol. 2007, 178, 4222–4229. [Google Scholar]
- Legrand, F.; Driss, V.; Delbeke, M.; Loiseau, S.; Hermann, E.; Dombrowicz, D.; Capron, M. Human eosinophils exert TNF-α and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J. Immunol. 2010, 185, 7443–7451. [Google Scholar] [CrossRef]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar]
- Lin, E.Y.; Li, J.-F.; Bricard, G.; Wang, W.; Deng, Y.; Sellers, R.; Porcelli, S.A.; Pollard, J.W. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 2007, 1, 288–302. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Bieber, T. Fc epsilon RI on human epidermal Langerhans cells: An old receptor with new structure and functions. Int. Arch. Allergy Immunol. 1997, 113, 30–34. [Google Scholar] [CrossRef]
- Lowin, B.; Hahne, M.; Mattmann, C.; Tschopp, J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature 1994, 370, 650–652. [Google Scholar] [CrossRef]
- Regnault, A.; Lankar, D.; Lacabanne, V.; Rodriguez, A.; Théry, C.; Rescigno, M.; Saito, T.; Verbeek, S.; Bonnerot, C.; Ricciardi-Castagnoli, P.; et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J. Exp. Med. 1999, 189, 371–380. [Google Scholar] [CrossRef]
- Platzer, B.; Dehlink, E.; Turley, S.J.; Fiebiger, E. How to connect an IgE-driven response with CTL activity? Cancer Immunol. Immunother. 2012, 61, 1521–1525. [Google Scholar] [CrossRef]
- Parish, C.R. Cancer immunotherapy: The past, the present and the future. Immunol. Cell Biol. 2003, 81, 106–113. [Google Scholar] [CrossRef]
- Sant, A.J.; Chaves, F.A.; Jenks, S.A.; Richards, K.A.; Menges, P.; Weaver, J.M.; Lazarski, C.A. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol. Rev. 2005, 207, 261–278. [Google Scholar] [CrossRef]
- Reali, E.; Greiner, J.W.; Corti, A.; Gould, H.J.; Bottazzoli, F.; Paganelli, G.; Schlom, J.; Siccardi, A.G. IgEs targeted on tumor cells: Therapeutic activity and potential in the design of tumor vaccines. Cancer Res. 2001, 61, 5517–5522. [Google Scholar]
- Nigro, E.A.; Brini, A.T.; Soprana, E.; Ambrosi, A.; Dombrowicz, D.; Siccardi, A.G.; Vangelista, L. Antitumor IgE adjuvanticity: Key role of Fc epsilon RI. J. Immunol. 2009, 183, 4530–4536. [Google Scholar] [CrossRef]
- Nigro, E.A.; Soprana, E.; Brini, A.T.; Ambrosi, A.; Yenagi, V.A.; Dombrowicz, D.; Siccardi, A.G.; Vangelista, L. An antitumor cellular vaccine based on a mini-membrane IgE. J. Immunol. 2012, 188, 103–110. [Google Scholar] [CrossRef]
- Nagy, E.; Berczi, I.; Sehon, A.H. Growth inhibition of murine mammary carcinoma by monoclonal IgE antibodies specific for the mammary tumor virus. Cancer Immunol. Immunother. 1991, 34, 63–69. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Darcy, P.K.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Tumor-specific IgE-mediated inhibition of human colorectal carcinoma xenograft growth. Oncol. Res. 1998, 10, 133–142. [Google Scholar]
- Gould, H.J.; Mackay, G.A.; Karagiannis, S.N.; O’Toole, C.M.; Marsh, P.J.; Daniel, B.E.; Coney, L.R.; Zurawski, V.R.; Joseph, M.; Capron, M.; et al. Comparison of IgE and IgG antibody-dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur. J. Immunol. 1999, 29, 3527–3537. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Wang, Q.; East, N.; Burke, F.; Riffard, S.; Bracher, M.G.; Thompson, R.G.; Durham, S.R.; Schwartz, L.B.; Balkwill, F.R.; et al. Activity of human monocytes in IgE antibody-dependent surveillance and killing of ovarian tumor cells. Eur. J. Immunol. 2003, 33, 1030–1040. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Bracher, M.G.; Hunt, J.; McCloskey, N.; Beavil, R.L.; Beavil, A.J.; Fear, D.J.; Thompson, R.G.; East, N.; Burke, F.; et al. IgE-antibody-dependent immunotherapy of solid tumors: Cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J. Immunol. 2007, 179, 2832–2843. [Google Scholar]
- Karagiannis, S.N.; Bracher, M.G.; Beavil, R.L.; Beavil, A.J.; Hunt, J.; McCloskey, N.; Thompson, R.G.; East, N.; Burke, F.; Sutton, B.J.; et al. Role of IgE receptors in IgE antibody-dependent cytotoxicity and phagocytosis of ovarian tumor cells by human monocytic cells. Cancer Immunol. Immunother. 2008, 57, 247–263. [Google Scholar]
- Karagiannis, P.; Singer, J.; Hunt, J.; Gan, S.K.E.; Rudman, S.M.; Mechtcheriakova, D.; Knittelfelder, R.; Daniels, T.R.; Hobson, P.S.; Beavil, A.J.; et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol. Immunother. 2009, 58, 915–930. [Google Scholar] [CrossRef]
- Daniels, T.R.; Leuchter, R.K.; Quintero, R.; Helguera, G.; Rodríguez, J.A.; Martínez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; Penichet, M.L. Targeting HER2/neu with a fully human IgE to harness the allergic reaction against cancer cells. Cancer Immunol. Immunother. 2012, 61, 991–1003. [Google Scholar] [CrossRef]
- Teo, P.Z.; Utz, P.J.; Mollick, J.A. Using the allergic immune system to target cancer: Activity of IgE antibodies specific for human CD20 and MUC1. Cancer Immunol. Immunother. 2012, 61, 2295–2309. [Google Scholar] [CrossRef]
- Spillner, E.; Plum, M.; Blank, S.; Miehe, M.; Singer, J.; Braren, I. Recombinant IgE antibody engineering to target EGFR. Cancer Immunol. Immunother. 2012, 61, 1565–1573. [Google Scholar] [CrossRef]
- Daniels-Wells, T.R.; Helguera, G.; Leuchter, R.K.; Quintero, R.; Kozman, M.; Rodríguez, J.A.; Ortiz-Sánchez, E.; Martínez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; et al. A novel IgE antibody targeting the prostate-specific antigen as a potential prostate cancer therapy. BMC Cancer 2013, 13, 195. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Darcy, P.K.; Trapani, J.A.; Smyth, M.J. The use of chimeric human Fc(epsilon) receptor I to redirect cytotoxic T lymphocytes to tumors. J. Leukoc. Biol. 1996, 60, 721–728. [Google Scholar]
- Teng, M.W.L.; Kershaw, M.H.; Jackson, J.T.; Smyth, M.J.; Darcy, P.K. Adoptive transfer of chimeric FcepsilonRI gene-modified human T cells for cancer immunotherapy. Hum. Gene Ther. 2006, 17, 1134–1143. [Google Scholar] [CrossRef]
- Riemer, A.B.; Untersmayr, E.; Knittelfelder, R.; Duschl, A.; Pehamberger, H.; Zielinski, C.C.; Scheiner, O.; Jensen-Jarolim, E. Active induction of tumor-specific IgE antibodies by oral mimotope vaccination. Cancer Res. 2007, 67, 3406–3411. [Google Scholar] [CrossRef]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef]
- Benigni, F.; Zimmermann, V.S.; Hugues, S.; Caserta, S.; Basso, V.; Rivino, L.; Ingulli, E.; Malherbe, L.; Glaichenhaus, N.; Mondino, A. Phenotype and homing of CD4 tumor-specific T cells is modulated by tumor bulk. J. Immunol. 2005, 175, 739–748. [Google Scholar]
- Dombrowicz, D.; Flamand, V.; Brigman, K.K.; Koller, B.H.; Kinet, J.P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 1993, 75, 969–976. [Google Scholar] [CrossRef]
- Yu, P.; Kosco-Vilbois, M.; Richards, M.; Köhler, G.; Lamers, M.C. Negative feedback regulation of IgE synthesis by murine CD23. Nature 1994, 369, 753–756. [Google Scholar] [CrossRef]
- Dombrowicz, D.; Brini, A.T.; Flamand, V.; Hicks, E.; Snouwaert, J.N.; Kinet, J.P.; Koller, B.H. Anaphylaxis mediated through a humanized high affinity IgE receptor. J. Immunol. 1996, 157, 1645–1651. [Google Scholar]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Erfle, V.; Staib, C.; Sutter, G.; Siccardi, A.G. Marker gene swapping facilitates recombinant modified vaccinia virus Ankara production by host-range selection. J. Virol. Methods 2009, 156, 37–43. [Google Scholar] [CrossRef]
- Di Lullo, G.; Soprana, E.; Panigada, M.; Palini, A.; Agresti, A.; Comunian, C.; Milani, A.; Capua, I.; Erfle, V.; Siccardi, A.G. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J. Virol. Methods 2010, 163, 195–204. [Google Scholar] [CrossRef]
- Riemer, A.B.; Klinger, M.; Wagner, S.; Bernhaus, A.; Mazzucchelli, L.; Pehamberger, H.; Scheiner, O.; Zielinski, C.C.; Jensen-Jarolim, E. Generation of peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J. Immunol. 2004, 173, 394–401. [Google Scholar]
- Untersmayr, E.; Schöll, I.; Swoboda, I.; Beil, W.J.; Förster-Waldl, E.; Walter, F.; Riemer, A.; Kraml, G.; Kinaciyan, T.; Spitzauer, S.; et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: A fish allergy model in BALB/c mice. J. Allergy Clin. Immunol. 2003, 112, 616–623. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Clark, M.R.; Greenwood, J.; Waldmann, H. Therapy with monoclonal antibodies. An in vivo model for the assessment of therapeutic potential. J. Immunol. 1992, 148, 3062–3071. [Google Scholar]
- Mount, P.F.; Sutton, V.R.; Li, W.; Burgess, J.; McKEnzie, I.F.; Pietersz, G.A.; Trapani, J.A. Chimeric (mouse/human) anti-colon cancer antibody c30.6 inhibits the growth of human colorectal cancer xenografts in scid/scid mice. Cancer Res. 1994, 54, 6160–6166. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Ford, A.C.; Grandis, J.R. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck 2003, 25, 67–73. [Google Scholar] [CrossRef]
- Mendelsohn, J. Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy. Clin. Cancer Res. 1997, 3, 2703–2707. [Google Scholar]
- Schiller, J.H. Developments in epidermal growth factor receptor-targeting therapy for solid tumors: Focus on matuzumab (EMD 72000). Cancer Invest. 2008, 26, 81–95. [Google Scholar] [CrossRef]
- Berlyn, K.A.; Schultes, B.; Leveugle, B.; Noujaim, A.A.; Alexander, R.B.; Mann, D.L. Generation of CD4(+) and CD8(+) T lymphocyte responses by dendritic cells armed with PSA/anti-PSA (antigen/antibody) complexes. Clin. Immunol. 2001, 101, 276–283. [Google Scholar]
- Neuchrist, C.; Kornfehl, J.; Grasl, M.; Lassmann, H.; Kraft, D.; Ehrenberger, K.; Scheiner, O. Distribution of immunoglobulins in squamous cell carcinoma of the head and neck. Int. Arch. Allergy Immunol. 1994, 104, 97–100. [Google Scholar] [CrossRef]
- Fu, S.L.; Pierre, J.; Smith-Norowitz, T.A.; Hagler, M.; Bowne, W.; Pincus, M.R.; Mueller, C.M.; Zenilman, M.E.; Bluth, M.H. Immunoglobulin E antibodies from pancreatic cancer patients mediate antibody-dependent cell-mediated cytotoxicity against pancreatic cancer cells. Clin. Exp. Immunol. 2008, 153, 401–409. [Google Scholar] [CrossRef]
- Untersmayr, E.; Bises, G.; Starkl, P.; Bevins, C.L.; Scheiner, O.; Boltz-Nitulescu, G.; Wrba, F.; Jensen-Jarolim, E. The high affinity IgE receptor Fc epsilonRI is expressed by human intestinal epithelial cells. PLoS One 2010, 5, e9023. [Google Scholar]
- Rudman, S.M.; Josephs, D.H.; Cambrook, H.; Karagiannis, P.; Gilbert, A.E.; Dodev, T.; Hunt, J.; Koers, A.; Montes, A.; Taams, L.; et al. Harnessing engineered antibodies of the IgE class to combat malignancy: initial assessment of FcεRI-mediated basophil activation by a tumour-specific IgE antibody to evaluate the risk of type I hypersensitivity. Clin. Exp. Allergy 2011, 41, 1400–1413. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Singer, J. Why could passive Immunoglobulin E antibody therapy be safe in clinical oncology? Clin. Exp. Allergy 2011, 41, 1337–1340. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Josephs, D.H.; Karagiannis, P.; Gilbert, A.E.; Saul, L.; Rudman, S.M.; Dodev, T.; Koers, A.; Blower, P.J.; Corrigan, C.; et al. Recombinant IgE antibodies for passive immunotherapy of solid tumours: from concept towards clinical application. Cancer Immunol. Immunother. 2012, 61, 1547–1564. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nigro, E.A.; Siccardi, A.G.; Vangelista, L. Role and Redirection of IgE against Cancer. Antibodies 2013, 2, 371-391. https://doi.org/10.3390/antib2020371
Nigro EA, Siccardi AG, Vangelista L. Role and Redirection of IgE against Cancer. Antibodies. 2013; 2(2):371-391. https://doi.org/10.3390/antib2020371
Chicago/Turabian StyleNigro, Elisa A., Antonio G. Siccardi, and Luca Vangelista. 2013. "Role and Redirection of IgE against Cancer" Antibodies 2, no. 2: 371-391. https://doi.org/10.3390/antib2020371
APA StyleNigro, E. A., Siccardi, A. G., & Vangelista, L. (2013). Role and Redirection of IgE against Cancer. Antibodies, 2(2), 371-391. https://doi.org/10.3390/antib2020371