X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
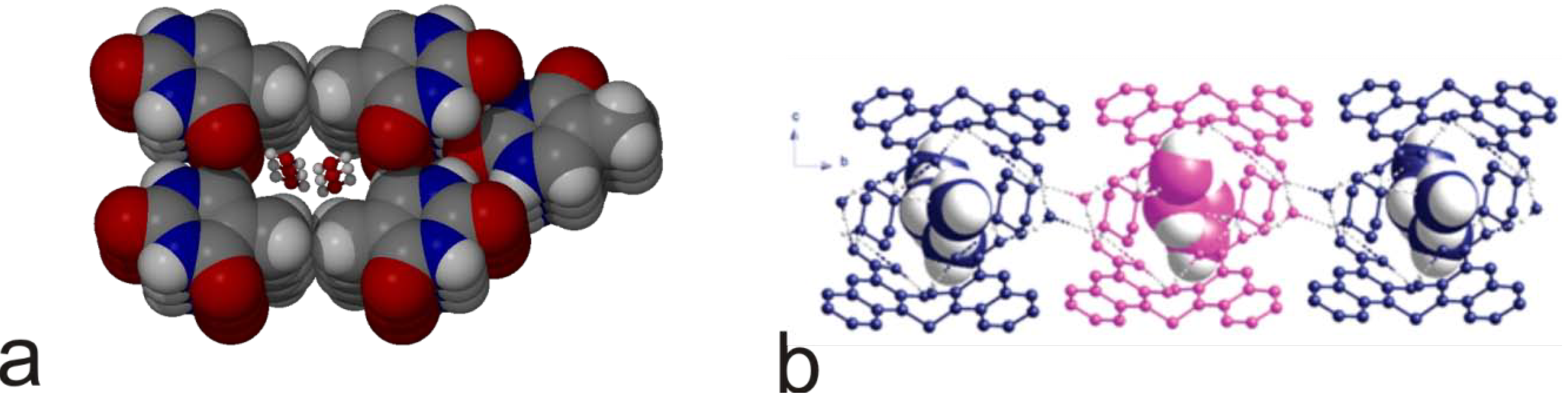{kind=link}
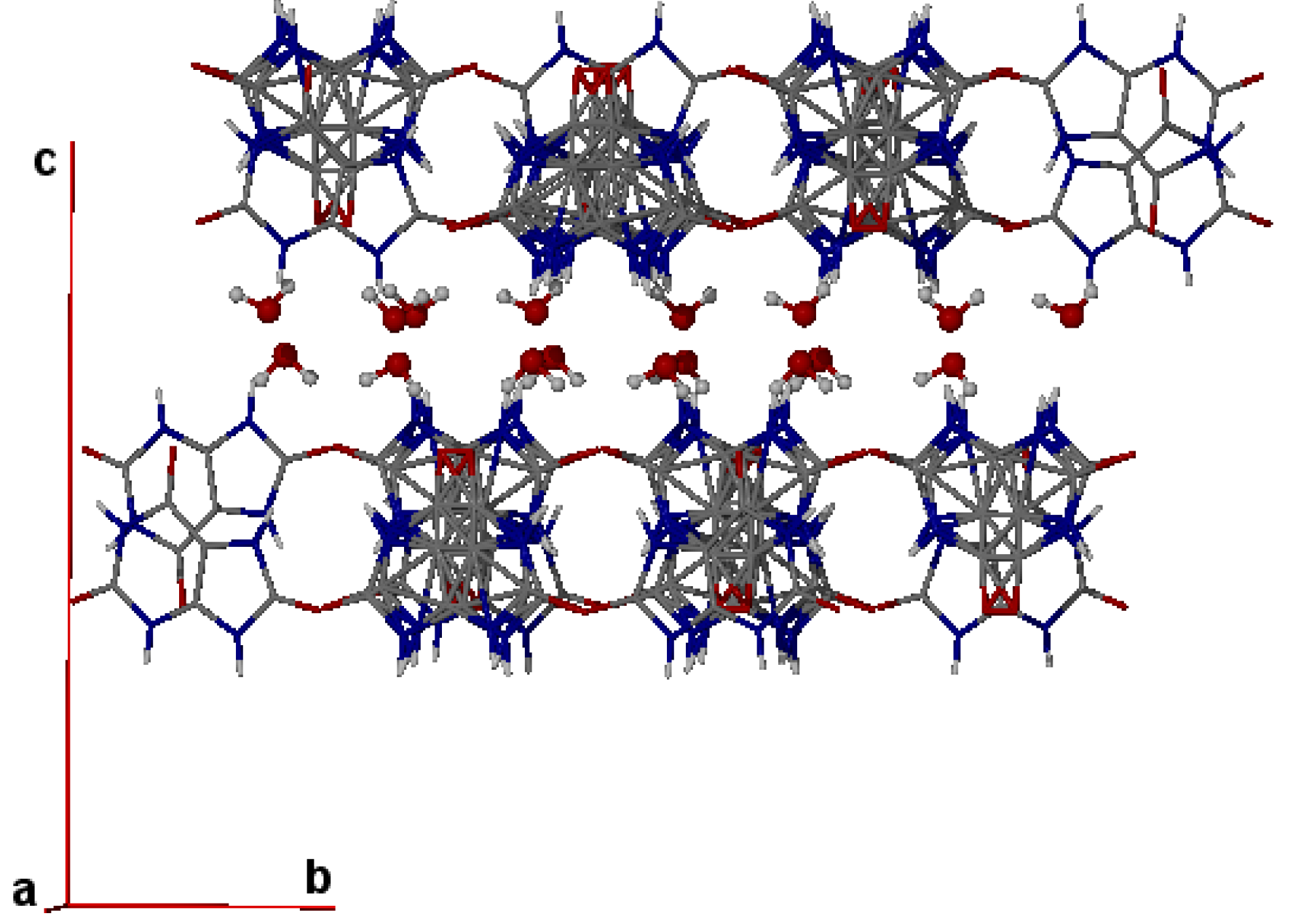{kind=link}
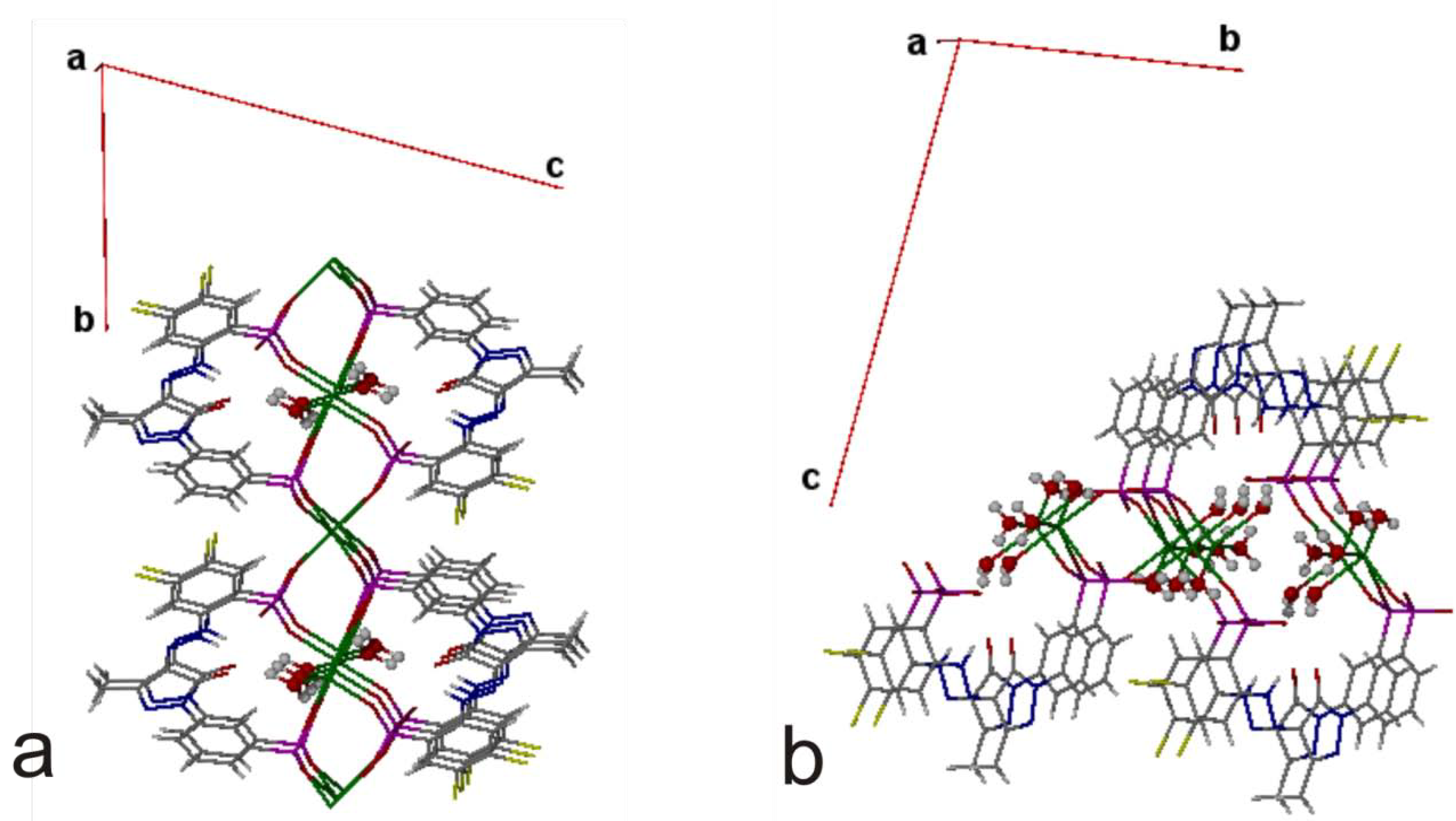{kind=link}
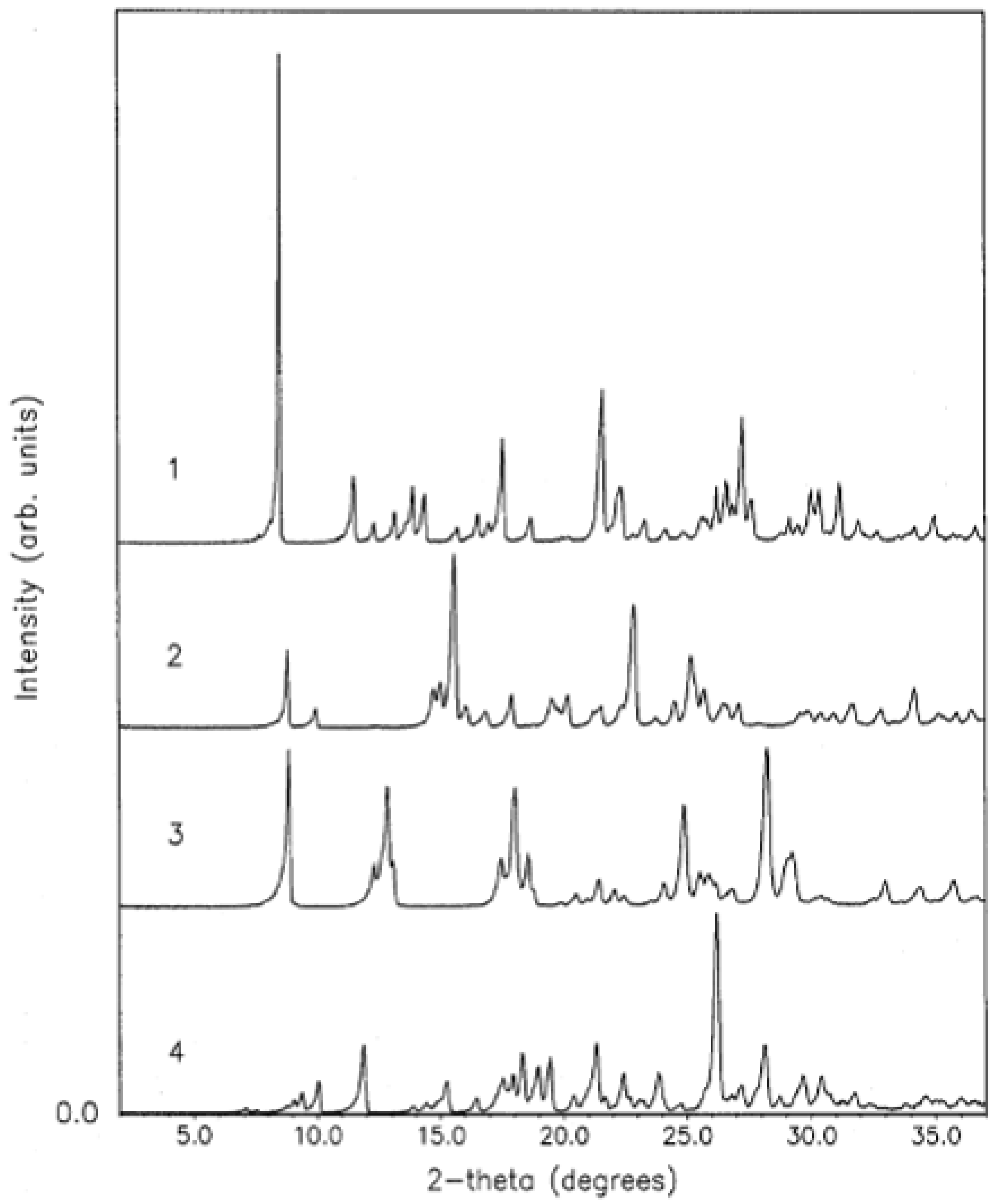{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
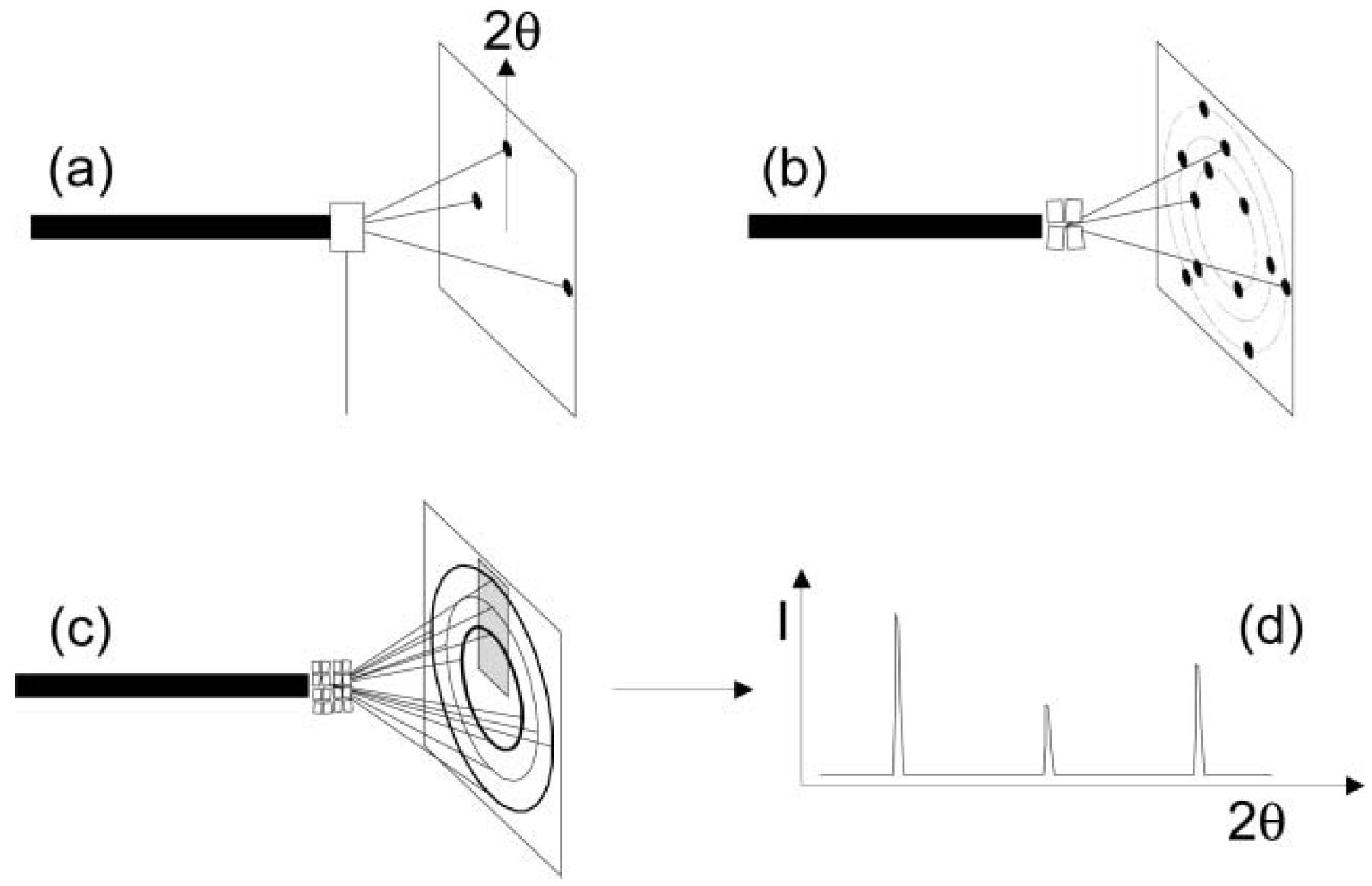
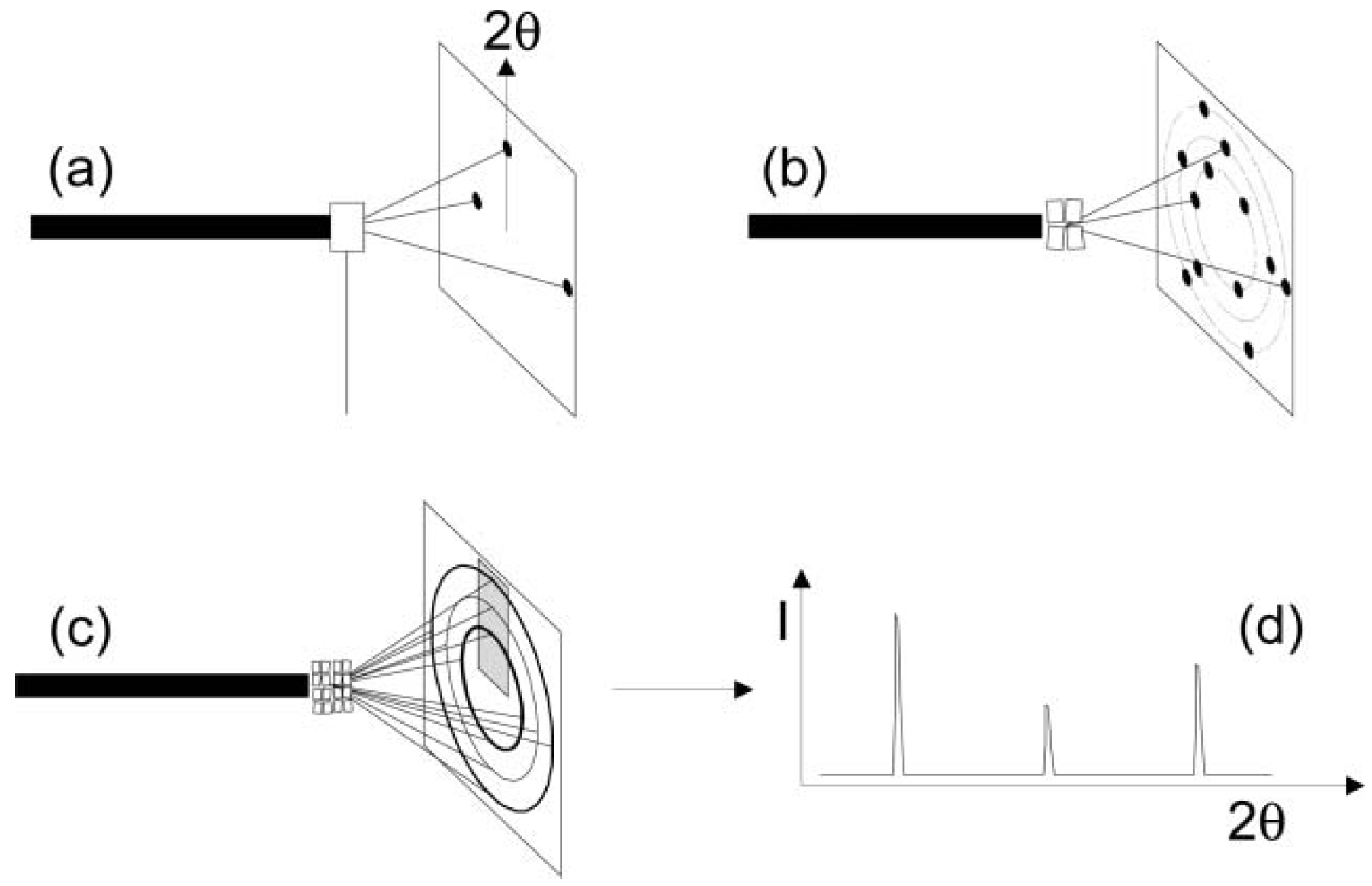
2. Diffraction Techniques—General Principles
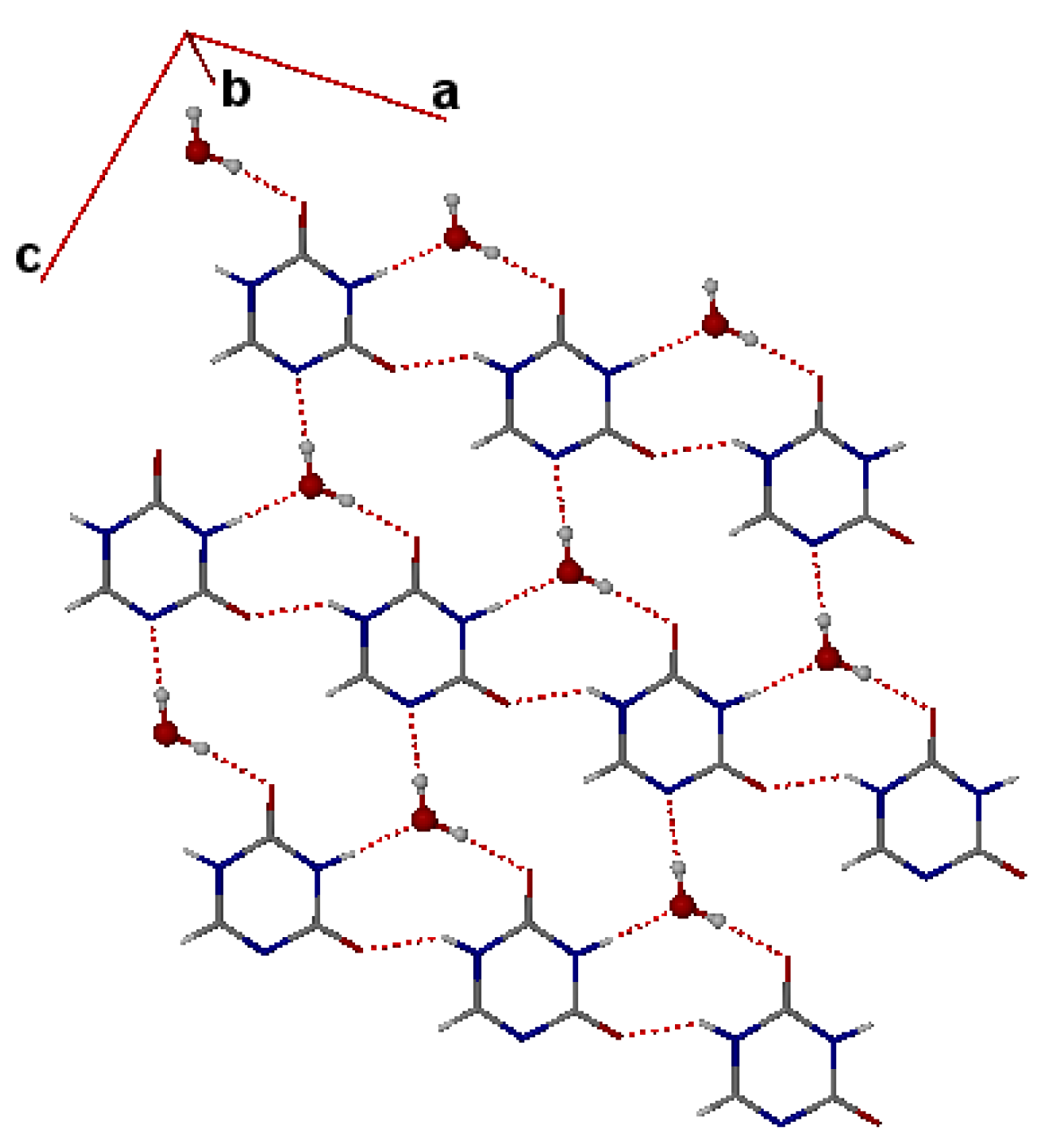
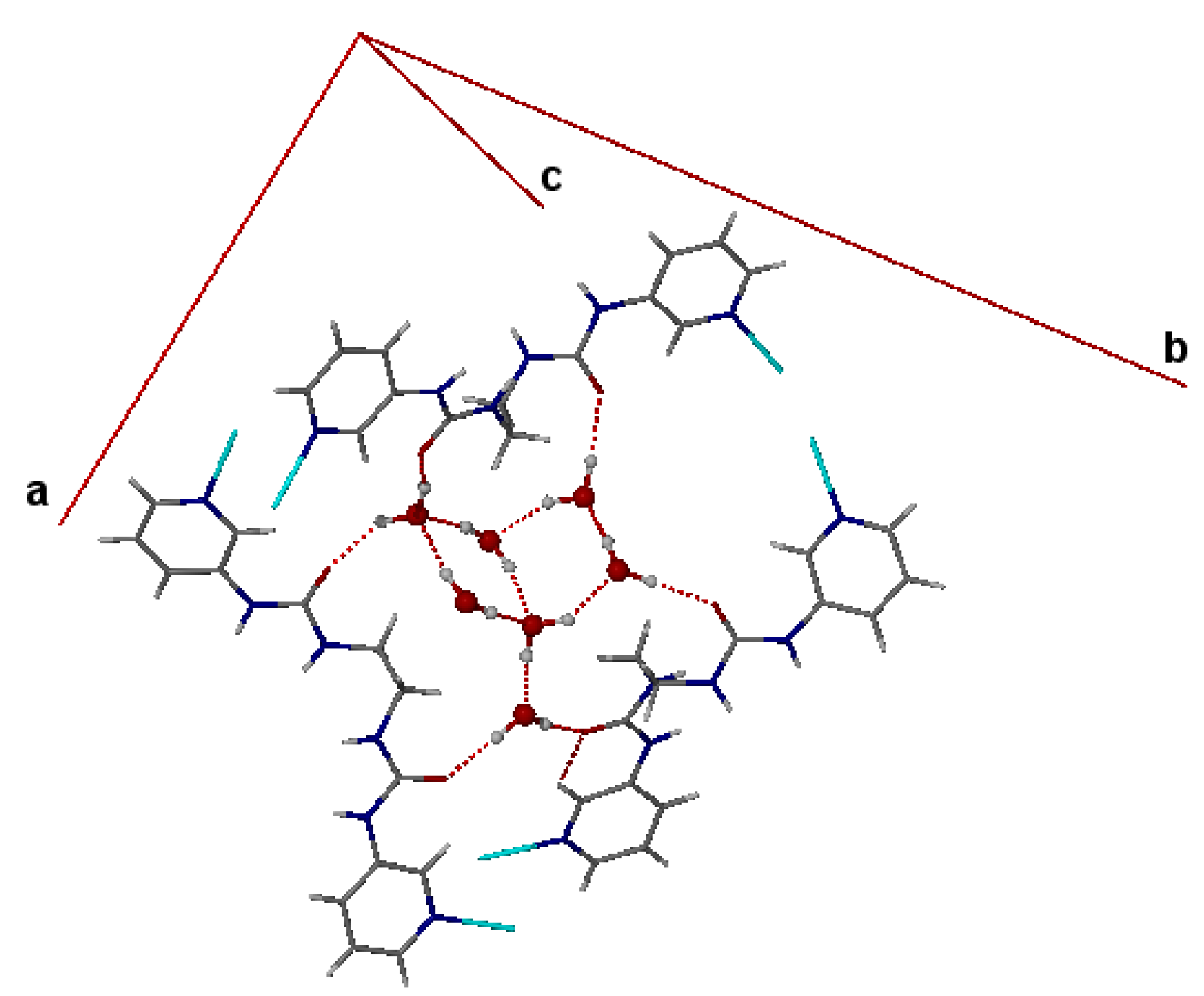
3. Single Crystal X-ray Structures of Hydrates
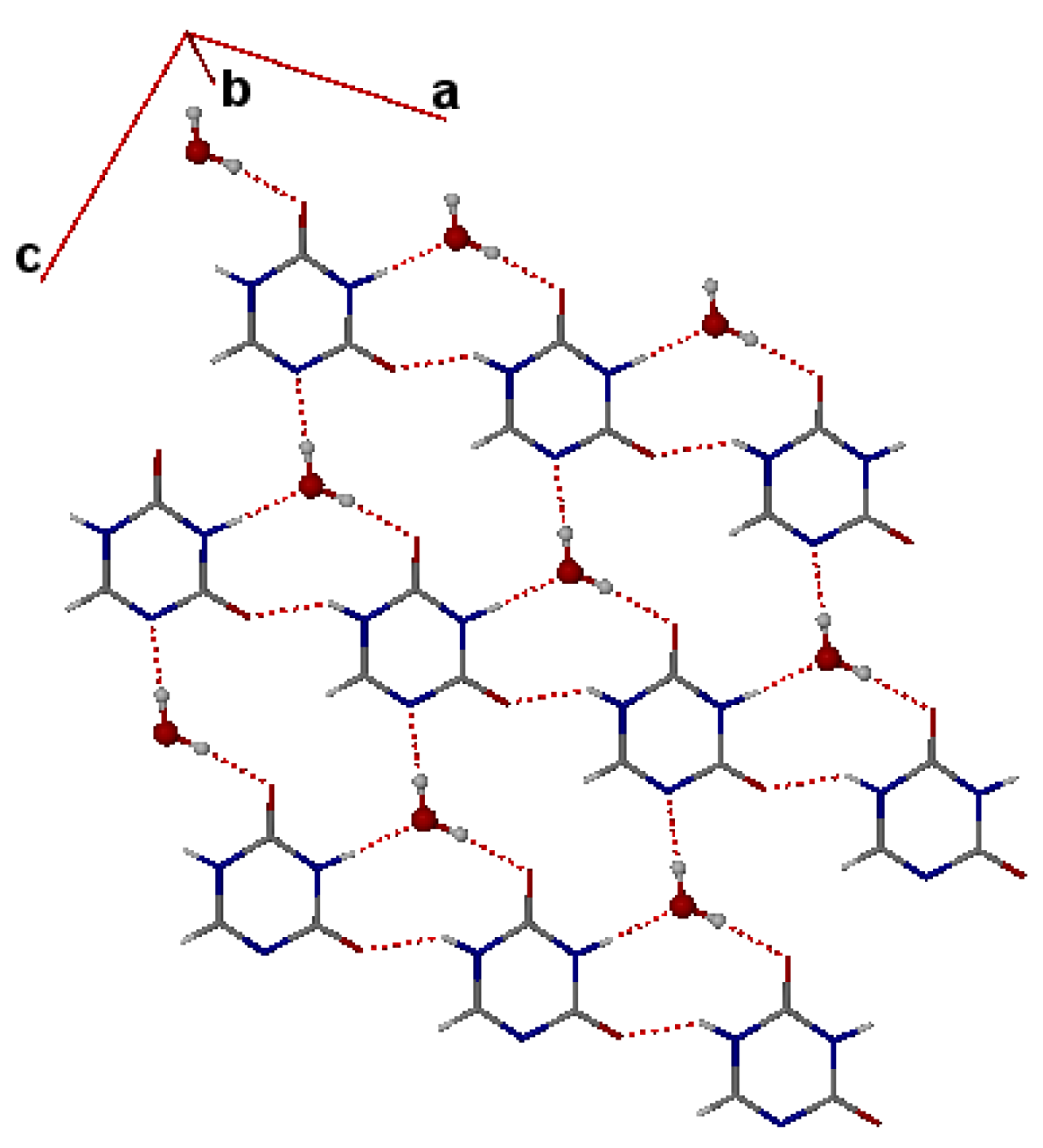
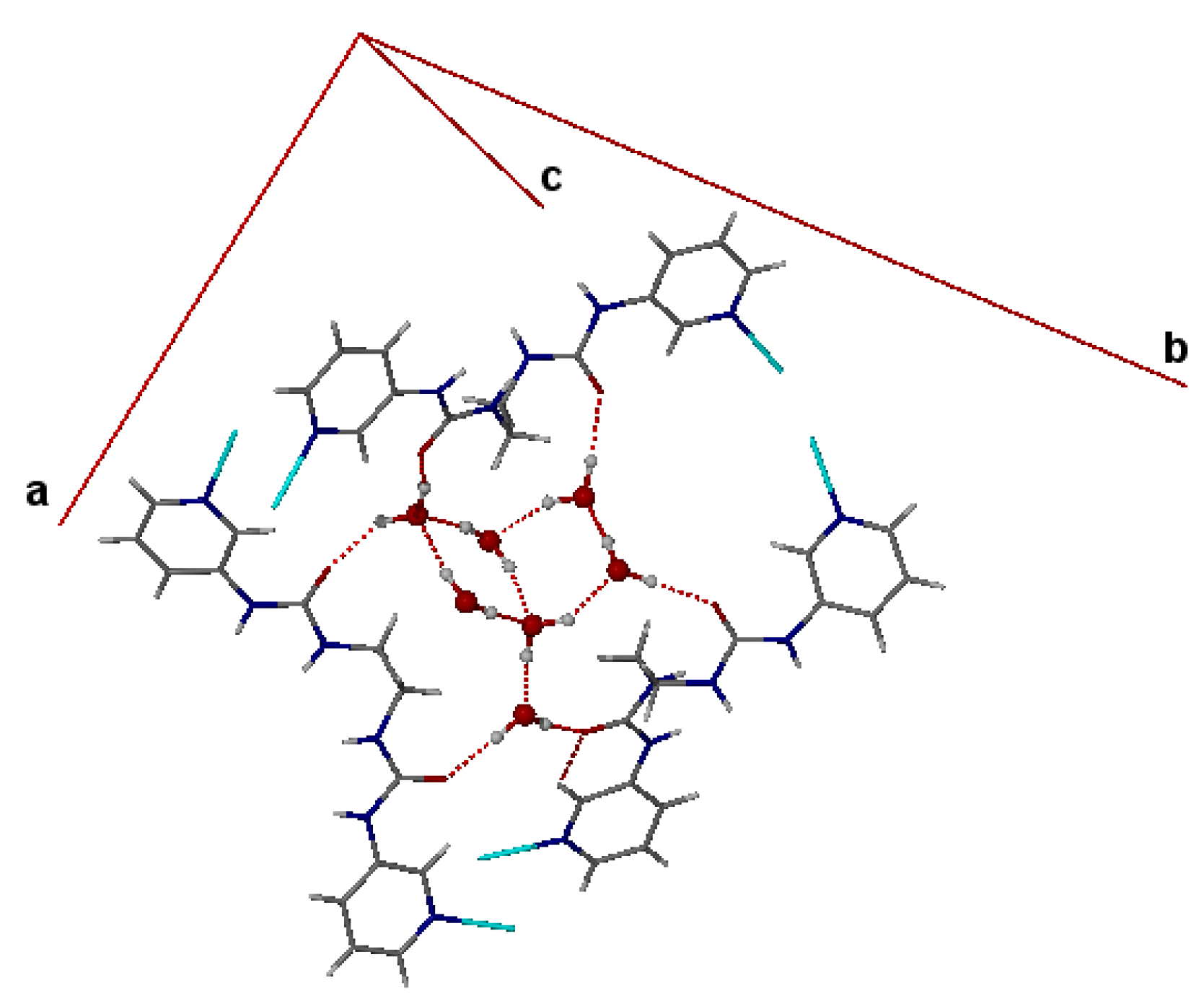
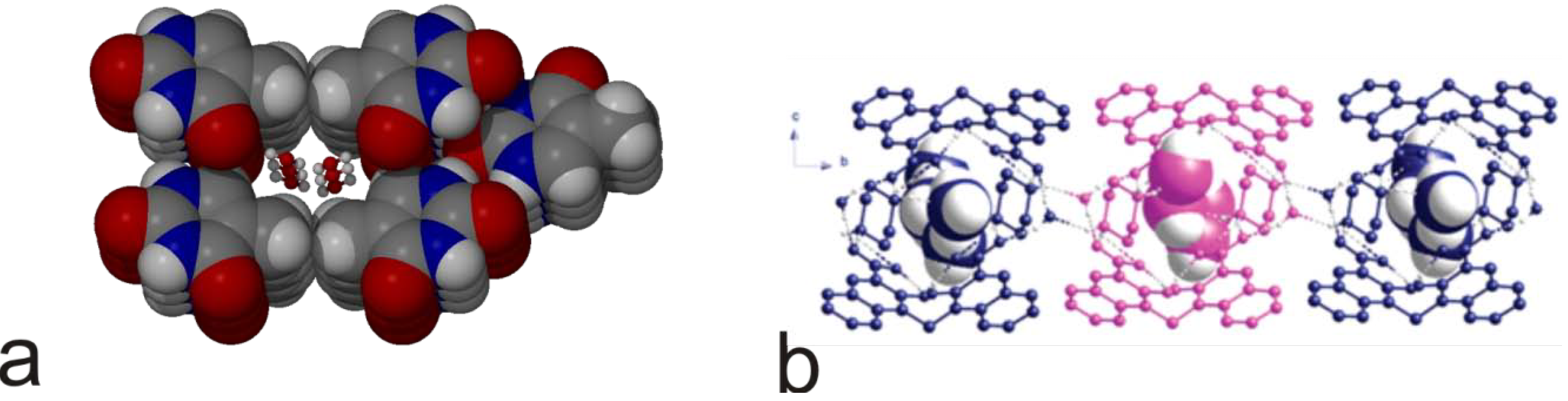
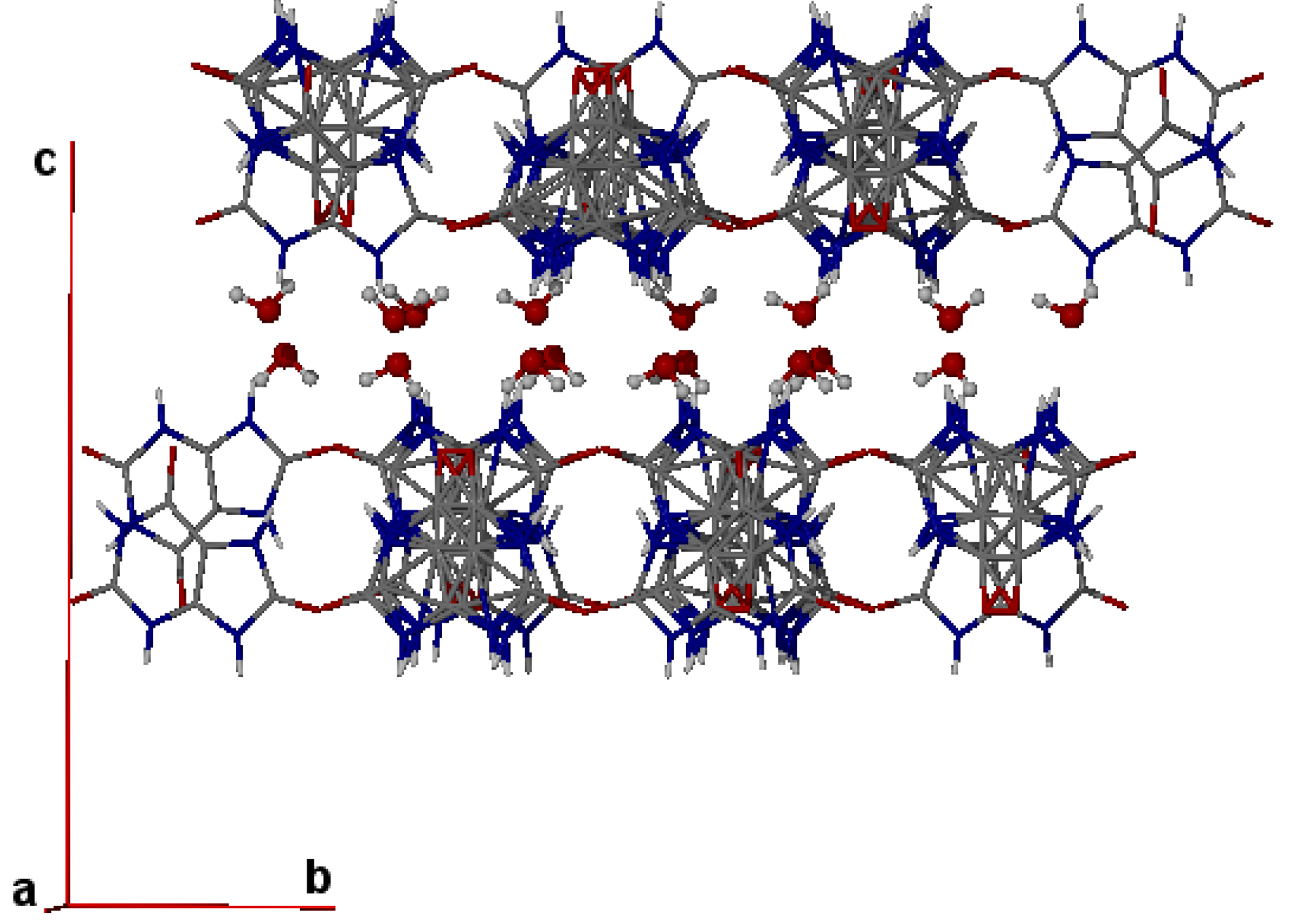
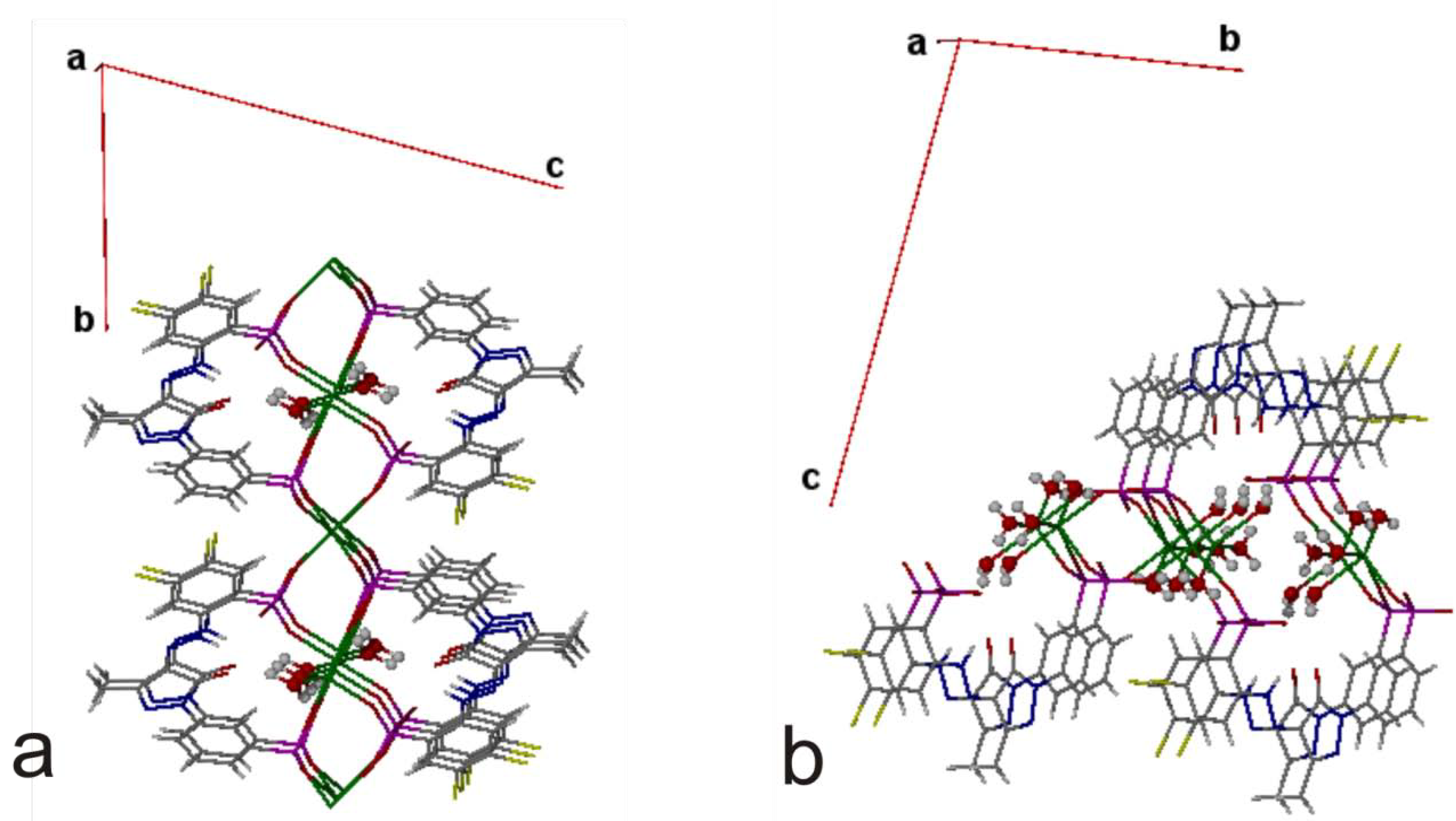
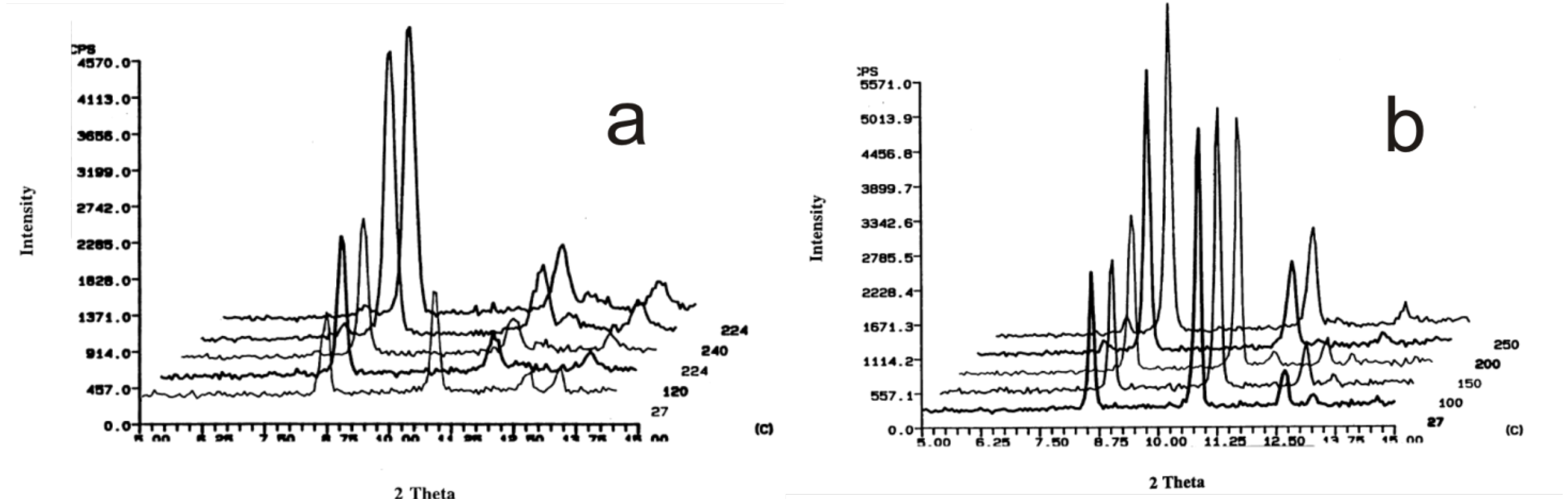
4. Powder X-ray Diffraction
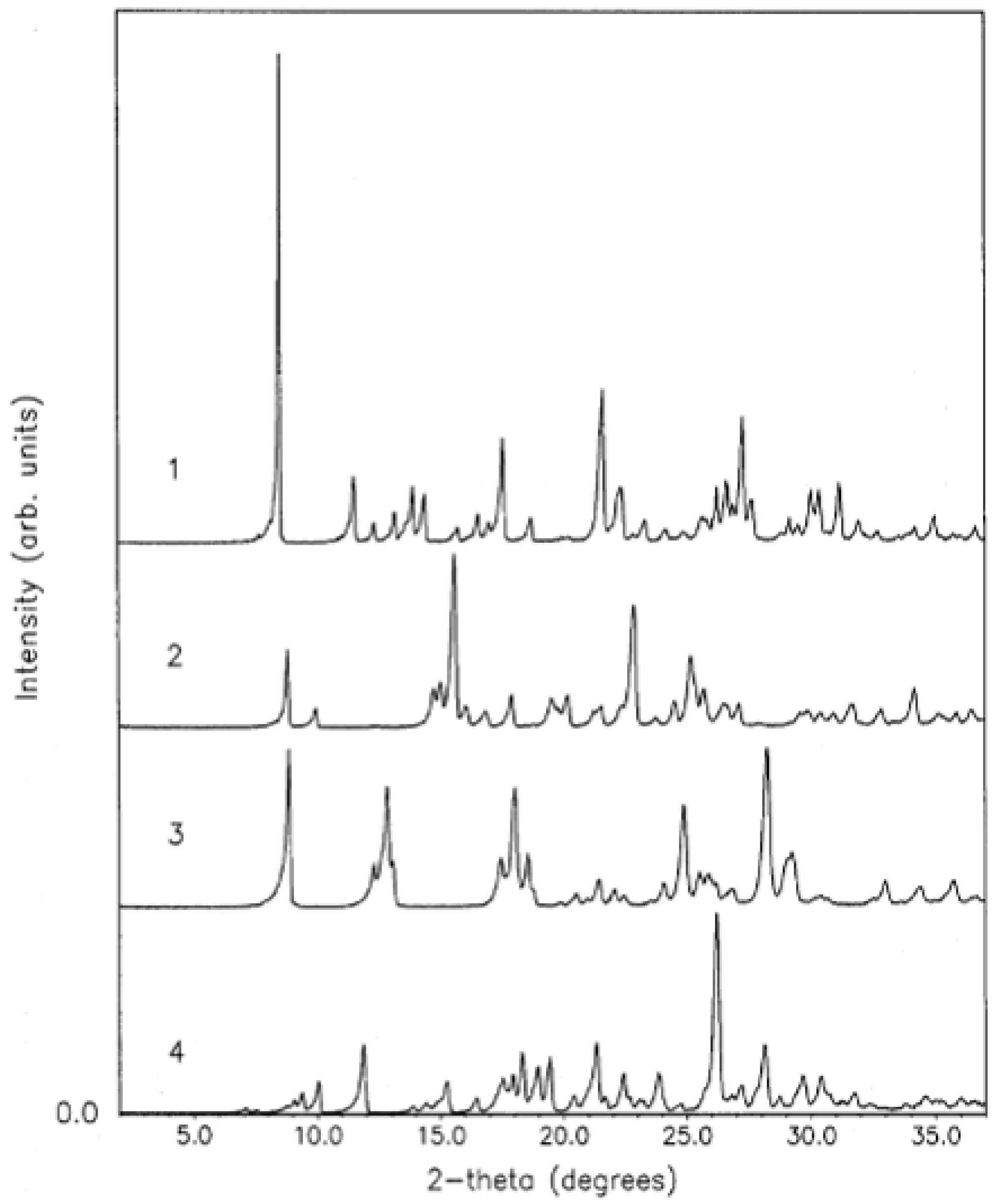

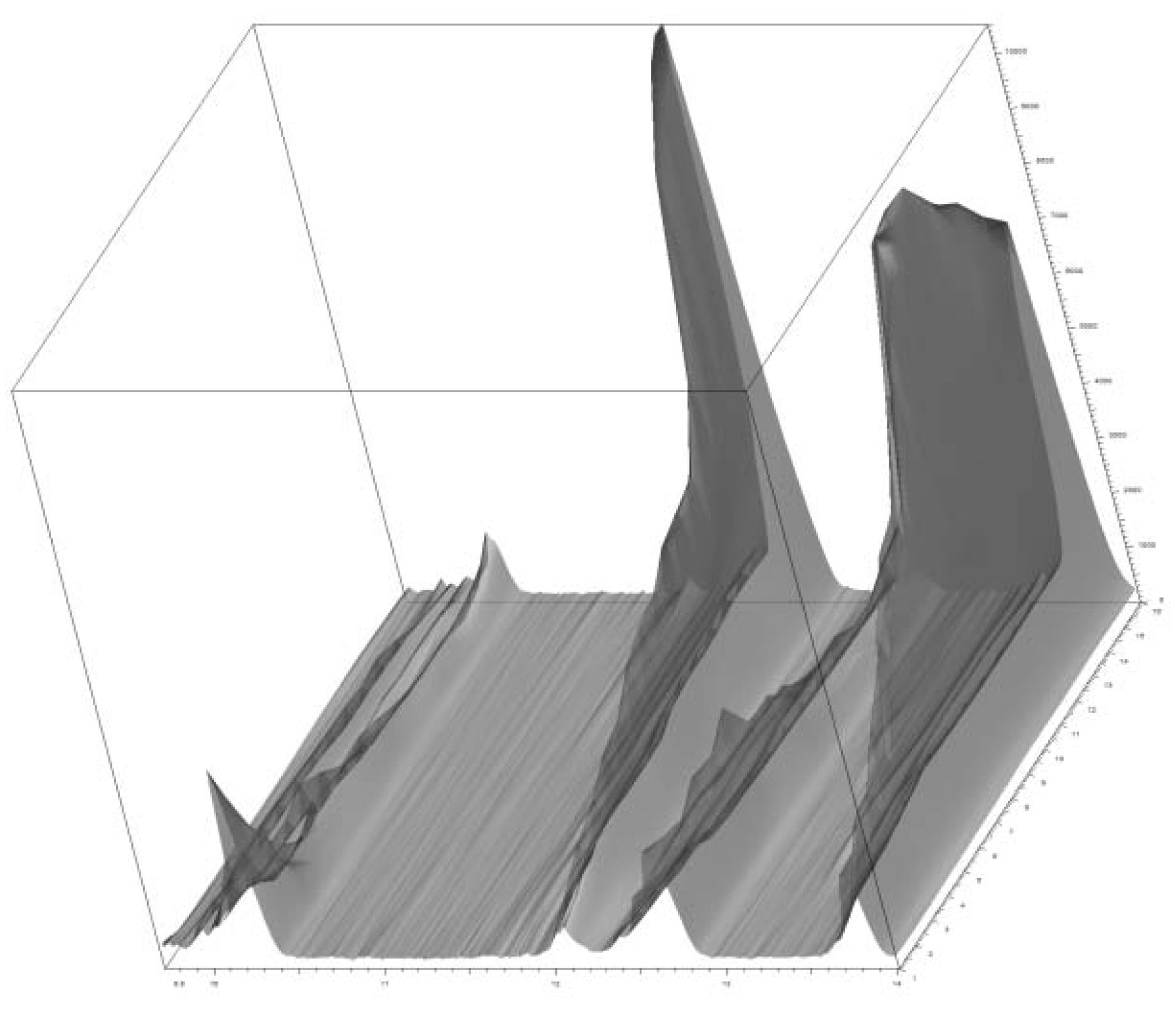
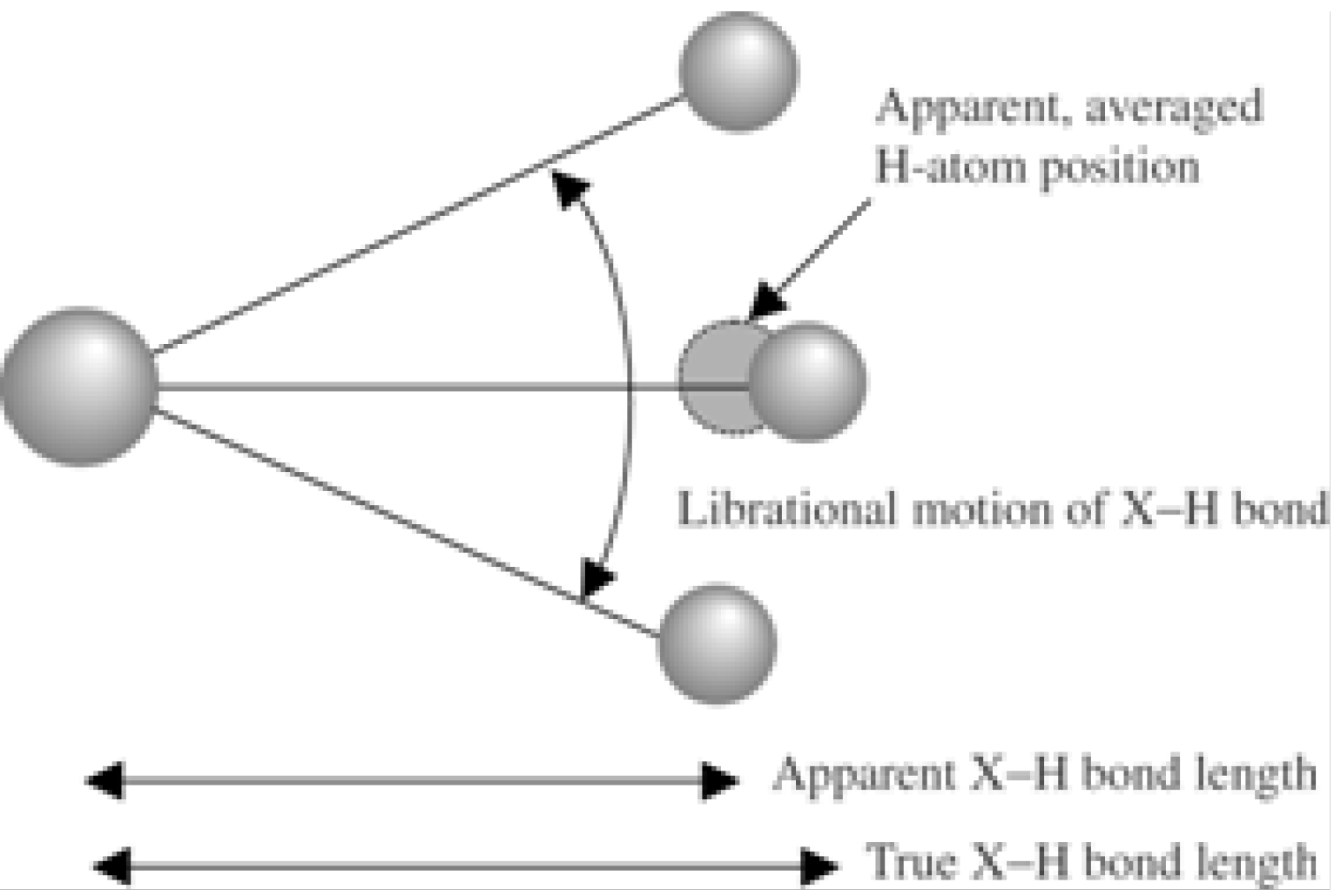
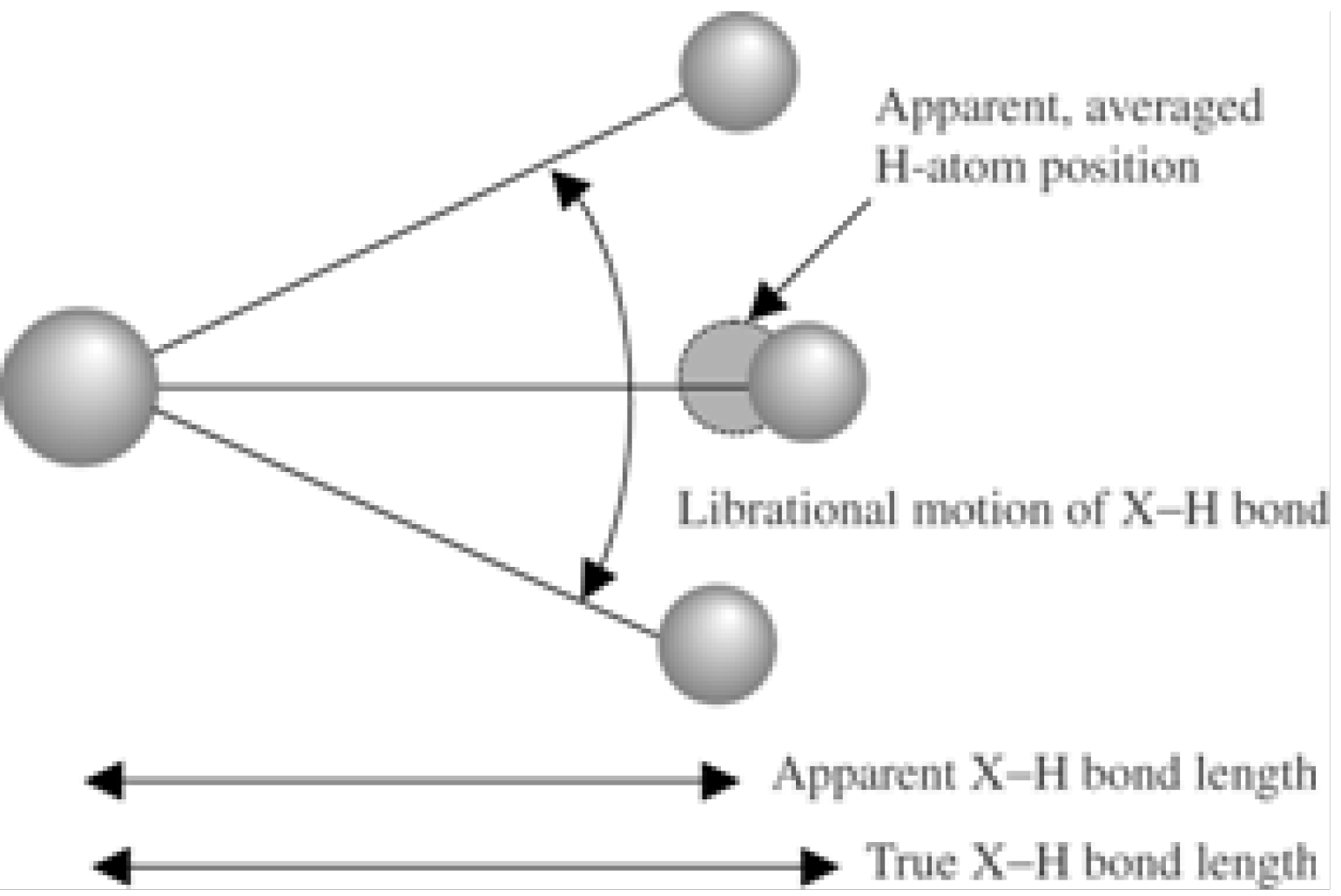
5. Neutron Diffraction
6. Conclusion and Perspective
Acknowledgement
References
- Yoshizawa, M.; Kusukawa, T.; Kawano, M.; Ohhara, T.; Tanaka, I.; Kurihara, K.; Niimura, N.; Fujita, M. Endohedral clusterization of ten water molecules into a “molecular ice” within the hydrophobic pocket of a self-assembled cage. J. Am. Chem. Soc. 2005, 127, 2798–2799. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Bogge, H.; Diemann, E.; Muller, A. Chameleon water: assemblies confined in nanocapsules. J. Mol. Liq. 2005, 118, 155–162. [Google Scholar] [CrossRef]
- Papoian, G.A.; Ulander, J.; Eastwood, M.P.; Luthey-Schulten, Z.; Wolynes, P.G. Water in protein structure prediction. Proc. Natl. Acad. Sci. USA 2004, 101, 3352–3357. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Taulelle, F.; Loiseau, T.; Beitone, L.; Ferey, G. Is water templating nanoporous materials? Chem. Eur. J. 2004, 10, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Koop, T. Homogeneous ice nucleation in water and aqueous solutions. Z. Phys. Chemie-Int. J. Res. Phys. Chem. Chem. Phys. 2004, 218, 1231–1258. [Google Scholar]
- Breakthrough of the Year—The Runners-Up. Science 2004, 306, 2013–2017.
- Soper, A.K.; Teixeira, J.; Head-Gordon, T. Is ambient water inhomogeneous on the nanometer-length scale? Proc. Natl. Acad. Sci. USA 2010, 107, E44. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, C.G.; Radaelli, P.G.; Mayer, E.; Finney, J.L. Ice XV: a new thermodynamically stable phase of water ice. Phys. Rev. Lett. 2009, 103, 105701. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O.; Radosavljevic Evans, I. Beyond classical applications of powder diffraction. Chem. Soc. Rev. 2004, 33, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.C.; Shankland, N.; Florence, A.J. A single-crystal neutron diffraction study of the temperature dependence of hydrogen-atom disorder in benzoic acid dimers. J. Chem. Soc.-Faraday Trans. 1996, 92, 5051–5057. [Google Scholar] [CrossRef]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry, 2nd ed.; Wiley: Chichester, UK, 2009; p. 449. [Google Scholar]
- Snowden, T.S.; Bisson, A.P.; Anslyn, E.V. A comparison of NH-π versus lone pair hydrogen bonding effects on carbon acid pKa shifts. J. Am. Chem. Soc. 1999, 121, 6324–6325. [Google Scholar] [CrossRef]
- Gillon, A.L.; Feeder, N.; Davey, R.J.; Storey, R. Hydration in Molecular Crystals—A Cambridge Structural Database Analysis. Crys. Growth Des. 2003, 3, 663–673. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997; pp. 15–16. [Google Scholar]
- Allen, F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydration in organic crystals: prediction from molecular structure. Chem. Commun. 1991, 6, 426–428. [Google Scholar] [CrossRef]
- Gorbitz, C.H.; Hersleth, H.-P. On the inclusion of solvent molecules in the crystal structures of organic compounds. Acta Crystallogr. Sect. B 2000, B56, 526–534. [Google Scholar] [CrossRef]
- Infantes, L.; Chisholm, J.; Motherwell, S. Extended motifs from water and chemical functional groups in organic molecular crystals. Cryst. Eng. Comm. 2003, 5, 480–486. [Google Scholar] [CrossRef]
- Mascal, M.; Infantes, L.; Chisholm, J. Water oligomers in crystal hydrates—What's news and what isn't? Angew. Chem.-Int. Ed. 2006, 45, 32–36. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Maluszynska, H. The stereochemistry of the water molecules in the hydrates of small biological molecules. Acta Crystallogr. Sect. B 1990, 46, 546–549. [Google Scholar] [CrossRef]
- Infantes, L.; Fabian, L.; Motherwell, W.D.S. Organic crystal hydrates: what are the important factors for formation. Cryst. Eng. Comm. 2007, 9, 65–71. [Google Scholar] [CrossRef]
- Haynes, D.A.; Jones, W.; Motherwell, W.D.S. Occurrence of pharmaceutically acceptable anions and cations in the Cambridge structural database. J. Pharm. Sci. 2005, 94, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Haynes, D.A.; Jones, W.; Motherwell, W.D.S. Hydrate formation in NH+-containing salts of pharmaceutically acceptable anions: A CSD survey. Cryst. Eng. Comm. 2005, 7, 342–345. [Google Scholar] [CrossRef]
- Sansam, B.C.R.; Anderson, K.M.; Steed, J.W. A Simple Strategy for Crystal Engineering Water Clusters. Cryst. Growth Des. 2007, 7, 2649–2653. [Google Scholar] [CrossRef]
- Hulme, A.T.; Price, S.L. Toward the Prediction of Organic Hydrate Crystal Structures. J. Chem. Theory Comput. 2007, 3, 1597–1608. [Google Scholar] [CrossRef]
- Morris, K.R. Structural Aspects of Hydrates and Solvates. In Polymorphism in Pharmaceutical Solids; Brittain, H.G., Ed.; Marcel Dekker Ltd: New York, NY, USA, 1999; Chapter 4; pp. 125–181. [Google Scholar]
- Potter, B.S.; Palmer, R.A.; Withnall, R.; Chowdhry, B.Z. Aza analogs of nucleic acid bases: infrared and Raman spectra of 5-azauracil and crystal structure of 5-azauracil monohydrate. New J. Chem. 1999, 23, 117–122. [Google Scholar] [CrossRef]
- Todd, A.M.; Anderson, K.M.; Byrne, P.; Goeta, A.E.; Steed, J.W. Helical or Polar Guest-Dependent Z' = 1.5 or Z' = 2 Forms of a Sterically Hindered Bis(urea) Clathrate. Cryst. Growth Des. 2006, 6, 1750–1752. [Google Scholar] [CrossRef]
- Byrne, P.; Lloyd, G.O.; Clarke, N.; Steed, J.W. A “compartmental” Borromean weave coordination polymer exhibiting saturated hydrogen bonding to anions and water cluster inclusion. Angew. Chemie-Int. Ed. 2008, 47, 5761–5764. [Google Scholar] [CrossRef]
- Barbour, L.J.; Orr, G.W.; Atwood, J.L. An intermolecular (H2O)10 cluster in a solid-state supramolecular complex. Nature 1998, 393, 671–673. [Google Scholar] [CrossRef]
- Konig, H. A cubic ice modification. Z. Kristallogr. 1944, 105, 279–286. [Google Scholar]
- Du, M.; Zhang, Z.-H.; Guo, W.; Fu, X.-J. Multi-Component Hydrogen-Bonding Assembly of a Pharmaceutical Agent Pamoic Acid with Piperazine or 4,4'-Bipyridyl: A Channel Hydrated Salt with Multiple-Helical Motifs vs a Bimolecular Cocrystal. Cryst. Growth Des. 2009, 9, 1655–1657. [Google Scholar] [CrossRef]
- Gerdil, R. The crystal structure of thymine monohydrate. Acta Crystallogr. 1961, 14, 333–344. [Google Scholar] [CrossRef]
- Perrier, P.; Byrn, S.R. Influence of crystal packing on the solid-state desolvation of purine and pyrimidine hydrates: loss of water of crystallization from thymine monohydrate, cytosine monohydrate, 5-nitrouracil monohydrate, and 2'-deoxyadenosine monohydrate. J. Org. Chem. 1982, 47, 4671–4676. [Google Scholar] [CrossRef]
- Sutor, D.J. The structures of the pyrimidines and purines. VII. The crystal structure of caffeine. Acta Crystallogr. 1958, 11, 453–4588. [Google Scholar] [CrossRef]
- Edwards, H.G.M.; Lawson, E.; de Matas, M.; Shields, L.; York, P. Metamorphosis of caffeine hydrate and anhydrous caffeine. J. Chem. Soc., Perkin Tran. 1997, 2, 1985–1990. [Google Scholar]
- Gerdil, R.; Marsh, R.E. Arrangement of the H2O molecules in the crystal structure of caffeine. Acta Crystallogr. 1960, 13, 166–167. [Google Scholar] [CrossRef]
- Sutor, D.J. The structures of the pyrimidines and purines. VI. The crystal structure of theophylline. Acta Crystallogr. 1958, 11, 83–87. [Google Scholar] [CrossRef]
- Sun, C.; Zhou, D.; Grant, D.J.W.; Young Junior, V.G. Theophylline monohydrate. Acta Crystallogr. Sect. E 2002, 58, o368–o370. [Google Scholar] [CrossRef]
- Griesser, U.J.; Burger, A. The effect of water vapor pressure on desolvation kinetics of caffeine 4/5-hydrate. Int. J. Pharm. 1995, 120, 83–93. [Google Scholar] [CrossRef]
- Ahlqvist, M.U.A.; Taylor, L.S. Water dynamics in channel hydrates investigated using H/D exchange. Int. J. Pharm. 2002, 241, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Suihko, E.; Ketolainen, J.; Poso, A.; Ahlgren, M.; Gynther, J.; Paronen, P. Dehydration of theophylline monohydrate-a two step process. Int. J. Pharm. 1997, 158, 47–55. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Okoth, M.O.; Sheen, D.B.; Sherwood, J.N.; Teat, S.J.; Vrcelj, R.M. Cephalexin: a channel hydrate. Acta Crystallog. Sect. C 2003, C59, o650–o652. [Google Scholar] [CrossRef]
- Stephenson, G.A.; Groleau, E.G.; Kleemann, R.L.; Xu, W.; Rigsbee, D.R. Formation of Isomorphic Desolvates: Creating a Molecular Vacuum. J. Pharm. Sci. 1998, 87, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.K.; Nangia, A. First example of an ice-like water hexamer boat tape structure in a supramolecular organic host. Chem. Commun. 2006, 1825–1827. [Google Scholar]
- Ringertz, H. Optical and Crystallographic Data of Uric Acid and its Dihydrate. Acta Crystallogr. 1965, 19, 286–287. [Google Scholar] [CrossRef]
- Artioli, G.; Masciocchi, N.; Galli, E. The elusive crystal structure of uric acid dihydrate: Implication for epitaxial growth during biomineralization. Acta Crystallogr. Sect. B 1997, 53, 498–503. [Google Scholar] [CrossRef]
- Parkin, S.; Hope, H. Uric acid dihydrate revisited. Acta Crystallogr. Sect. B 1998, 54, 339–344. [Google Scholar] [CrossRef]
- Bock, H.; Schoedel, H.; Van, T.T.H.; Dienelt, R.; Gluth, M. Interactions in crystals. 136. Protonated dipyridylamine salts with different anions. Monomeric tetraphenylborate as well as bis(trifluoromethylsulfonate), dimeric squarate, and polymeric chloride dihydrate. J. Prakt. Chem./Chem.-Ztg. 1998, 340, 722–732. [Google Scholar]
- Ivashevskaya, S.N.; van de Streek, J.; Djanhan, J. E.; Bruening, J.; Alig, E.; Bolte, M.; Schmidt, M.U.; Blaschka, P.; Hoeffken, H.W.; Erk, P. Structure determination of seven phases and solvates of Pigment Yellow 183 and Pigment Yellow 191 from X-ray powder and single-crystal data. Acta Crystallogr. Sect. B 2009, B65, 212–222. [Google Scholar] [CrossRef]
- Gorbitz, C.H.; Hersleth, H.-P. On the inclusion of solvent molecules in the crystal structures of organic compounds. Acta Crystallogr. Sect. B 2000, B56, 526–534. [Google Scholar] [CrossRef]
- Herbst, W.; Hunger, K. Industrial Organic Pigments: Production, Properties, Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Holleman, A.F. Text Book of Inorganic Chemistry; Veit and Co: Leipzig, Germany, 1906. [Google Scholar]
- Oxtoby, N.S.; Blake, A.J.; Champness, N.R.; Wilson, C. Water superstructures within organic arrays; Hydrogen-bonded water sheets, chains and clusters. Chem. Eur. J. 2005, 11, 4643–4654. [Google Scholar] [CrossRef] [PubMed]
- Vrecer, F.; Vrbinc, M.; Meden, A. Characterization of piroxicam crystal modifications. Int. J. Pharm. 2003, 256, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.D.M. Structure determination of molecular crystals directly from powder diffraction data. Rigaku J. 2001, 18, 23–32. [Google Scholar]
- Harris, K.D.M.; Johnston, R.L.; Turner, G.W.; Tedesco, E.; Cheung, E.Y.; Kariuki, B.M. Recent advances in the opportunities for solving molecular crystal structures directly from powder diffraction data. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 2002, 389, 123–129. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K. Structure determination from powder diffraction data. Acta Crystallogr. Sect. A 2008, A64, 52–64. [Google Scholar] [CrossRef]
- Redman-Furey, N.; Dicks, M.; Bigalow-Kern, A.; Cambron, R.T.; Lubey, G.; Lester, C.; Vaughn, D. Structural and analytical characterization of three hydrates and an anhydrate form of risedronate. J. Pharm. Sci. 2005, 94, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garibay, M.A. Molecular crystals on the move: from single-crystal-to-single-crystal photoreactions to molecular machinery. Angew. Chem.-Intl. Ed. 2007, 46, 8945–8947. [Google Scholar] [CrossRef]
- Das, D.; Engel, E.; Barbour, L.J. Reversible single-crystal to single-crystal polymorphic phase transformation of an organic crystal. Chem. Commun. 2010, 46, 1676–1678. [Google Scholar] [CrossRef]
- Yin, S.X.; Scaringe, R.P.; Malley, M.F.; Gougoutas, J.Z. In-situ variable temperature powder X-ray diffraction and thermal analysis -applications in the pharmaceutical industry. Am. Pharm. Rev. 2005, 8, 56-58, 60, 62, 67. [Google Scholar]
- Tong, H.H.Y.; Chow, A.S.F.; Chan, H.M.; Chow, A.H.L.; Wan, Y.K.Y.; Williams, I.D.; Shek, F.L.Y.; Chan, C.K. Process-induced phase transformation of berberine chloride hydrates. J. Pharm. Sci. 2010, 99, 1942–1954. [Google Scholar] [PubMed]
- Zhu, H.J.; Grant, D.J.W. Dehydration behavior of nedocromil magnesium pentahydrate. Int. J. Pharm. 2001, 215, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Kachrimanis, K.; Fucke, K.; Noisternig, M.; Siebenhaar, B.; Griesser, U.J. Effects of Moisture and Residual Solvent on the Phase Stability of Orthorhombic Paracetamol. Pharm.Res. 2008, 25, 1440–1449. [Google Scholar] [CrossRef]
- Anderson, K.M.; Day, G.M.; Paterson, M.J.; Byrne, P.; Clarke, N.; Steed, J.W. Structure calculation of an elastic hydrogel from sonication of rigid small molecule components. Angew. Chem.-Intl. Ed. 2008, 47, 1058–1062. [Google Scholar] [CrossRef]
- Weller, M.T.; Henry, P.F.; Ting, V.P.; Wilson, C.C. Crystallography of hydrogen-containing compounds: realizing the potential of neutron powder diffraction. Chem. Commun. 2009, 2973–2989. [Google Scholar]
- Wilson, C.C.; Thomas, L.H. The short hydrogen bond in molecular materials: neutron diffraction and complementary studies. C. R. Chim. 2005, 8, 1434–1443. [Google Scholar] [CrossRef]
- Adam, M.S.; Gutmann, M.J.; Leech, C.K.; Middlemiss, D.S.; Parkin, A.; Thomas, L.H.; Wilson, C.C. Stability and cooperativity of hydrogen bonds in dihydroxybenzoic acids. New J. Chem. 2010, 34, 85–91. [Google Scholar] [CrossRef]
- Thomas, L.H.; Florence, A.J.; Wilson, C.C. Hydrogen atom behaviour imaged in a short intramolecular hydrogen bond using the combined approach of X-ray and neutron diffraction. New J. Chem. 2009, 33, 2486–2490. [Google Scholar] [CrossRef]
- Wilson, C.C.; Xu, X.; Florence, A.J.; Shankland, N. Temperature dependence of proton transfer in 4-chlorobenzoic acid. New J. Chem. 2006, 30, 979–981. [Google Scholar] [CrossRef]
- Turner, D.R.; Henry, M.; Wilkinson, C.; McIntyre, G.J.; Mason, S.A.; Goeta, A.E.; Steed, J.W. Cooperative Hydrogen-Bonding Effects in a Water Square: A Single-Crystal Neutron and Partial Atomic Charges and Hardness Analysis Study. J. Am. Chem. Soc. 2005, 127, 11063–11074. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, G.; Franchini-Angela, M. Survey of the geometry and environment of water molecules in crystalline hydrates studied by neutron diffraction. Acta Crystallogr. Sect. B 1972, 28, 3572–3583. [Google Scholar] [CrossRef]
- Bouquiere, J.P.; Finney, J.L.; Lehmann, M.S.; Lindley, P.F.; Savage, H.F.J. High-resolution Neutron Study of Vitamin B12 Coenzyme at 15 K: Structure Analysis and Comparison with thte Structure at 279 K. Acta Crystallogr. Sect. B 1993, 49, 79–89. [Google Scholar] [CrossRef]
- Bouquiere, J.P.; Finney, J.L.; Savage, H.F.J. High-Resolution Neutron Study of Vitamin B12 Coenzyme at 15 K: Solvent Structure. Acta Crystallogr. Sect. B 1994, 50, 566–578. [Google Scholar] [CrossRef]
- Bilton, C.; Howard, J.A.K.; Madhavi, N.N.L.; Desiraju, G.R.; Allen, F.H.; Wilson, C.C. Crystal engineering in the gem-alkynol family: the key role of water in the structure of 2,3,5,6-tetrabromo-trans-1,4-diethynyl-cyclohexa-2,5-diene-1,4-diol dihydrate determined by X-ray and neutron diffraction at 150 K. Acta Crystallogr. Sect. B 2001, 57, 560–566. [Google Scholar] [CrossRef]
- Krebs, F.C.; Jorgensen, M.; Lebech, B.; Frydenvang, K. A perdeuterated cryoprotectant for neutron studies and a demonstration of its use for neutron powder diffraction on L-(–)-ephedrine hemihydrate. J. Appl. Crystallogr. 2001, 34, 203–207. [Google Scholar] [CrossRef]
- Bordeneuve, H.; Tenailleau, C.; Guillemet-Fritsch, S.; Smith, R.; Suard, E.; Rousset, A. Structural variations and cation distributions in Mn3-xCoxO4 (0 ≤ x ≤ 3) dense ceramics using neutron diffraction data. Solid State Sci. 2010, 12, 379–386. [Google Scholar] [CrossRef]
- Bull, D.J.; Weidner, E.; Shabalin, I.L.; Telling, M.T.F.; Jewell, C.M.; Gregory, D.H.; Ross, D.K. Pressure-dependent deuterium reaction pathways in the Li-N-D system. Phys. Chem. Chem. Phys. 2010, 12, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Telepeni, I.; Yang, S.; Lin, X.; Kockelmann, W.; Dailly, A.; Blake, A.J.; Lewis, W.; Walker, G.S.; Allan, D.R.; Barnett, S.A.; Champness, N.R.; Schroder, M. Metal-Organic Polyhedral Frameworks: High H2 Adsorption Capacities and Neutron Powder Diffraction Studies. J. Am. Chem. Soc. 2010, 132, 4092–4094. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, A.M.; Price, J.W.H.; Finlayson, T.R.; Lienert, U.; Ibrahim, R. Comparison of Neutron and Synchrotron Diffraction Measurements of Residual Stress in Bead-on-Plate Weldments. J. Pressure Vessel Technol. 2010, 132, 011502/1–011502/8. [Google Scholar] [CrossRef]
- Paradowska, A.M.; Price, J.W.H.; Kerezsi, B.; Dayawansa, P.; Zhao, X.L. Stress relieving and its effect on life of welded tubular joints. Eng. Failure Anal. 2010, 17, 320–327. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in single- and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fucke, K.; Steed, J.W. X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates. Water 2010, 2, 333-350. https://doi.org/10.3390/w2030333
Fucke K, Steed JW. X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates. Water. 2010; 2(3):333-350. https://doi.org/10.3390/w2030333
Chicago/Turabian StyleFucke, Katharina, and Jonathan W. Steed. 2010. "X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates" Water 2, no. 3: 333-350. https://doi.org/10.3390/w2030333
APA StyleFucke, K., & Steed, J. W. (2010). X-ray and Neutron Diffraction in the Study of Organic Crystalline Hydrates. Water, 2(3), 333-350. https://doi.org/10.3390/w2030333