
Role of Specific Autoantibodies in Neurodegenerative Diseases: Pathogenic Antibodies or Promising Biomarkers for Diagnosis




{kind=link}
Highlights
- Emerging evidence suggests that autoantibodies, traditionally not associated with classic neurodegenerative diseases, may serve as novel biomarkers for these conditions.
- In diseases like Alzheimer's and Parkinson's, previously unrecognized pathogenic roles of specific autoantibodies are becoming clearer.
- This review underlines the potential of using these antibodies in diagnostics and therapeutic strategies for neurodegenerative diseases.
Abstract
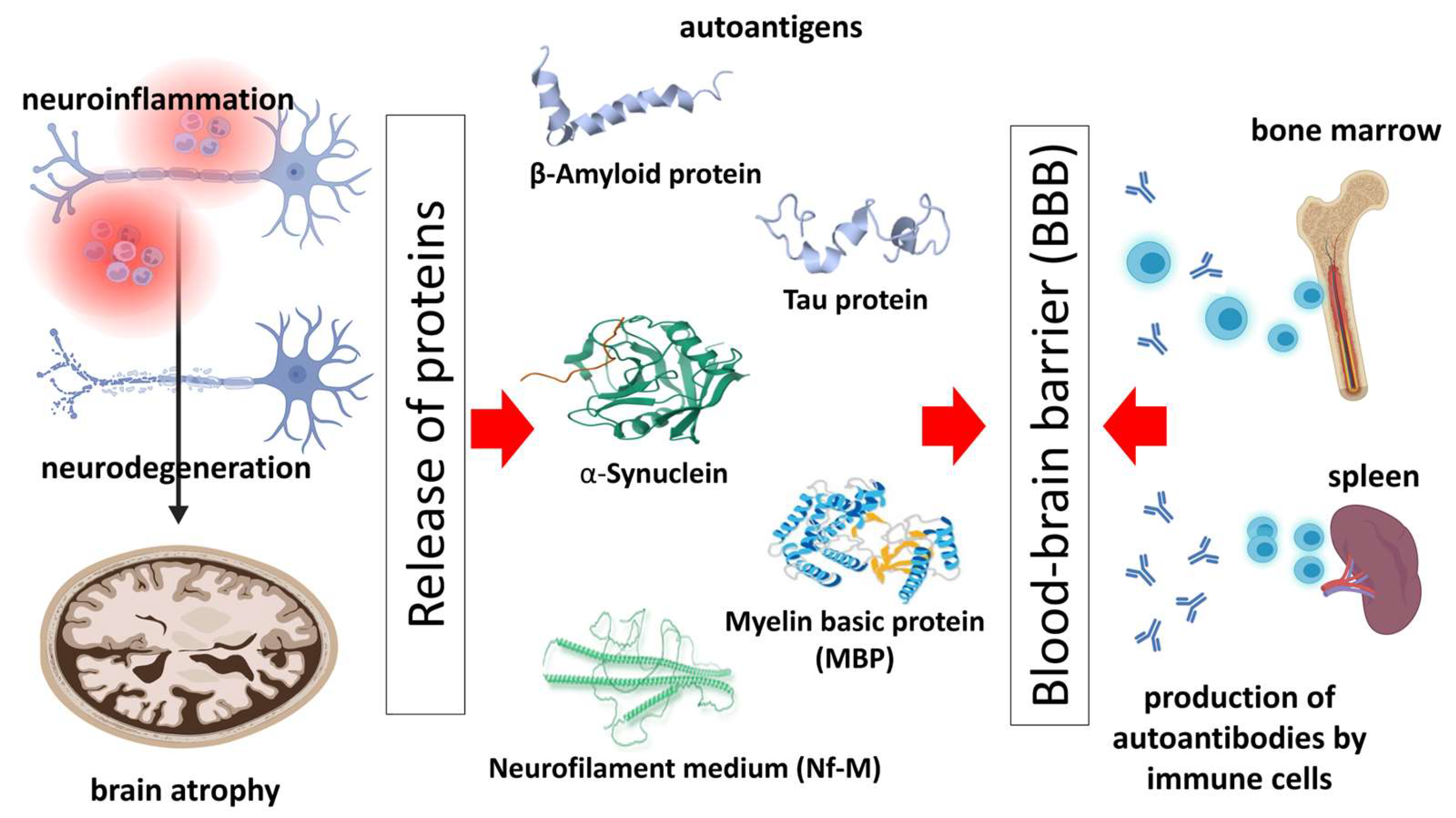
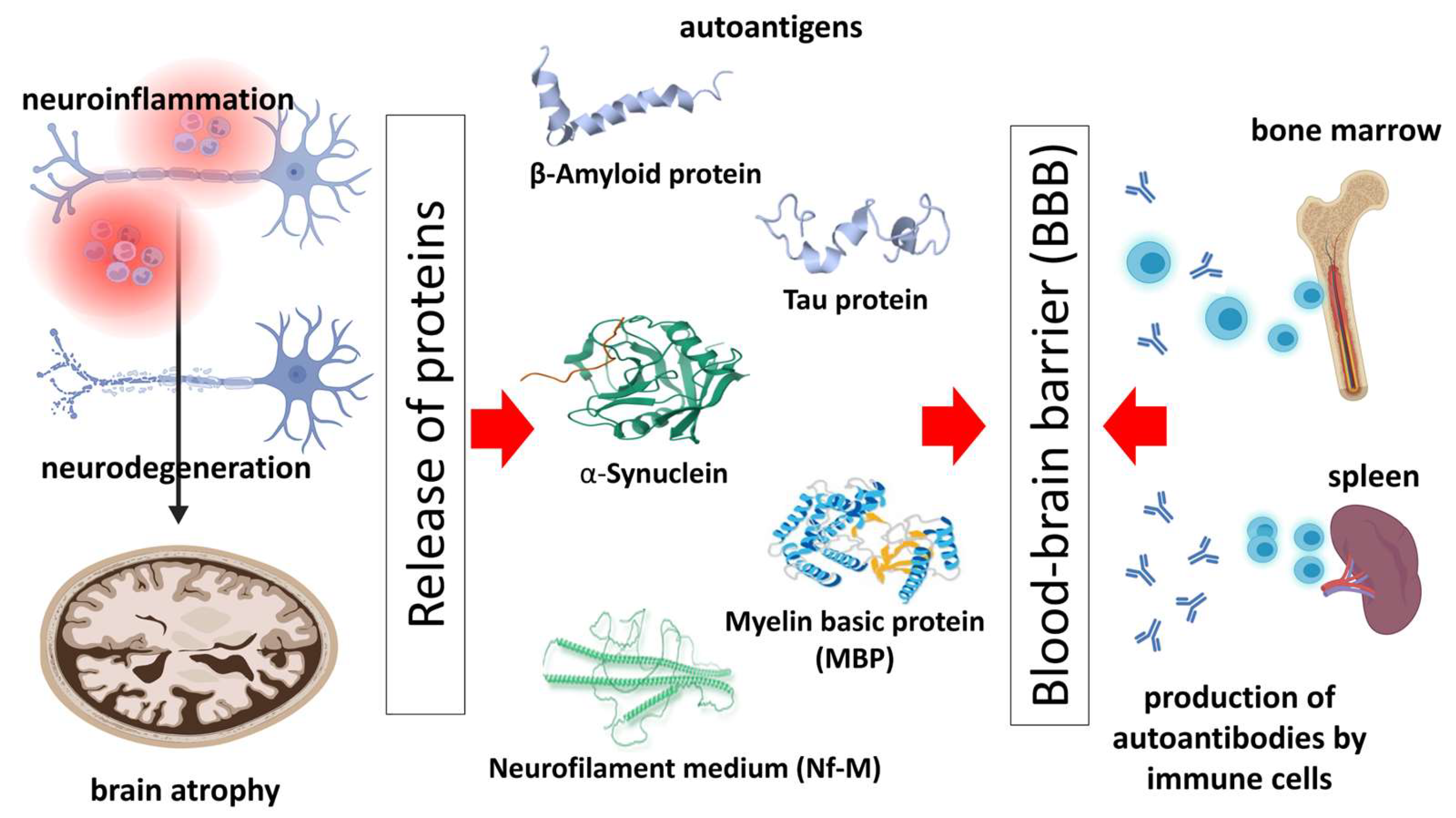
:1. Introduction: Neurodegenerative Diseases and the Role of the Immune System
2. Autoantibodies—Biology and Pathogenic Role in Neurodegenerative Diseases
3. Autoantibodies against Pathology-Related Molecules of the Most Common Neurodegenerative Diseases
3.1. Amyloid-β Autoantibodies and Microtubule Protein Tau Autoantibodies in Alzheimer’s Disease
3.2. α-Synuclein Autoantibodies in Parkinson’s Disease
3.3. Anti-Myelin Basic Protein (-MBP) and Anti-Myelin Oligodendrocyte Glycoprotein (MOG) Autoantibodies in Multiple Sclerosis
3.4. Neurofilament Autoantibodies in AD, PD, MS, and ALS
4. Autoantibodies as Biomarkers for Neurodegenerative Disease Diagnosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Hentz, J.G.; Adler, C.H.; Sabbagh, M.N.; Shill, H.A.; Jacobson, S.; Caviness, J.N.; Belden, C.; Driver-Dunckley, E.; Davis, K.J.; et al. Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J. Neuropathol. Exp. Neurol. 2014, 73, 244–252, Erratum in J. Neuropathol. Exp. Neurol. 2014, 73, 732.. [Google Scholar] [CrossRef] [PubMed]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, A.M.; Alberghina, L.; Papa, M. Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci. Lett. 2014, 565, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Meng, X. Vitamin D and neurodegenerative diseases. Heliyon 2023, 9, e12877. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Verde, F.; Morelli, L.; Rizzo, G.; Ricciardiello, F.; Liguori, R. Neural Surface Antibodies and Neurodegeneration: Clinical Commonalities and Pathophysiological Relationships. Biomedicines 2023, 11, 666. [Google Scholar] [CrossRef]
- Kocurova, G.; Ricny, J.; Ovsepian, S.V. Autoantibodies targeting neuronal proteins as biomarkers for neurodegenerative diseases. Theranostics 2022, 12, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- Prüss, H. Autoantibodies in neurological disease. Nat. Rev. Immunol. 2021, 21, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Marquetand, J.; van Lessen, M.; Bender, B.; Reimold, M.; Elsen, G.; Stoecker, W.; Synofzik, M. Slowly progressive LGI1 encephalitis with isolated late-onset cognitive dysfunction: A treatable mimic of Alzheimer’s disease. Eur. J. Neurol. 2016, 23, e28–e29. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Marnane, M.; O’connell, M.; Stack, J.P.; Kelly, P.J.; Lynch, T. Potassium channel antibody associated encephalopathy presenting with a frontotemporal dementia like syndrome. Arch. Neurol. 2007, 64, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, H.; Meyer, T.; Prüss, H. GABAB receptor encephalitis in a patient diagnosed with amyotrophic lateral sclerosis. BMC Neurol. 2019, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- May, C.; Nordhoff, E.; Casjens, S.; Turewicz, M.; Eisenacher, M.; Gold, R.; Brüning, T.; Pesch, B.; Stephan, C.; Woitalla, D.; et al. Highly immunoreactive IgG antibodies directed against a set of twenty human proteins in the sera of patients with amyotrophic lateral sclerosis identified by protein array. PLoS ONE 2014, 9, e89596. [Google Scholar] [CrossRef] [PubMed]
- Tüzün, E.; Gezen-Ak, D.; Tzartos, J.; Dursun, E.; Giriş, M.; Zisimopoulou, P.; Karagiorgou, K.; Yetimler, B.; Küçükali, C.I.; Idrisoğlu, H.A. LRP4 antibody positive amyotrophic lateral sclerosis patients display neuropil-reactive IgG and enhanced serum complement levels. Immunol. Lett. 2018, 203, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Landa, J.; Gaig, C.; Planagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef]
- Rocchi, A.; Sacchetti, S.; De Fusco, A.; Giovedi, S.; Parisi, B.; Cesca, F.; Höltje, M.; Ruprecht, K.; Ahnert-Hilger, G.; Benfenati, F. Autoantibodies to synapsin I sequestrate synapsin I and alter synaptic function. Cell Death Dis. 2019, 10, 864. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P.; Nomenclature Committee of the International Society of Amyloidosis. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2012, 19, 167–170. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.; Brennan, S.; Keon, M.; Ooi, L. The role of amyloid oligomers in neurodegenerative pathologies. Int. J. Biol. Macromol. 2021, 181, 582–604. [Google Scholar] [CrossRef] [PubMed]
- Katsinelos, T.; Tuck, B.J.; Mukadam, A.S.; McEwan, W.A. The Role of Antibodies and Their Receptors in Protection Against Ordered Protein Assembly in Neurodegeneration. Front. Immunol. 2019, 10, 1139. [Google Scholar] [CrossRef] [PubMed]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.L.; Wang, L.H.; Yang, T.; Sun, J.Y.; Mao, L.L.; Yang, M.F.; Yuan, H.; Colvin, R.A.; Yang, X.Y. Lymphatic drainage system of the brain: A novel target for intervention of neurological diseases. Prog. Neurobiol. 2018, 163–164, 118–143. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Cho, H.; Kim, J.-H.; Kim, S.H.; Ham, J.-S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.-H.; Hong, Y.-K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Kheirkhah, R.; DeMarshall, C.; Sieber, F.; Oh, E.; Nagele, R.G. The origin and nature of the complex autoantibody profile in cerebrospinal fluid. Brain Behav. Immun. Health. 2020, 2, 100032. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Gaskin, F.; Finley, J.; Fang, Q.; Xu, S.; Fu, S.M. Human antibodies reactive with beta-amyloid protein in Alzheimer’s disease. J. Exp. Med. 1993, 177, 1181–1186. [Google Scholar] [CrossRef]
- Du, Y.; Dodel, R.; Hampel, H.; Buerger, K.; Lin, S.; Eastwood, B.; Bales, K.; Gao, F.; Moeller, H.J.; Oertel, W.; et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology 2001, 57, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Weksler, M.E.; Relkin, N.; Turkenich, R.; LaRusse, S.; Zhou, L.; Szabo, P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontol. 2002, 37, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Baril, L.; Nicolas, L.; Croisile, B.; Crozier, P.; Hessler, C.; Sassolas, A.; McCormick, J.B.; Trannoy, E. Immune response to Abeta-peptides in peripheral blood from patients with Alzheimer’s disease and control subjects. Neurosci. Lett. 2004, 355, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.X.; Gong, Y.; Moore, C.; Fu, M.; German, D.C.; Chang, L.Y.; Rosenberg, R.; Diaz-Arrastia, R. Beta-amyloid auto-antibodies are reduced in Alzheimer’s disease. J. Neuroimmunol. 2014, 274, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, S.; Morgenthaler, N.G.; Teipel, S.J.; Fischer-Schulz, C.; Bürger, K.; Dodel, R.; Du, Y.; Möller, H.J.; Bergmann, A.; Hampel, H. Decreased serum amyloid β1-42 autoantibody levels in Alzheimer’s disease, determined by a newly developed immuno-precipitation assay with radiolabeled amyloid β1-42 peptide. Biol. Psychiatry 2005, 57, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Yao, Z.; Quan, W.; Cao, Z.; Xu, J.; Luo, J. Low avidity and level of serum anti-Abeta antibodies in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 127–132. [Google Scholar]
- Sohn, J.H.; So, J.O.; Hong, H.J.; Kim, J.W.; Na, D.R.; Kim, M.; Kim, H.; Nam, E.; Ha, H.J.; Kim, Y.H.; et al. Identification of autoantibody against beta-amyloid peptide in the serum of elderly. Front. Biosci. (Landmark Ed.) 2009, 14, 3879–3883. [Google Scholar] [CrossRef]
- Nath, A.; Hall, E.; Tuzova, M.; Dobbs, M.; Jones, M.; Anderson, C.; Woodward, J.; Guo, Z.; Fu, W.; Kryscio, R.; et al. Autoantibodies to amyloid beta-peptide (Abeta) are increased in Alzheimer’s disease patients and Abeta antibodies can enhance Abeta neurotoxicity: Implications for disease pathogenesis and vaccine development. Neuromolecular Med. 2003, 3, 29–40. [Google Scholar] [CrossRef]
- McMahon, M.J.; O’kennedy, R. Polyreactivity as an acquired artefact, rather than a physiologic property, of antibodies: Evidence that monoreactive antibodies may gain the ability to bind to multiple antigens after exposure to low pH. J. Immunol. Methods 2000, 241, 1–10. [Google Scholar] [CrossRef]
- Li, X.-W.; Li, X.-X.; Liu, Q.-S.; Cheng, Y. Blood and Cerebrospinal Fluid Autoantibody to Aβ Levels in Patients with Alzheimer’s Disease: A Meta-Analysis Study. J. Mol. Neurosci. 2020, 70, 1208–1215. [Google Scholar] [CrossRef]
- Bartos, A.; Fialová, L.; Švarcová, J. Lower Serum Antibodies Against Tau Protein and Heavy Neurofilament in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Bartos, A.; Fialová, L.; Švarcová, J.; Ripova, D. Patients with Alzheimer disease have elevated intrathecal synthesis of antibodies against tau protein and heavy neurofilament. J. Neuroimmunol. 2012, 252, 100–105. [Google Scholar] [CrossRef]
- Klaver, A.C.; Coffey, M.P.; Bennett, D.A.; Loeffler, D.A. Specific serum antibody binding to phosphorylated and non-phosphorylated tau in non-cognitively impaired, mildly cognitively impaired, and Alzheimer’s disease subjects: An exploratory study. Transl. Neurodegener. 2017, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Krestova, M.; Hromadkova, L.; Bilkova, Z.; Bartos, A.; Ricny, J. Characterization of Isolated Tau-Reactive Antibodies From the Ivig Product, Plasma of Patients with Alzheimer’s Disease and Cognitively Normal Individuals. J. Neuroimmunol. 2017, 313, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Rogosch, T.; Schindler, T.I.; Tackenberg, B.; Zemlin, M.; Maier, R.F.; Dodel, R.; Kronimus, Y. Serum titers of autoantibodies against α-synuclein and tau in child- and adulthood. J. Neuroimmunol. 2018, 315, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Skillbäck, T.; Farahmand, B.Y.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; Schott, J.M.; et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015, 138, 2716–2731. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef]
- Hromadkova, L.; Kolarova, M.; Jankovicova, B.; Bartos, A.; Ricny, J.; Bilkova, Z.; Ripova, D. Identification and characterization of natural antibodies against tau protein in an intravenous immunoglobulin product. J. Neuroimmunol. 2015, 289, 121–129. [Google Scholar] [CrossRef]
- Rosenmann, H.; Meiner, Z.; Geylis, V.; Abramsky, O.; Steinitz, M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer’s disease and healthy subjects. Neurosci. Lett. 2006, 410, 90–93. [Google Scholar] [CrossRef]
- Pascual, G.; Wadia, J.S.; Zhu, X.; Keogh, E.; Kükrer, B.; van Ameijde, J.; Inganäs, H.; Siregar, B.; Perdok, G.; Diefenbach, O.; et al. Immunological memory to hyperphosphorylated tau in asymptomatic individuals. Acta Neuropathol. 2017, 133, 767–783. [Google Scholar] [CrossRef]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.E.; Blurton-Jones, M. Examining the mechanisms that link β-amyloid and α-synuclein pathologies. Alzheimer’s Res. Ther. 2012, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872, Erratum in Neurology 2005, 65, 1992. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.; Koga, S.; O’leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp. Neurol. 2002, 176, 98–104. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.-I.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Miller, D.W.; Hague, S.M.; Clarimon, J.; Baptista, M.; Gwinn-Hardy, K.; Cookson, M.R.; Singleton, A.B. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 2004, 62, 1835–1838. [Google Scholar] [CrossRef]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Park. Dis. 2017, 7, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Roy, S. A-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of α-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Park. Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K. The Role of Alpha-Synuclein Autoantibodies in the Induction of Brain Inflammation and Neurodegeneration in Aged Humans. Front. Aging Neurosci. 2022, 14, 902191. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; Van Der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2019, 36, 1–12. [Google Scholar] [CrossRef]
- Chahine, L.M.; Beach, T.G.; Adler, C.H.; Hepker, M.; Kanthasamy, A.; Appel, S.; Pritzkow, S.; Pinho, M.; Mosovsky, S.; Serrano, G.E.; et al. Central and peripheral α-synuclein in Parkinson disease detected by seed amplification assay. Ann. Clin. Transl. Neurol. 2023, 10, 696–705. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Growdon, W.; Gomez-Isla, T.; Newell, K.; George, J.M.; Clayton, D.F.; Hyman, B.T. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain alpha-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol. 1998, 57, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Orr, C.F.; Rowe, D.B.; Mizuno, Y.; Mori, H.; Halliday, G.M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 2005, 128, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Gold, M.; Deuschle, C.; Bernhard, F.; Maetzler, W.; Berg, D.; Dodel, R. Naturally occurring alpha-synuclein autoantibodies in Parkinson’s disease: Sources of (error) variance in biomarker assays. PLoS ONE 2014, 9, e114566. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, J.M.; Duke, R.; Middeldorp, J.M.; Stevens, S.; Vervoort, M.; Hashimoto, M.; Masliah, E.; Chan, P.; Di Monte, D.A.; Langston, J.W.; et al. Absence of elevated anti-alpha-synuclein and anti-EBV latent membrane protein antibodies in PD. Neurology 2002, 58, 1435. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Schiess, M.C.; Coffey, M.P.; Klaver, A.C.; Loeffler, D.A. A-Synuclein and anti-α-synuclein antibodies in Parkinson’s disease, atypical Parkinson syndromes, REM sleep behavior disorder, and healthy controls. PLoS ONE 2012, 7, e52285. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Gruden, M.A.; Casaite, V.; Meskys, R.; Forsgren, L.; Morozova-Roche, L.A. A-Synuclein Reactive Antibodies as Diagnostic Biomarkers in Blood Sera of Parkinson’s Disease Patients. PLoS ONE 2011, 6, e18513. [Google Scholar] [CrossRef]
- Horvath, I.; Iashchishyn, I.A.; Forsgren, L.; Morozova-Roche, L.A. Immunochemical Detection of α-Synuclein Autoantibodies in Parkinson’s Disease: Correlation between Plasma and Cerebrospinal Fluid Levels. ACS Chem. Neurosci. 2017, 8, 1170–1176. [Google Scholar] [CrossRef]
- Bryan, T.; Luo, X.; Forsgren, L.; Morozova-Roche, L.A.; Davis, J.J. The robust electrochemical detection of a Parkinson’s disease marker in whole blood sera. Chem. Sci. 2012, 3, 3468–3473. [Google Scholar] [CrossRef]
- Besong-Agbo, D.; Wolf, E.; Jessen, F.; Oechsner, M.; Hametner, E.; Poewe, W.; Reindl, M.; Oertel, W.H.; Noelker, C.; Bacher, M.; et al. Naturally occurring α-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology 2013, 80, 169–175. [Google Scholar] [CrossRef]
- Maetzler, W.; Berg, D.; Synofzik, M.; Brockmann, K.; Godau, J.; Melms, A.; Gasser, T.; Hörnig, S.; Langkamp, M. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of Lewy body-associated dementias. J. Alzheimer’s Dis. 2011, 26, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Gruden, M.A.; Sewell, R.D.; Yanamandra, K.; Davidova, T.V.; Kucheryanu, V.G.; Bocharov, E.V.; Bocharova, O.A.; Polyschuk, V.V.; Sherstnev, V.V.; Morozova-Roche, L.A. Immunoprotection against toxic biomarkers is retained during Parkinson’s disease progression. J. Neuroimmunol. 2011, 233, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, R.S.; Licata, J.P.; Luk, K.C.; Shaw, L.M.; Trojanowski, J.Q.; Lee, V.M. Measurements of auto-antibodies to α-synuclein in the serum and cerebral spinal fluids of patients with Parkinson’s disease. J. Neurochem. 2018, 145, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Koehler, N.K.; Stransky, E.; Shing, M.; Gaertner, S.; Meyer, M.; Schreitmüller, B.; Leyhe, T.; Laske, C.; Maetzler, W.; Kahle, P.; et al. Altered serum IgG levels to α-synuclein in dementia with Lewy bodies and Alzheimer’s disease. PLoS ONE 2013, 8, e64649. [Google Scholar] [CrossRef] [PubMed]
- Mey, G.M.; Mahajan, K.R.; DeSilva, T.M. Neurodegeneration in multiple sclerosis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2023, 15, e1583. [Google Scholar] [CrossRef]
- Sandi, D.; Fricska-Nagy, Z.; Bencsik, K.; Vécsei, L. Neurodegeneration in Multiple Sclerosis: Symptoms of Silent Progression, Biomarkers and Neuroprotective Therapy-Kynurenines Are Important Players. Molecules 2021, 26, 3423. [Google Scholar] [CrossRef] [PubMed]
- Pette, M.; Fujita, K.; Kitze, B.; Whitaker, J.N.; Albert, E.; Kappos, L.; Wekerle, H. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology 1990, 40, 1770. [Google Scholar] [CrossRef]
- O’Connor, K.C.; Chitnis, T.; E Griffin, D.; Piyasirisilp, S.; Bar-Or, A.; Khoury, S.; Wucherpfennig, K.W.; A Hafler, D. Myelin basic protein-reactive autoantibodies in the serum and cerebrospinal fluid of multiple sclerosis patients are characterized by low-affinity interactions. J. Neuroimmunol. 2003, 136, 140–148. [Google Scholar] [CrossRef]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef]
- Angelucci, F.; Mirabella, M.; Frisullo, G.; Caggiula, M.; Tonali, P.A.; Batocchi, A.P. Serum levels of anti-myelin antibodies in relapsing-remitting multiple sclerosis patients during different phases of disease activity and immunomodulatory therapy. Dis. Markers 2005, 21, 49–55. [Google Scholar] [CrossRef]
- Olsson, T.; Baig, S.; Hojeberg, B.; Link, H. Antimyelin basic protein and antimyelin antibody-producing cells in multiple sclerosis. Ann. Neurol. 1990, 27, 132–136. [Google Scholar] [CrossRef]
- Reindl, M.; Linington, C.; Brehm, U.; Egg, R.; Dilitz, E.; Deisenhammer, F.; Poewe, W.; Berger, T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain 1999, 122, 2047–2056. [Google Scholar] [CrossRef]
- Warren, K.G.; Catz, I.; Johnson, E.; Mielke, B. Anti-myelin basic protein and anti-proteolipid protein specific forms of multiple sclerosis. Ann. Neurol. 1994, 35, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef]
- Tran, G.T.; Hodgkinson, S.J.; Carter, N.; Killingsworth, M.; Spicer, S.T.; Hall, B.M. Attenuation of experimental allergic encephalomyelitis in complement component 6-deficient rats is associated with reduced complement C9 deposition, P-selectin expression, and cellular infiltrate in spinal cords. J. Immunol. 2002, 168, 4293–4300. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.; Cudrici, C.; Niculescu, F. C5b-9 complement complex in autoimmune demyelination and multiple sclerosis: Dual role in neuroinflammation and neuroprotection. Ann. Med. 2005, 37, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.W. Early cellular events in multiple sclerosis. Intimations of an extrinsic myelinolytic antigen. Clin. Neurol. Neurosurg. 2006, 108, 234–240. [Google Scholar] [CrossRef]
- Coyle, P.K.; Procyk-Dougherty, Z. Multiple sclerosis immune complexes: An analysis of component antigens and antibodies. Ann. Neurol. 1984, 16, 660–667. [Google Scholar] [CrossRef]
- Geffard, M.; Boullerne, A.; Brochet, B. Seric immune complexes in multiple sclerosis do not contain MBP epitopes. Brain Res. Bull. 1993, 30, 365–368. [Google Scholar] [CrossRef]
- Hedegaard, C.J.; Chen, N.; Sellebjerg, F.; Sørensen, P.S.; Leslie, R.G.Q.; Bendtzen, K.; Nielsen, C.H. Autoantibodies to myelin basic protein (MBP) in healthy individuals and in patients with multiple sclerosis: A role in regulating cytokine responses to MBP. Immunology 2009, 128 (Suppl. S1), e451–e461. [Google Scholar] [CrossRef]
- Villar, L.; Garcia-Barragan, N.; Espiño, M.; Roldán, E.; Sadaba, M.; Gómez-Rial, J.; Gonzalez-Porque, P.; Alvarez-Cermeno, J. Influence of oligoclonal IgM specificity in multiple sclerosis disease course. Mult. Scler. J. 2008, 14, 183–187. [Google Scholar] [CrossRef]
- Ambrosius, W.; Michalak, S.; Kozubski, W.; Kalinowska, A. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. Int. J. Mol. Sci. 2020, 22, 100. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; de Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. MOG encephalomyelitis: International recommendations on diagnosis and antibody testing. J. Neuroinflammation 2018, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.; Menon, S.; Lu, J.Q. Neurofilaments in neurologic disorders and beyond. J. Neurol. Sci. 2022, 441, 120380. [Google Scholar] [CrossRef] [PubMed]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the multiple aspects of neurofilament functions and their possible contribution to neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Gafson, A.R.; Barthélemy, N.R.; Bomont, P.; Carare, R.O.; Durham, H.D.; Julien, J.P.; Kuhle, J.; Leppert, D.; Nixon, R.A.; Weller, R.O.; et al. Neurofilaments: Neurobiological foundations for biomarker applications. Brain 2020, 143, 1975–1998. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Gentil, B.J.; Tibshirani, M.; Durham, H.D. Neurofilament dynamics and involvement in neurological disorders. Cell Tissue Res. 2015, 360, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, A.; Berges, R.; Frank, R.; Robert, P.; Peterson, A.C.; Eyer, J. Neurofilaments bind tubulin and modulate its polymerization. J. Neurosci. 2009, 29, 11043–11054. [Google Scholar] [CrossRef]
- Schwartz, M.L.; Shneidman, P.S.; Bruce, J.; Schlaepfer, W.W. Stabilization of neurofilament transcripts during postnatal development. Brain Res. Mol. Brain Res. 1994, 27, 215–220. [Google Scholar] [CrossRef]
- Yuan, A.; Nixon, R.A. Specialized roles of neurofilament proteins in synapses: Relevance to neuropsychiatric disorders. Brain Res. Bull. 2016, 126, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Novakova, L.; Axelsson, M.; Velayudhan, L.; Rabinovici, G.D.; Miller, B.; Pariante, C.; Nikkheslat, N.; Resnick, S.M.; Thambisetty, M.; Schöll, M.; et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat. Commun. 2021, 12, 3400. [Google Scholar] [CrossRef]
- Hansson, O.; Janelidze, S.; Hall, S.; Magdalinou, N.; Lees, A.J.; Andreasson, U.; Norgren, N.; Linder, J.; Forsgren, L.; Constantinescu, R.; et al. Blood-based NfL: A biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017, 88, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Ehling, R.; Lutterotti, A.; Wanschitz, J.; Khalil, M.; Gneiss, C.; Deisenhammer, F.; Reindl, M.; Berger, T. Increased frequencies of serum antibodies to neurofilament light in patients with primary chronic progressive multiple sclerosis. Mult. Scler. J. 2004, 10, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Fialová, L.; Bartos, A.; Švarcová, J.; Zimova, D.; Kotoucova, J.; Malbohan, I. Serum and cerebrospinal fluid light neurofilaments and antibodies against them in clinically isolated syndrome and multiple sclerosis. J. Neuroimmunol. 2013, 262, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; van der Star, B.J.; Bosca, I.; Raffel, J.; Gnanapavan, S.; Watchorn, J.; Kuhle, J.; Giovannoni, G.; Baker, D.; Malaspina, A.; et al. Neurofilament light antibodies in serum reflect response to natalizumab treatment in multiple sclerosis. Mult. Scler. J. 2014, 20, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Barro, C.; Disanto, G.; Mathias, A.; Soneson, C.; Bonnier, G.; Yaldizli, Ö.; Regeniter, A.; Derfuss, T.; Canales, M.; et al. Serum neurofilament light chain in early relapsing remitting MS is increased and correlates with CSF levels and with MRI measures of disease severity. Mult. Scler. J. 2016, 22, 1550–1559. [Google Scholar] [CrossRef]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring disease activity in multiple sclerosis. Using serum neurofilament light protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli, Ö.; et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Piehl, F.; Kockum, I.; Khademi, M.; Blennow, K.; Lycke, J.; Zetterberg, H.; Olsson, T. Plasma neurofilament light chain levels in patients with MS switching from injectable therapies to fingolimod. Mult. Scler. 2018, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Chen, X.X.; Huang, L.D.; Zhu, C.Q.; Gu, Y.Y.; Ye, S. Anti-alpha-internexin autoantibody from neuropsychiatric lupus induce cognitive damage via inhibiting axonal elongation and promote neuron apoptosis. PLoS ONE 2010, 5, e11124. [Google Scholar] [CrossRef] [PubMed]
- Oron, L.; Dubovik, V.; Novitsky, L.; Eilam, D.; Michaelson, D.M. Animal model and in vitro studies of anti neurofilament antibodies mediated neurodegeneration in Alzheimer’s disease. J. Neural. Transm. Suppl. 1997, 49, 77–84. [Google Scholar] [PubMed]
- Stubbs, E.B.; Lawlor, M.W.; Richards, M.P.; Siddiqui, K.; Fisher, M.A.; Bhoopalam, N.; Siegel, G.J. Anti-neurofilament antibodies in neuropathy with monoclonal gammopathy of undetermined significance produce experimental motor nerve conduction block. Acta Neuropathol. 2003, 105, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Soussan, L.; Tchernakov, K.; Bachar-Lavi, O.; Yuvan, T.; Wertman, E.; Michaelson, D.M. Antibodies to different isoforms of the heavy neurofilament protein (NF-H) in normal aging and Alzheimer’s disease. Mol. Neurobiol. 1994, 9, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Elizan, T.S.; Casals, J.; Yahr, M.D. Antineurofilament antibodies in postencephalitic and idiopathic Parkinson’s disease. J. Neurol. Sci. 1983, 59, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Karcher, D.; Federsppiel, B.S.S.; Lowenthal, F.D.; Frank, F.; Lowenthal, A. Anti-neurofilament antibodies in blood of patients with neurological diseases. Acta Neuropathol. 1986, 72, 82–85. [Google Scholar] [CrossRef]
- Kronimus, Y.; Albus, A.; Balzer-Geldsetzer, M.; Straub, S.; Semler, E.; Otto, M.; Klotsche, J.; Dodel, R.; Landscape Consortium; Mengel, D. Naturally Occurring Autoantibodies against Tau Protein Are Reduced in Parkinson’s Disease Dementia. PLoS ONE 2016, 11, e0164953. [Google Scholar] [CrossRef]
- Liu, T.W.; Chen, C.M.; Chang, K.H. Biomarker of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4148. [Google Scholar] [CrossRef]
- Couratier, P.; Yi, F.H.; Preud’homme, J.L.; Clavelou, P.; White, A.; Sindou, P.; Vallat, J.M.; Jauberteau, M.O. Serum autoantibodies to neurofilament proteins in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 154, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Fialová, L.; Švarcová, J.; Bartos, A.; Ridzoň, P.; Malbohan, I.; Keller, O.; Rusina, R. Cerebrospinal fluid and serum antibodies against neurofilaments in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2010, 17, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Puentes, F.; Topping, J.; Kuhle, J.; van der Star, B.J.; Douiri, A.; Giovannoni, G.; Baker, D.; Amor, S.; Malaspina, A. Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Niebroj-Dobosz, I.; Dziewulska, D.; Janik, P. Auto-antibodies against proteins of spinal cord cells in cerebrospinal fluid of patients with amyotrophic lateral sclerosis (ALS). Folia Neuropathol. 2006, 44, 191–196. [Google Scholar] [PubMed]
- Szychowski, K.A.; Skóra, B.; Wójtowicz, A.K. Elastin-Derived Peptides in the Central Nervous System: Friend or Foe. Cell. Mol. Neurobiol. 2022, 42, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- DeMarshall, C.; Sarkar, A.; Nagele, E.P.; Goldwaser, E.; Godsey, G.; Acharya, N.K.; Nagele, R.G. Utility of autoantibodies as biomarkers for diagnosis and staging of neurodegenerative diseases. Int. Rev. Neurobiol. 2015, 122, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Stover, C.M. The potential of circulating autoantibodies in the early diagnosis of Alzheimer’s disease. AIMS Allergy Immunol. 2017, 1, 62–70. [Google Scholar] [CrossRef]
- Gao, V.; Briano, J.A.; Komer, L.E.; Burré, J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J. Mol. Biol. 2023, 435, 167714. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Obrocki, P.; Khatun, A.; Ness, D.; Senkevich, K.; Hanrieder, J.; Capraro, F.; Mattsson, N.; Andreasson, U.; Portelius, E.; Ashton, N.J.; et al. Perspectives in fluid biomarkers in neurodegeneration from the 2019 biomarkers in neurodegenerative diseases course-a joint PhD student course at University College London and University of Gothenburg. Alzheimer’s Res. Ther. 2020, 12, 20. [Google Scholar] [CrossRef]
- Nagele, E.; Han, M.; DeMarshall, C.; Belinka, B.; Nagele, R. Diagnosis of Alzheimer’s Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS ONE 2011, 6, e23112. [Google Scholar] [CrossRef] [PubMed]
- Nagele, E.P.; Han, M.; Acharya, N.K.; DeMarshall, C.; Kosciuk, M.C.; Nagele, R.G. Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS ONE 2013, 8, e60726. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.M.; Wilson, R.; Wilson, J.; Connell, S.; Gocke, A.; Hynan, L.; German, D.; Kodadek, T. Identification of Candidate IgG Antibody Biomarkers for Alzheimer’s Disease Through Screening of Synthetic Combinatorial Libraries. Cell 2011, 144, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Leslie, D.; Lipsky, P.; Notkins, A.L. Autoantibodies as predictors of disease. J. Clin. Investig. 2001, 108, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Bingley, P.J.; Bonifacio, E.; Williams, A.J.K.; Genovese, S.; Bottazzo, G.F.; Gale, E.A.M. Prediction of IDDM in the general population: Strategies based on combinations of autoantibody markers. Diabetes 1997, 46, 1701–1710. [Google Scholar] [CrossRef]
- Cummings, J. Anti-Amyloid Monoclonal Antibodies are Transformative Treatments that Redefine Alzheimer’s Disease Therapeutics. Drugs 2023, 83, 569–576. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimer’s Res. Ther. 2022, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Gklinos, P.; Papadopoulou, M.; Stanulovic, V.; Mitsikostas, D.D.; Papadopoulos, D. Monoclonal Antibodies as Neurological Therapeutics. Pharmaceuticals 2021, 14, 92. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miteva, D.; Vasilev, G.V.; Velikova, T. Role of Specific Autoantibodies in Neurodegenerative Diseases: Pathogenic Antibodies or Promising Biomarkers for Diagnosis. Antibodies 2023, 12, 81. https://doi.org/10.3390/antib12040081
Miteva D, Vasilev GV, Velikova T. Role of Specific Autoantibodies in Neurodegenerative Diseases: Pathogenic Antibodies or Promising Biomarkers for Diagnosis. Antibodies. 2023; 12(4):81. https://doi.org/10.3390/antib12040081
Chicago/Turabian StyleMiteva, Dimitrina, Georgi V. Vasilev, and Tsvetelina Velikova. 2023. "Role of Specific Autoantibodies in Neurodegenerative Diseases: Pathogenic Antibodies or Promising Biomarkers for Diagnosis" Antibodies 12, no. 4: 81. https://doi.org/10.3390/antib12040081
APA StyleMiteva, D., Vasilev, G. V., & Velikova, T. (2023). Role of Specific Autoantibodies in Neurodegenerative Diseases: Pathogenic Antibodies or Promising Biomarkers for Diagnosis. Antibodies, 12(4), 81. https://doi.org/10.3390/antib12040081