Reactive Transport Modeling for Mobilization of Arsenic in a Sediment Downgradient from an Iron Permeable Reactive Barrier


Abstract
:1. Introduction
2. Methods
2.1. Laboratory Column Experiments
2.1.1. Materials
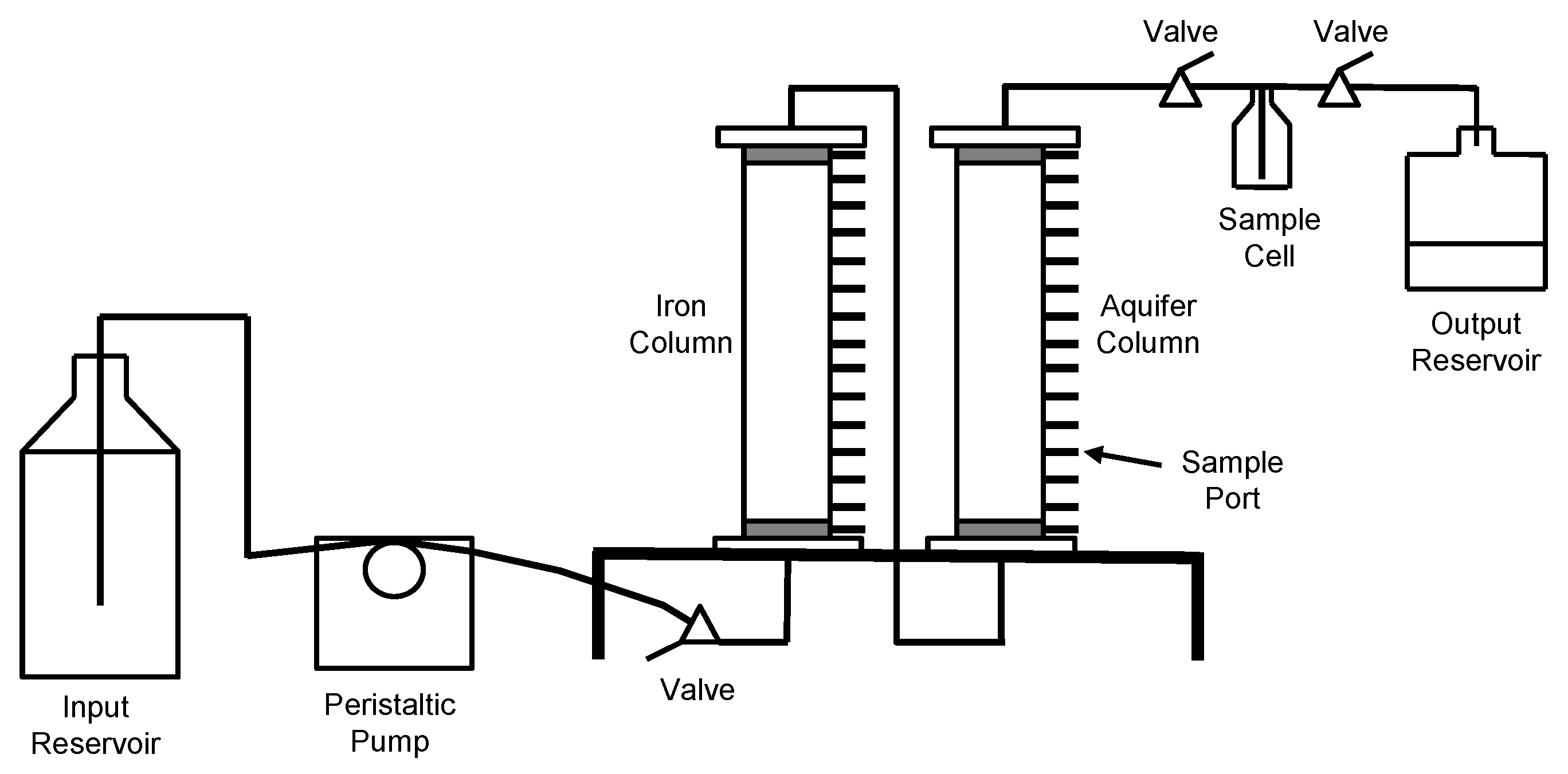
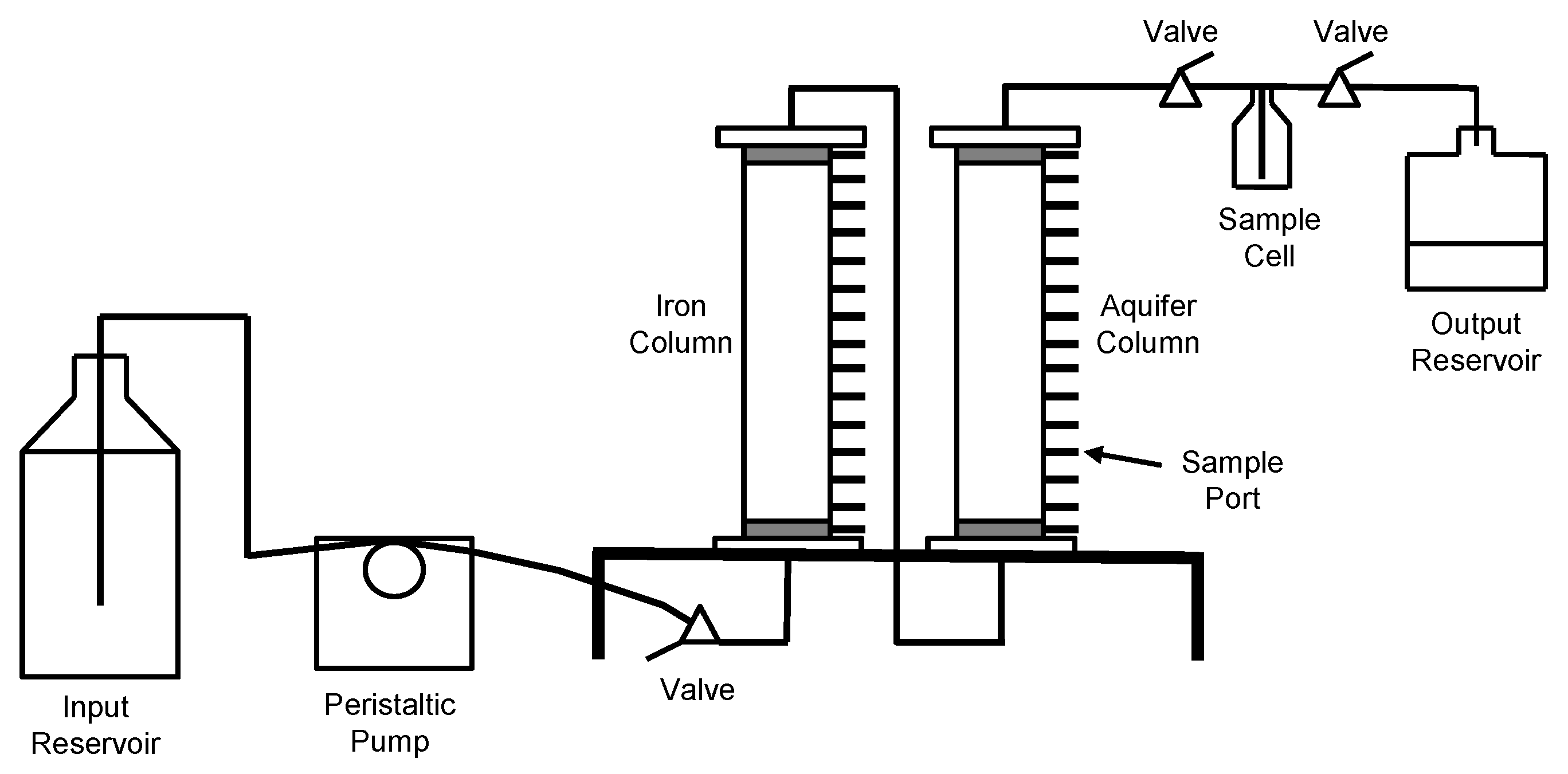
2.1.2. Column System
2.1.3. Column Operation
2.1.4. Sampling and Analyses
2.2. Reactive Transport Modeling
3. Results and Discussion
3.1. Column Experiments
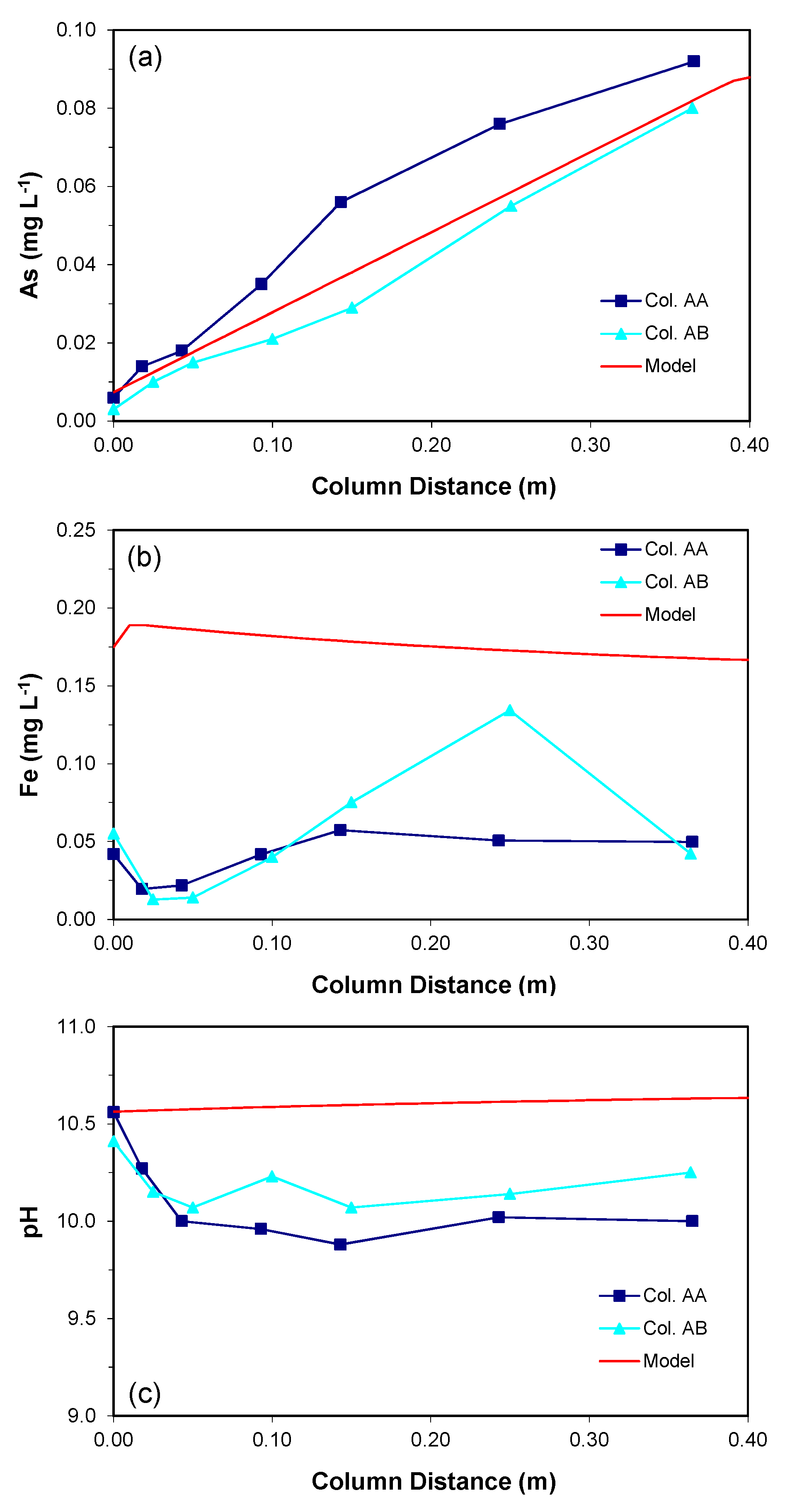
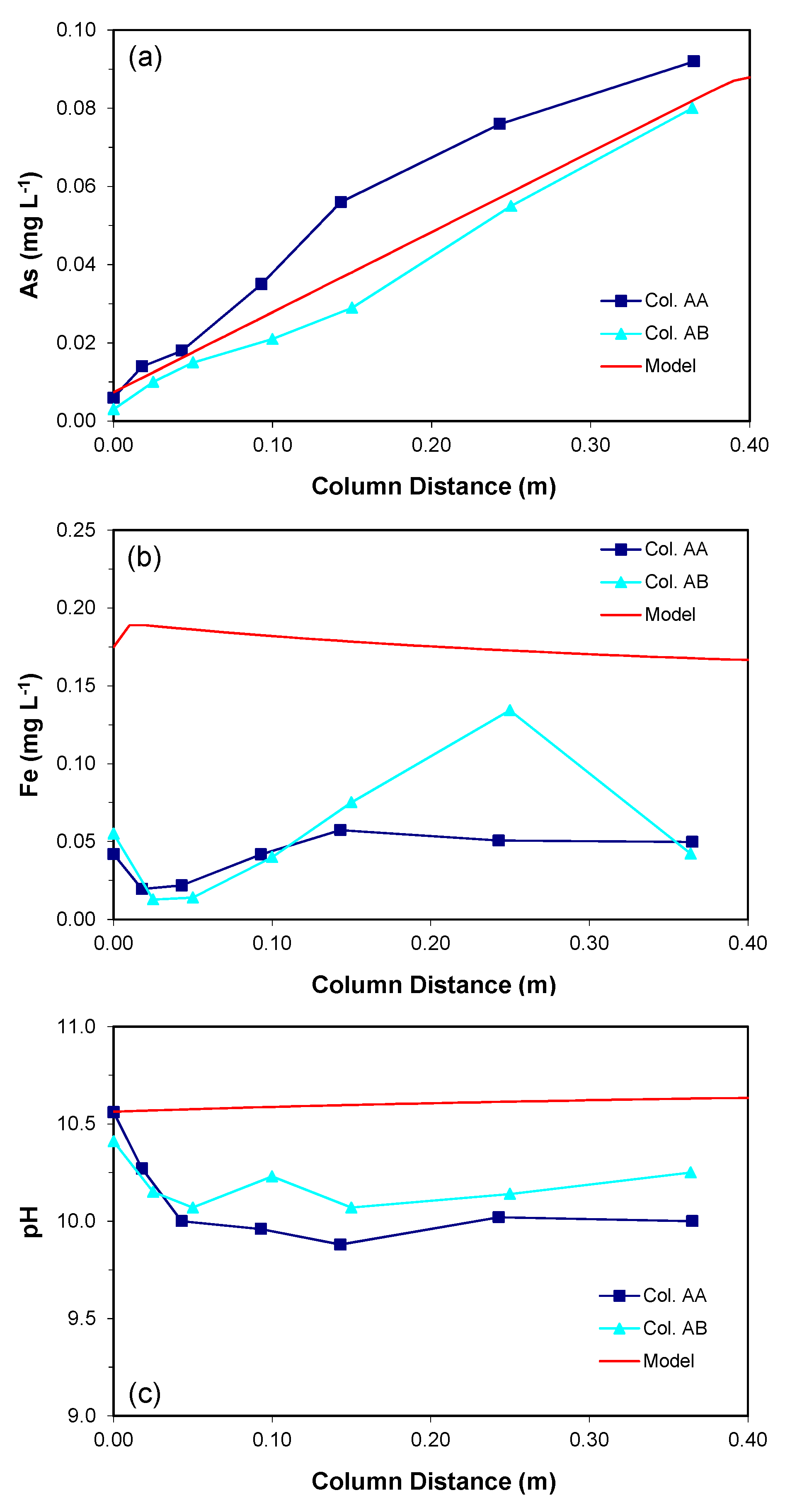
3.1.1. Arsenic Concentrations
3.1.2. Water Chemistry
3.2. Reactive Transport Modeling
3.2.1. Column Data
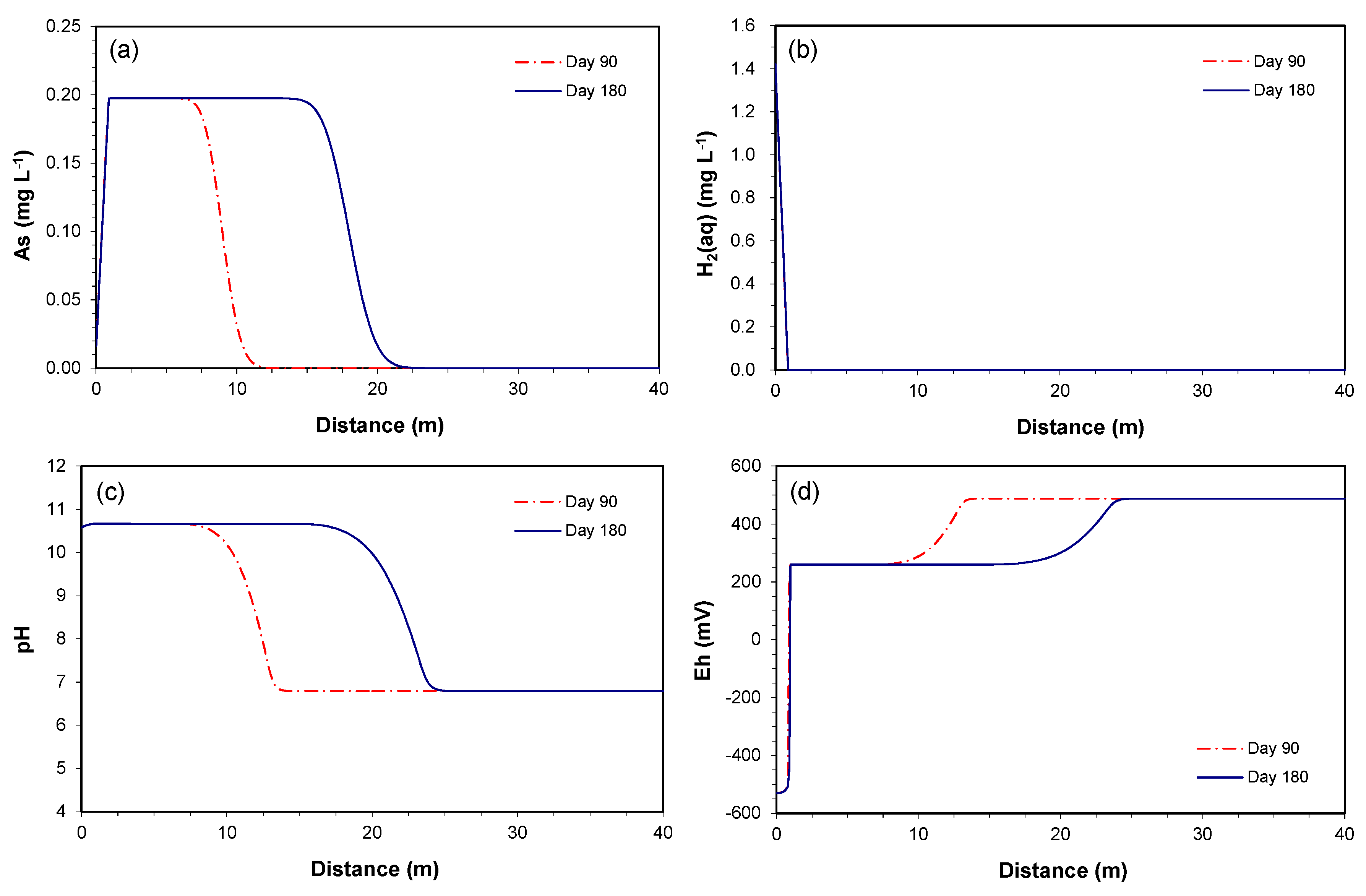
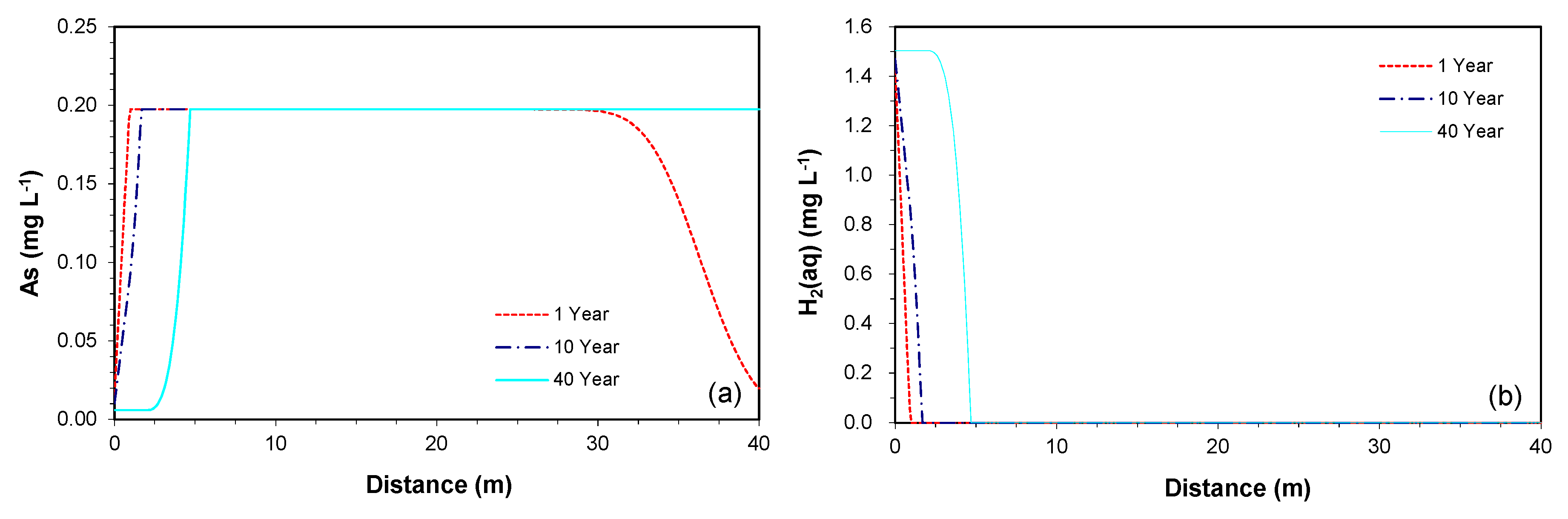
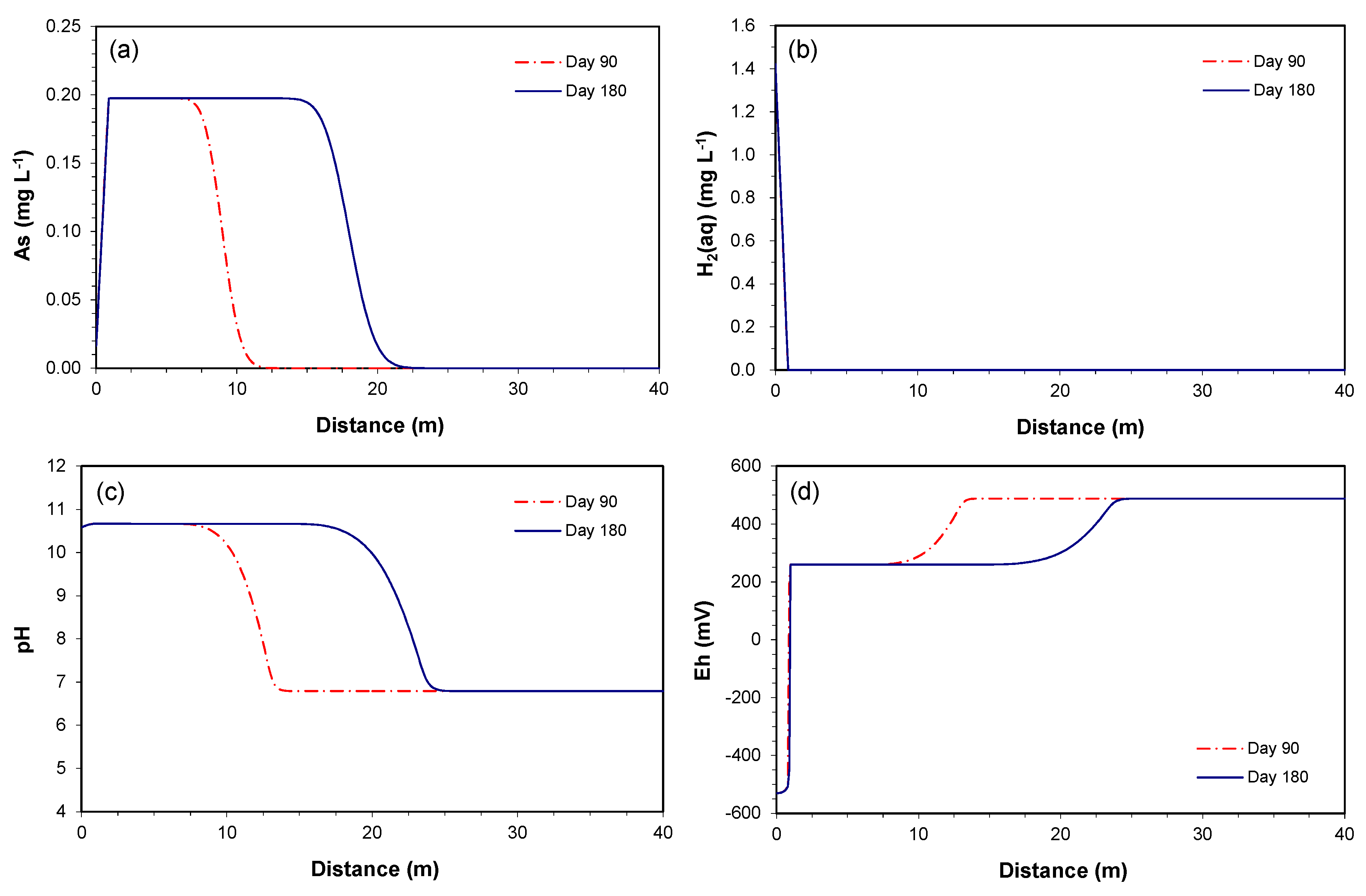
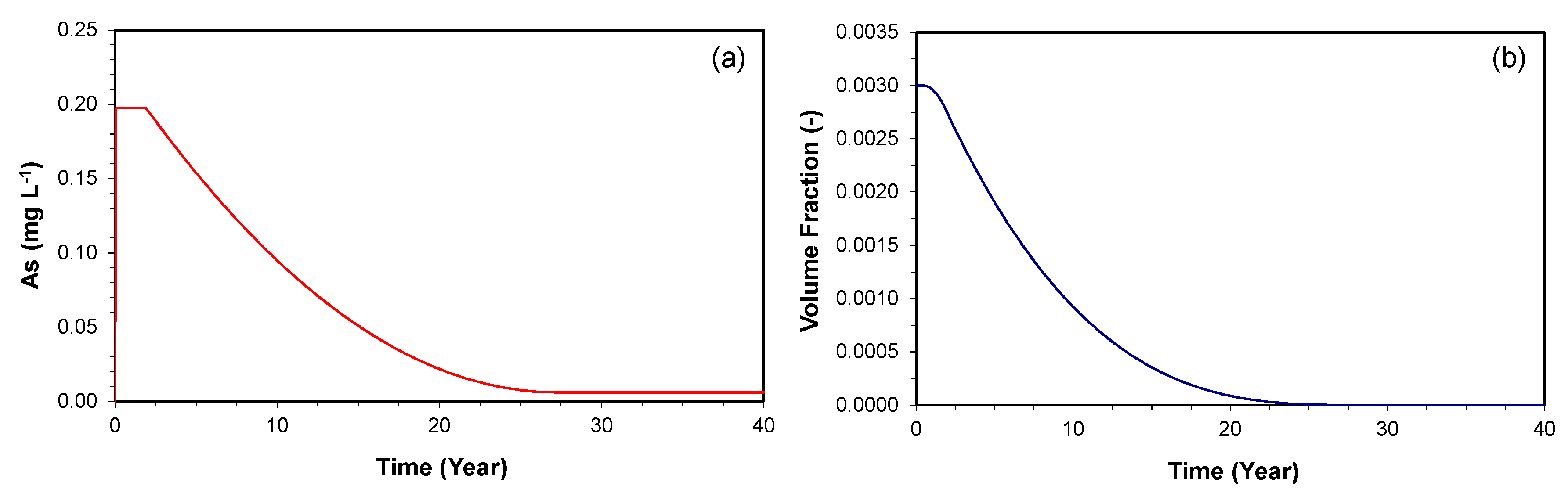
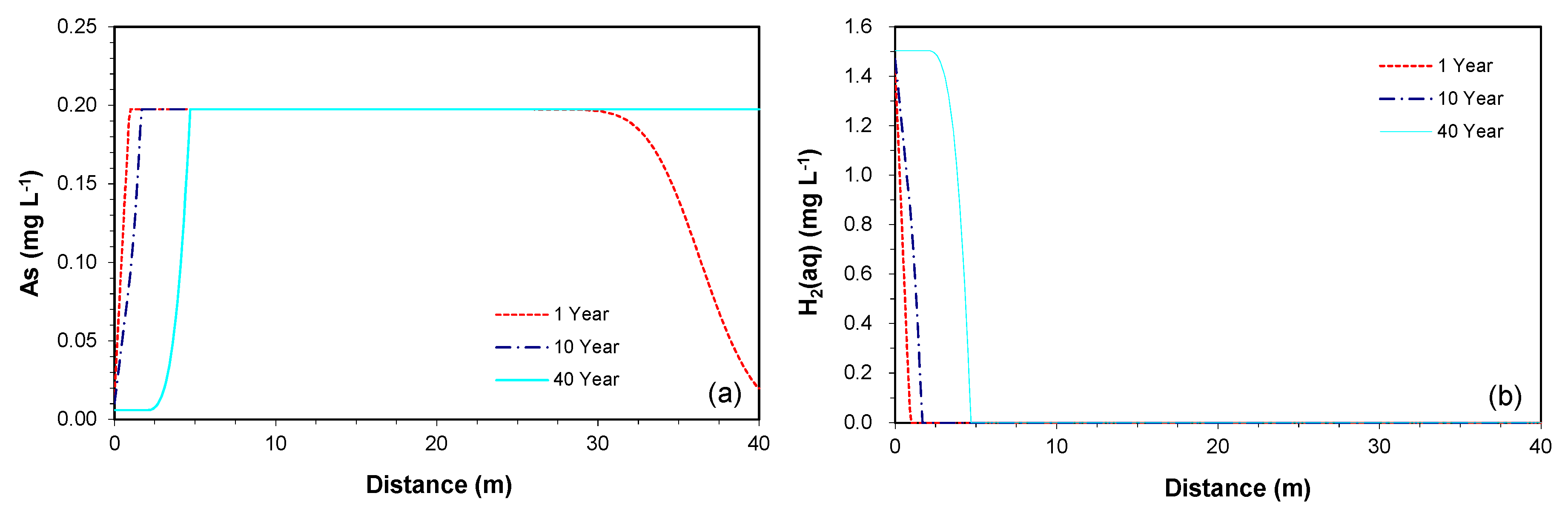
3.2.2. Prediction along the Downgradient of the Proposed PRB
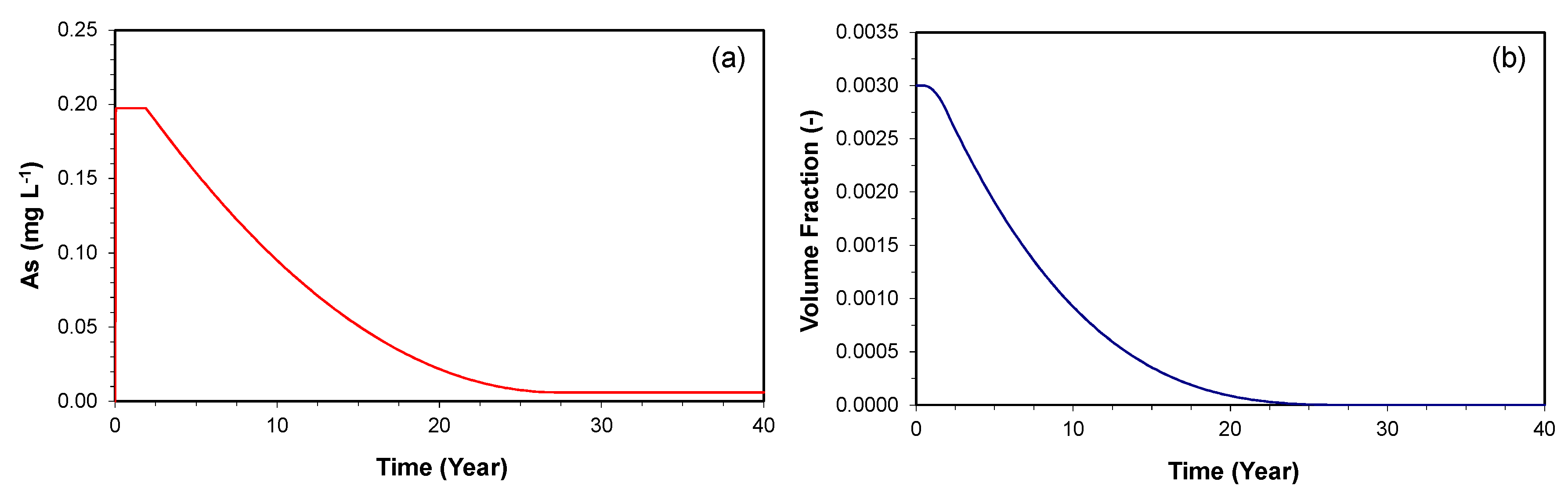
3.2.3. Consumption of As-Bearing Iron Oxides
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jain, C.; Ali, I. Arsenic: Occurrence, toxicity and speciation techniques. Water Res. 2000, 34, 4304–4312. [Google Scholar] [CrossRef]
- Garelick, H.; Jones, H.; Dybowska, A.; Valsami-Jones, E. Arsenic pollution sources. Rev. Environ. Contam. Toxicol. 2008, 197, 17–60. [Google Scholar] [PubMed]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Kim, K.; Kim, S.-H.; Jeong, G.Y.; Kim, R.-H. Relations of As concentrations among groundwater, soil, and bedrock in Chungnam, Korea: Implications for as mobilization in groundwater according to the as-hosting mineral change. J. Hazard. Mater. 2012, 199–200, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Anawar, H.M.; Akai, J.; Mihaljevič, M.; Sikder, A.M.; Ahmed, G.; Tareq, S.M.; Rahman, M.M. Arsenic contamination in groundwater of Bangladesh: Perspectives on geochemical, microbial and anthropogenic issues. Water 2011, 3, 1050–1076. [Google Scholar] [CrossRef]
- McBean, E.A.; Rajib, M.A.; Rahman, M.M. Improved sustainability of water supply options in areas with arsenic-impacted groundwater. Water 2013, 5, 1941–1951. [Google Scholar] [CrossRef]
- Tian, Y.; Yu, C.; Zha, X.; Wu, J.; Gao, X.; Feng, C.; Luo, K. Distribution and potential health risks of arsenic, selenium, and fluorine in natural waters in Tibet, China. Water 2016, 8, 568. [Google Scholar] [CrossRef]
- Thakur, J.K.; Thakur, R.K.; Ramanathan, A.L.; Kumar, M.; Singh, S.K. Arsenic contamination of groundwater in Nepal—An overview. Water 2011, 3, 1–20. [Google Scholar] [CrossRef]
- Kapaj, S.; Peterson, H.; Liber, K.; Bhattacharya, P. Human health effects from chronic arsenic poisoning—A review. J. Environ. Sci. Health A 2006, 41, 2399–2428. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Gan, Y.; Wang, Y.; Liu, C.; Yu, K.; Deng, Y.; Zhao, K.; Dong, C. Arsenic speciation in aquifer sediment under varying groundwater regime and redox conditions at Jianghan Plain of Central China. Sci. Total Environ. 2017, 607–608, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, Y.; Su, C.; Liu, H.; Duan, M.; Xie, Z. Arsenic mobilization in shallow aquifers of Datong Basin: Hydrochemical and mineralogical evidences. J. Geochem. Explor. 2008, 98, 107–115. [Google Scholar] [CrossRef]
- Kim, K.; Moon, J.-T.; Kim, S.-H.; Ko, K.-S. Importance of surface geologic condition in regulating As concentration of groundwater in the alluvial plain. Chemosphere 2009, 77, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Donselaar, M.E.; Bhatt, A.G.; Ghosh, A.K. On the relation between fluvio-deltaic flood basin geomorphology and the wide-spread occurrence of arsenic pollution in shallow aquifers. Sci. Total Environ. 2017, 574, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, R.T.; Acree, S.D.; Ross, R.R.; Beak, D.G.; Lee, T.R. Performance of a zerovalent iron reactive barrier for the treatment of arsenic in groundwater: Part 1. Hydrogeochemical studies. J. Contam. Hydrol. 2009, 106, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Puls, R.W. Arsenate and arsenite removal by zerovalent iron: Kinetics, redox transformation, and implications for in situ groundwater remediation. Environ. Sci. Technol. 2001, 35, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Blowes, D.W.; Ptacek, C.J.; Benner, S.G.; McRae, C.W.T.; Puls, R.W. Treatment of dissolved metals and nutrients using permeable reactive barriers. J. Contam. Hydrol. 2000, 45, 123–137. [Google Scholar] [CrossRef]
- Morrison, S.J.; Metzler, D.R.; Dwyer, B.P. Removal of As, Mn, Mo, Se, U, V and Zn from groundwater by zero-valent iron in a passive treatment cell: Reaction progress modeling. J. Contam. Hydrol. 2002, 56, 99–116. [Google Scholar] [CrossRef]
- Luwig, R.D.; Smyth, D.J.A.; Blowes, D.W.; Spink, L.E.; Wilkin, R.T.; Jewett, D.G.; Weisener, C.J. Treatment of arsenic, heavy metals, and acidity using a mixed ZVI-compost PRB. Environ. Sci. Technol. 2009, 43, 1970–1976. [Google Scholar] [CrossRef]
- Phillips, D.H.; Van Nooten, T.; Bastiaens, L.; Russell, M.I.; Dickson, K.; Plant, S.; Ahad, J.M.E.; Newton, T.; Elliot, T.; Kalin, R.M. Ten year performance evaluation of a field-scale zero-valent iron permeable reactive barrier installed to remediate trichloroethene contaminated groundwater. Environ. Sci. Technol. 2010, 44, 3861–3869. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Sun, Y.; Qin, H.; Li, J.; Lo, I.M.C.; He, D.; Dong, H. The limitations of applying zero-valent iron technology in contaminants sequestration and the corresponding countermeasures: The development in zero-valent iron technology in the last two decades (1994–2014). Water Res. 2015, 75, 224–248. [Google Scholar] [CrossRef] [PubMed]
- Beak, D.; Wilkin, R.T. Performance of a zerovalent iron reactive barrier for the treatment of arsenic in groundwater: Part 2. Geochemical modeling and solid phase studies. J. Contam. Hydrol. 2009, 106, 15–28. [Google Scholar] [CrossRef] [PubMed]
- O’Hannesin, S.F.; Gillham, R.W. Long-term performance of an in-situ “iron wall” for remediation of VOCs. Ground Water 1998, 36, 164–170. [Google Scholar] [CrossRef]
- Mayer, K.U.; Blowes, D.W.; Frind, E.O. Reactive transport modeling of an in situ reactive barrier for the treatment of hexavalent chromium and trichloroethylene in groundwater. Water Resour. Res. 2001, 37, 3091–3103. [Google Scholar] [CrossRef]
- Mayer, K.U.; Frind, E.O.; Blowes, D.W. Multicomponent reactive transport modeling in variably saturated porous media using a generalized formulation for kinetically controlled reactions. Water Resour. Res. 2002, 38, 1174. [Google Scholar] [CrossRef]
- Bilardi, S.; Amos, R.T.; Blowes, D.W.; Calabrò, P.S.; Moraci, N. Reactive transport modeling of ZVI column experiments for nickel remediation. Ground Water Monit. Remediat. 2013, 33, 97–104. [Google Scholar] [CrossRef]
- Jeen, S.-W.; Yang, Y.; Gui, L.; Gillham, R.W. Treatment of trichloroethene and hexavalent chromium by granular iron in the presence of dissolved CaCO3. J. Contam. Hydrol. 2013, 144, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Jeen, S.-W.; Gui, L.; Gillham, R.W. Nitrate reduction and its effects on trichloroethylene degradation by granular iron. Water Res. 2017, 112, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Jeen, S.-W. Geochemical interactions of mine seepage water with an aquifer: Laboratory tests and reactive transport modeling. Environ. Earth Sci. 2016, 75, 1333. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1996. [Google Scholar]
- Allison, J.D.; Brown, D.S.; Novo-Gradac, K.J. MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.0 User’s Manual; EPA-600/3-91-021; U.S. Environmental Protection Agency: Athens, GA, USA, 1991.
- Freeze, R.A.; Cherry, J.A. Groundwater; Prentice-Hall: Englewood Cliffs, NJ, USA, 1979. [Google Scholar]
- Jeen, S.-W.; Mayer, K.U.; Gillham, R.W.; Blowes, D.W. Reactive transport modeling of trichloroethene treatment with declining reactivity of iron. Environ. Sci. Technol. 2007, 41, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Blowes, D.W.; Puls, R.W.; Gillham, R.W.; Ptacek, C.J.; Bennett, T.A.; Bain, J.G.; Hanton-Fong, C.J.; Paul, C.J. An In Situ Permeable Reactive Barrier for Treatment of Hexavalent Chromium and Trichloroethene in Groundwater: Volume 2 Performance Monitoring; EPA 600/R-99/095b; U.S. Environmental Protection Agency: Washington, DC, USA, 1999.
- Wilkin, R.T.; Puls, R.W.; Sewell, G.W. Long-Term Performance of Permeable Reactive Barriers Using Zero-Valent Iron: An Evaluation at Two Sites; Report EPA/600/S-02/001; U.S. Environmental Protection Agency: Cincinnati, OH, USA, 2002.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Height (g) | Mass (g) | Sand (g) | Iron/Aquifer (g) | Pore Volume (mL) | Porosity | |
|---|---|---|---|---|---|---|
| Granular Iron Columns | ||||||
| Column CA | ||||||
| Sand | 2.4 | 82.54 | ||||
| Granular Iron | 39 | 2106.19 | ||||
| Sand | 41 | 80.3 | ||||
| Column Total | 162.8 | 2106.2 | 440 | 0.53 | ||
| Column CB | ||||||
| Sand | 3.1 | 101.86 | ||||
| Granular Iron | 39.6 | 1282.44 | ||||
| Sand | 41 | 57.54 | ||||
| Column Total | 159.4 | 1282.4 | 448 | 0.54 | ||
| Aquifer Columns | ||||||
| Column AA | ||||||
| Sand | 3.1 | 101.86 | ||||
| Aquifer | 39.6 | 1353.6 | ||||
| Moisture a | 95.43 | |||||
| Sand | 41 | 70.62 | ||||
| Column Total | 172.5 | 1258.2 | 233 | 0.28 | ||
| Column AB | ||||||
| Sand | 3 | 99.1 | ||||
| Aquifer | 39.4 | 1407.27 | ||||
| Moisture a | 161.30 | |||||
| Sand | 41 | 49.04 | ||||
| Column Total | 148.1 | 1245.9 | 268 | 0.32 | ||
| Parameter | Concentration (mg L−1) | |||||
|---|---|---|---|---|---|---|
| Influent for Iron Columns | Aquifer Column AA | Aquifer Column AB | Detection Limit | |||
| Influent | Effluent | Influent | Effluent | |||
| As (total) | 0.66 | 0.006 | 0.092 | 0.003 | 0.080 | 0.001 |
| Al | <0.037 | <0.037 | 0.33 | 0.24 | 0.45 | 0.037 |
| Ca | 1.7 | 0.49 | 0.88 | <0.10 | 0.59 | 0.10 |
| Fe | 0.273 | 0.041 | 0.049 | 0.054 | 0.042 | 0.003 |
| K | 1.25 | 1.38 | 1.12 | 1.52 | 0.91 | 0.25 |
| Mg | 0.64 | <0.025 | 0.06 | <0.025 | <0.025 | 0.025 |
| Na | 117 | 116 | 115 | 117 | 125 | 0.05 |
| SiO2 | 20 | 1.9 | 9.9 | 3.8 | 11.6 | 0.25 |
| F | 32 | 32 | 32 | 32 | 32 | 0.40 |
| Cl | 14 | 15 | 16 | 17 | 16 | 1.0 |
| SO4 | 32 | 34 | 36 | 31 | 31 | 1.0 |
| Alkalinity (as CaCO3) | 165 | 154 | 132 | 170 | 169 | 0.10 |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeen, S.-W. Reactive Transport Modeling for Mobilization of Arsenic in a Sediment Downgradient from an Iron Permeable Reactive Barrier. Water 2017, 9, 890. https://doi.org/10.3390/w9110890
Jeen S-W. Reactive Transport Modeling for Mobilization of Arsenic in a Sediment Downgradient from an Iron Permeable Reactive Barrier. Water. 2017; 9(11):890. https://doi.org/10.3390/w9110890
Chicago/Turabian StyleJeen, Sung-Wook. 2017. "Reactive Transport Modeling for Mobilization of Arsenic in a Sediment Downgradient from an Iron Permeable Reactive Barrier" Water 9, no. 11: 890. https://doi.org/10.3390/w9110890
APA StyleJeen, S.-W. (2017). Reactive Transport Modeling for Mobilization of Arsenic in a Sediment Downgradient from an Iron Permeable Reactive Barrier. Water, 9(11), 890. https://doi.org/10.3390/w9110890