Abstract
As one of several types of pollutants in water, chlorinated compounds have been routinely subjected to sonochemical analysis to check the environmental applications of this technology. In this review, an extensive study of the influence of the initial concentration, ultrasonic intensity and frequency on the kinetics, degradation efficiency and mechanism has been analyzed. The sonochemical degradation follows a radical mechanism which yields a very wide range of chlorinated compounds in very low concentrations. Special attention has been paid to the mass balance comparing the results from several analytical techniques. As a conclusion, sonochemical degradation alone is not an efficient treatment to reduce the organic pollutant level in waste water.
1. Introduction
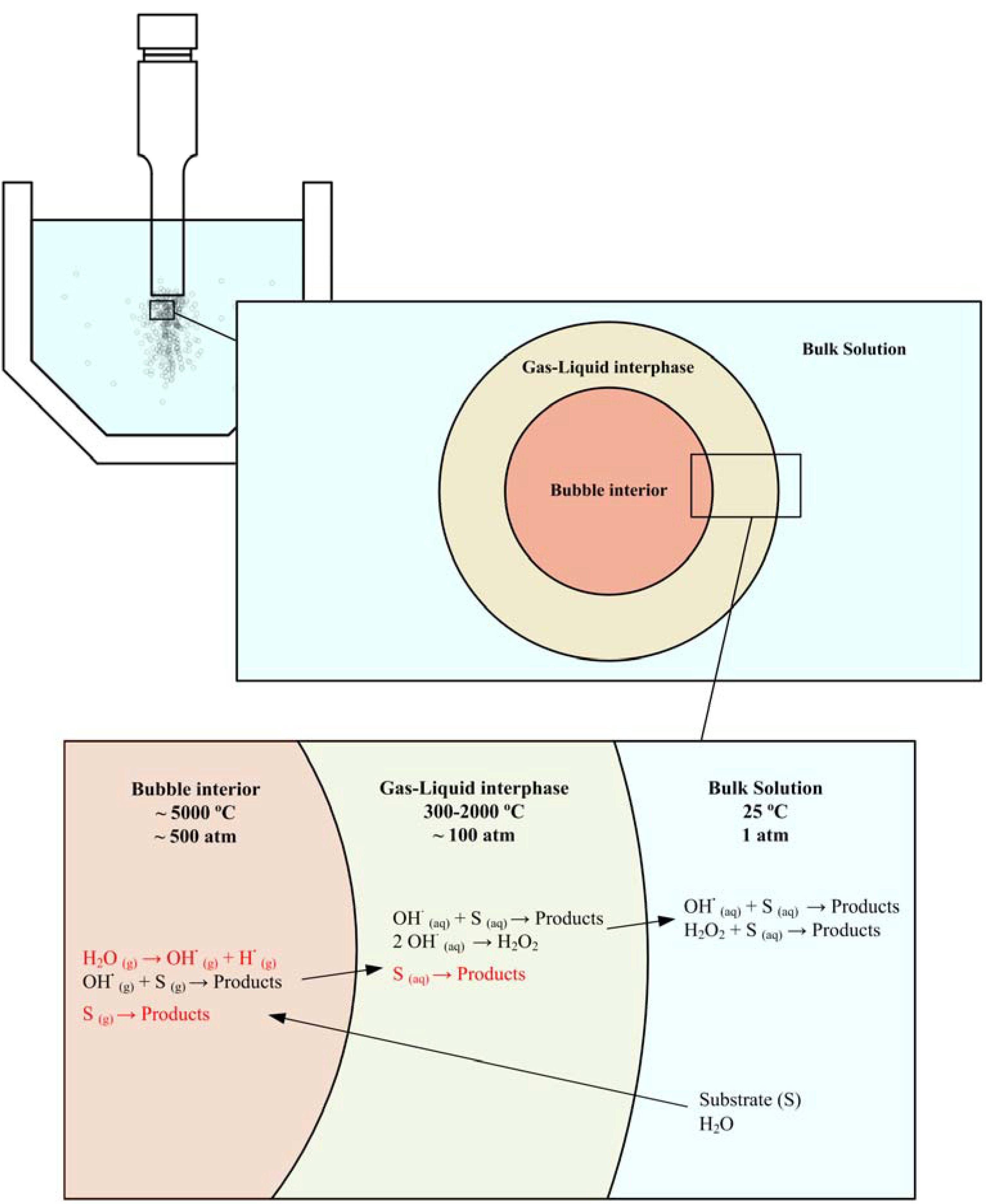
Chlorinated compounds are one of the most widespread pollutant groups in any media in our environment [1] and, because of this, a large number of techniques, including traditional techniques [2], sequential combinations of techniques [3], hybrid [4] and new technologies [5], are continuously being developed to provide an efficient solution to this important issue. Among these methods, sonochemical treatment has received a lot of attention due to its special features. Ultrasound is a unique means of interacting energy and matter, and differs from traditional energy sources such as light, heat or ionizing radiation in duration, pressure and energy per molecule [6]. The chemical effects of ultrasound derive from acoustic cavitation, which is a nonlinear process that serves as a means of concentration of the diffuse energy of sound in liquids. This high-energy microenvironment is induced by the extreme conditions during the cavitation event by means of the generation of high-energy species (radicals), at least from the solvent, and/or the high temperature and pressure in the cavitating bubble [7,8]. A three-region site for chemical reactions in ultrasonically irradiated liquids has been proposed: the cavitation bubble itself, the solution bulk and the bubble/solution interface [9]. A pictorial representation of these sites has been described [10]. The degradation mechanism of a specific molecule depends on the region where the chemical reaction takes place, finding a destructive thermal pathway (pyrolysis) and/or a destructive radically oxidative pathway. The cavitation process is dependent on the vapor pressure, viscosity and surface tension of the bulk liquid, and, therefore, on the temperature. However, apart from the nature of the solvent, there are other aspects to take into account when a sonochemical process takes place. For example, pollutant properties such as vapor pressure, solubility, Henry’s Law constant and the octanol-water partition coefficient (Kow), which depend again on the temperature, will also determine the chemical reaction site and therefore the performance of the degradation. In addition to pollutant and phase features, we have to take into account not only the traditional chemical operational variables, such as dissolved gas, static pressure, pH and pollutant concentration, but also specific ultrasonic operations variables, such as frequency, power and the system geometry. Therefore, the development of a sonochemical process for the degradation of a pollutant dissolved in water implies a careful selection and design of the experimental procedure. In this review, first, the different types of experimental set-up proposed in the literature for sonochemical degradation are briefly presented. Next, the effect upon the sonochemical degradation processes of the different ultrasonic operational variables is discussed, and later, the specific results found in the literature for the sonochemical degradation of chlorinated organocompounds are summarized, highlighting the advantages and drawbacks of this technology. Finally, general remarks to keep in mind in any experiment design are presented.
2. Experimental devices and methodology
One of the main drawbacks often reported in literature for sonochemical processes is the lack of reproducibility in different reactors [11,12,13], please see Figure 1. The cheapest and most readily available experimental set-up for carrying out sonochemical reactions is the ultrasonic bath, which is currently used not only at laboratory level but also on a larger scale [14]. This experimental set-up presents as drawbacks the use of fixed working frequencies and powers, the low power transmitted and the lack of reproducibility previously mentioned, due to the fact that the results depend strongly on the position of the beaker inside the bath. For example, Weissler et al. [12], working with vessels dipped in an ultrasonic system, pointed out some difficulty in getting quantitatively reproducible results, and analyzed the influence of some “experimental” variables, such as vessel material, vessel geometric dimensions (diameter, wall thickness), reaction volume and exact vertical and horizontal location of the reaction vessels in relation to the crystal source of ultrasound. As a result of their preliminary work, reproducibility between 5 and 10% was considered adequate. From then on, this necessity for careful control of experimental conditions has been systematically claimed by many authors in their works. Further developments provided the use of transducers, which are directly dipped in the working solution [15]. This configuration has been used widely at laboratory scale because it presents more advantages (it allows the ultrasonic waves to be directed in the working solution and provides much more efficient power control) than drawbacks (contamination, materials incompatibility). The successful use of this approach at laboratory scale has promoted its scale-up at pilot plant and industrial facilities, in applications such as disintegration of sewage sludge for digestion [16] and for contaminated soil remediation [17]. Nowadays, new different approaches using transducers can be found in literature, such as the high power stepped-plate ultrasonic transducer [18] and the parallel-plate near field sonoreactor working at two different frequencies and with the possibility for working at different power densities and ultrasonic intensities [19]. In addition, the full scale method has been developed for some interesting applications in environmental concerns such as sludge treatment [20,21,22] with some equipment commercially available [23]. In the development of all of these devices, two main targets must be addressed: (i) optimization of the energetic efficiency and (ii) maximization of the effective volume. For that, experimental [24] and theoretical studies [25] are continuously found in literature utilizing numerical analysis using finite element method (FEM) codes [26], visualization methods [27] and chemical dosimeters [28], sonoreactor design thus becoming one of the most active research areas in the field of Sonochemistry.
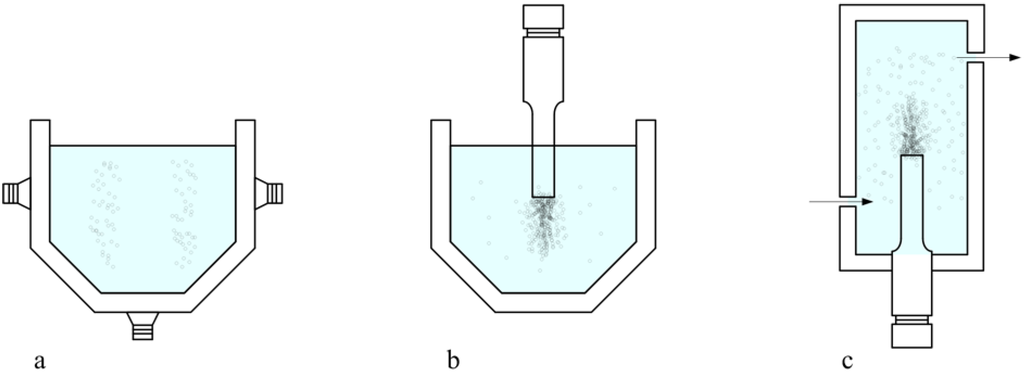
Figure 1.
Different sonochemical set-ups: (a) Ultrasonic bath configuration. (b) Transducer in batch configuration. (c) Transducer in a flow reactor.
Figure 1.
Different sonochemical set-ups: (a) Ultrasonic bath configuration. (b) Transducer in batch configuration. (c) Transducer in a flow reactor.
3. Influence of the operational variables
Fortunately, some of the deeper studies analyzing the influence of some operational variables in the sonochemical degradation of pollutants have been carried out using a chlorinated compound as the target molecule. For example, CCl4 has been extensively studied by several authors [7,8,12,13,29,30,31,32], analyzing the mechanism, degradation products and kinetic aspects and, especially, the influence of the frequency [33,34,35,36] on the process. Other C1 chlorinated organocompounds such as CHCl3, [13] CH2Cl2 and CCl4, or C2 compounds such as 1,1,1-trichloroethane, tetrachloroethylene, hexachloroethane, 1,1,2,2-tetrachloroethane and mainly trichloroethylene and also mixtures of them [37], have also been target molecules used in the analysis of the influence of the frequency [36,38] on the process and its kinetics. In addition, specific studies, such as the evolution of the pH [39] to acidic values, or the correlation of the molecule properties (vapor pressure) with the kinetics of the process and degradation mechanism [13,40,41,42,43] have been reported. Not only aliphatic compounds but also chlorinated aromatic compounds such as chlorotoluenes [44], chlorobenzenes [45,46,47,48,49] mixtures of chlorobenzene with trichloroethylene [50,51,52], p-chlorobenzoic acid [53] PCBs [54], chlorophenols [3,35,48,55,56,57,58,59,60,61] and chlorinated pesticides [58,62,63] have also been the subjects of study, providing a wide range of examples from volatile, nonvolatile, hydrophobic and hydrophilic compounds and their respective behaviors. In the following, the general trends of the operational variables reported in literature and those specifically observed for chlorinated compounds are presented.
3.1. Influence of the initial concentration
The study of the initial concentration of the pollutant in a sonochemical process has mainly been focused on its influence on the kinetics of the process. The information reported from analyses with non-chlorinated compounds reveals the complexity of the matter. Petrier et al. [64], working with solutions of phenol (hydrophilic and non-volatile compound), observed that the rate constant increases with the concentration until reaching a constant value. Similar behavior was reported by Kotronarou et al. [65] working with solution of hydrogen sulfide. However, these last same authors [66], working again with a hydrophilic and non-volatile compound such as p-nitrophenol, reported an opposite behavior with rate constant decreasing as the initial concentration increases. Hart et al. [67], investigating the sonolysis of volatile nonchlorinated compounds such as methane and ethane in aqueous solution at 300 kHz ultrasound, found a considerable decrease of the reaction rate at high concentrations. By comparing the yield of ethane to the sum of the yields of ethylene and acetylene, they showed that the temperature of cavitation decreased with increasing methane concentration. Some authors have reported that, for the sonolysis of aqueous solutions of non-volatile solutes, pyrolysis in the interfacial region is prevalent at high solute concentration, while at low solute concentration free-radical reactions are likely to predominate [66,68,69].
When chlorinated compounds are the subject of the study of the influence of initial concentration, some controversy remains about whether the initial concentration affects the destruction kinetics. Experimentally, some authors have reported no influence in studies carried out with chlorinated compounds such 1,1,1-trichloroetane at concentrations between 0.16 and 1.27 mM [41] or carbon tetrachloride [31]. However, other authors have found an influence not only for volatile [44,47] but also for non-volatile [53,57] halogenated compounds. Yiang et al. [49] have detected pseudo-first-order reaction kinetics with a significant effect of the initial substrate concentration on the decomposition rate of volatile compounds such as chlorobenzenes, although pyrolytic reactions are expected to follow simple first-order kinetics. Some authors have explained the dependence of the rate constant on the initial concentration by the fact that reaction conditions change as the concentration of organic compounds increases [44]. The temperature in the collapsing cavitation bubbles is determined by the average specific heat ratio γ (also named as polytropic index) [38] of the gas present in the cavitation bubbles. Moreover, the same authors, working with solution of trichloroethylene and chlorobenzene, have distinguished between ranges of concentrations. At mM scale, no influence of the concentration was detected, but at μM range they reported an influence as a consequence of a relevant role of the radical induced pathway in addition to the pyrolysis mechanism [50,51]. Another possible explanation for this behavior was reported by Hoffmann et al. [31], establishing differences between the reaction sites. The volume of the gas phase is estimated to be ≈2 x 104 times larger than the volume of the interfacial region [70]. If the compound is degraded inside the bubble in front of the interphase, more molecules can react during a single cavitation event. So, volatile and hydrophobic compounds, whose destruction occurs mostly in the cavitation bubble under pyrolysis conditions, can present independence from the initial concentration. However, non-volatile compounds, such as p-chlorobenzoic acid at micromolar concentrations, react in the shell region [53]. These explanations are in agreement with those reported above for non-volatile and non-chlorinated compounds.
3.2. Influence of the ultrasonic frequency
The frequency is the most important operational variable in a sonochemical process. In literature it is routinely reported that irradiation at higher frequencies (e.g., ≥100 kHz) leads to greater enhancements in reaction rates than irradiation at lower frequencies (e.g., ≤20 kHz). Sonoluminescence, which is the emission of light from acoustic cavitation bubbles, is also affected by variations in ultrasonic frequency. Both the intensity and the emission spectra of sonoluminescence are altered when cavitation is induced at different ultrasonic frequencies. The mechanism by which ultrasonic frequency enhances reaction rates has not been definitively established [54] and normally the explanations found in literature are partial and specifically related to the rate constants of OH· production and the formation of H2O2 by coupling of OH· radicals [64,71]. A critical qualitative analysis on the influence of the frequency has been proposed by Hua and Hoffmann [72]. These references suggest that the ultrasonic frequency will affect the reaction environment by determining the resonant bubble radius, due to the fact that acoustic cavitation is induced by the excitation of a population of bubble nuclei to a resonant radius, at which point the bubbles efficiently absorb energy from the sound waves. Although the radius is slightly influenced by the nature of the dissolved gases, the most significant variable is the frequency. Differences in bubble radii will result in variable bubble collapse times as well as surface area to volume ratios. As an example, the resonant radius ranges from 7 μm at 515 kHz to 170 μm at 20 kHz. However, it should be noted that the real situation is that a cavitating solution will contain a bubble population with a distribution of radii. Following the classical theory, the bubbles of this population grow by the process of rectified diffusion to reach the resonant size in complex bubble dynamics [73], please see Figure 2.
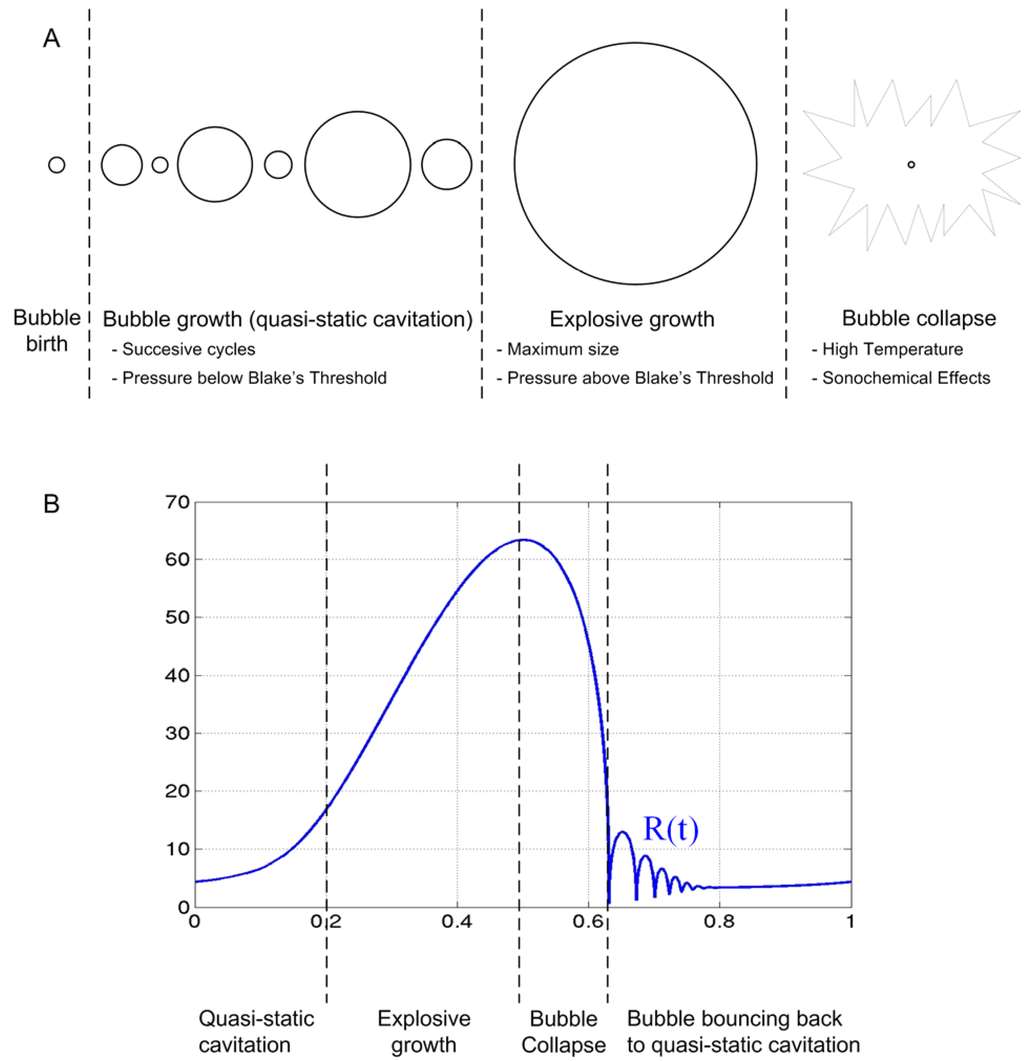
Figure 2.
(A) Formation, growth and collapse of the cavitation bubble. (B) Radius vs. time during the explosive growth and collapse of the cavitation bubble.
Figure 2.
(A) Formation, growth and collapse of the cavitation bubble. (B) Radius vs. time during the explosive growth and collapse of the cavitation bubble.
At higher frequencies, the resonant radius of a bubble is calculated to be smaller, and therefore fewer acoustic cycles are required before the bubble reaches its resonant size. With a greater number of acoustic cycles per unit time at higher frequencies, rectified diffusion occurs more rapidly. Thus, a greater number of nuclei can reach resonance more quickly than at lower frequencies. This implies that higher frequencies will be favored for the degradation of the chemicals which undergo pyrolysis inside the bubble. In addition to this enhancement of the collapse intensity of individual bubbles, there are other reasons for enhanced sonochemistry at higher frequencies. Differences in microstreaming at higher frequencies may result in a greater number of cavitating bubbles. Finally, some authors have detected experimentally some optimum frequency at which the rates of degradation are maxima [30], values which depend on the type of reactants and system geometry.
As we have previously mentioned, partial explanations of the influence of the frequency on the degradation process are routinely found in literature and the following statements can be summarized: (i) The radical attack mechanism has often been related to a better release of OH· radicals in solution at higher frequencies [64]. The resonance radius and duration of the collapse of a bubble are lower at higher frequencies, so, in such conditions, the OH· radicals could be ejected more efficiently in the solution before they have time to recombine in the bubble of cavitation [71]. (ii) Low ultrasonic frequencies are expected to induce destructive effects only for hydrophobic solutes, which easily diffuse into their long-lived cavity bubbles to undergo pyrolytic destruction inside the collapsing bubble, or hydroxylation and thermal decomposition at the interface, where pressure gradients and temperatures are still high enough to induce thermal effects [74]. For low frequencies, radical scavenging and recombination reactions can inhibit the mass injection of the radicals into the solution [75]. (iii) Finally, medium frequency gives rise to short-lived cavitational bubbles, which develop much higher pressures and temperatures and larger energies are released into the surrounding liquid during their more rapid and violent collapse [76]. These cavitation bubbles are so short lived and their collapse so rapid that the degree of the radical scavenging reactions in the bubble or at its interface is insufficient, and so the destructive effect of ultrasound in this condition is due to the high probability that long-lived hydroxyl radicals can be injected into the surrounding liquid [77].
Despite the fact that these discrepancies can be found in literature, especially related to the intensity of the bubble collapse at low frequencies, it is also often established that lower frequency sonication results in a greater degree of vapor-phase pyrolysis due to the high temperatures that are achieved during bubble collapse conditions, while higher frequencies favor OH· production [71]. The action of OH· radicals is, in fact, difficult to assess because it depends not only on the production of these radicals, but also on their dissemination in the liquid. The production of radicals itself is frequency-dependent already in a complicated manner. Bubble radial-dynamics simulations show that low frequencies yield stronger collapses, which would be more efficient breaking water molecules. Moreover, the quantity of water evaporated in the bubble, which is the “fuel” for OH· production, increases with the duration of the expansion phase, which is larger at low frequency [78]. Conversely, even if the low-frequency collapse is more productive, there are less collapses per second at low frequency than at high frequency. An optimum is therefore expected, and is indeed observed experimentally [79]. This is of course dependent on the size of the bubbles involved. This issue is far from being closed, but modern experimental observations [80] and theoretical results [81,82] suggest that the chemically active bubbles are in fact much smaller than the resonant radius, at least at low frequency. Concerning the dissemination of OH· radicals, this strongly depends on whether or not the bubble breaks up in the final stages of the collapse. A breaking-up bubble is more likely to inject radicals in the bulk liquid. Experimental hints suggest that some collapsing bubbles may survive several collapses, but may also undergo jetting [83], depending on the travelling or standing character of the wave they are excited by. This is confirmed by recent theory [84]. Besides, simulations show that a low-frequency bubble keeping its spherical shape during collapse is heated by adiabatic compression mainly around its center, so that the OH· radicals produced are important near the center, and relatively low near the bubble wall [85]. This would suggest that in this case, the chemical action of the radicals would be governed by the characteristic time of heat diffusion in the bubble interior, which in turn is frequency-dependent.
It should therefore be understood that the effect of frequency on the number of OH radicals produced, and/ or injected in the liquid, is closely related to complicated, yet unsolved problems of cavitation physics, and is difficult to predict with simple general rules.
In literature there are many studies analyzing the influence of the frequency on the chlorinated compounds degradation, especially with volatile compounds. Some controversy is again found in the literature. An increase of the rate of degradation with frequency has been routinely reported for the degradation of volatile compounds such as CCl4 [34], trichloroethylene [38] and chlorobenzene [45]. However, if the range of studied frequencies reaches values higher than 1 MHz, some authors have detected a maximum around ranging 350-850 kHz [30,36] while other authors have reported a continuous increase [35]. On the other hand, degradation efficiency is also superior at higher (500 kHz) compared to lower frequencies (20 kHz) but with the same speciation for both frequencies [34]. Comparative studies with volatile (chlorobenzene) and nonvolatile (4-chlorophenol) compounds have also reported an increase of the degradation rate with frequency [52]. However, no controversy has been found in the case of nonvolatile compounds and the increase of the degradation rate with frequency has been routinely reported for PCBs [54], 4-chlorophenol [55], pentachlorophenol [58], trichlorophenol [61] or alachlor [62] and, if the range of studied frequencies reaches values higher than 1 MHz, a maximum around 350 kHz has also been detected. Even so, different tendencies have been reported for degradation efficiency: on one hand, slight independence for PCBs, i.e., no tendency [54] and, on the other hand, a maximum around 360 kHz in the case of the trichlorophenol degradation [61].
3.3. Influence of the ultrasonic power
The other ultrasonic operational variable is related to the acoustic power. The power can be characterized by two parameters: (i) ultrasonic intensity, Ia, defined as the ratio between the power input to the irradiated medium to the transmitting area, or (ii) the power density, Π, defined as the ratio between the power input to the irradiated medium to the sonicated volume. There exists a threshold value of intensity at which the beneficial effects of cavitation start to occur [7,8,9,12]. Some authors have demonstrated [9,12,73,86] that the increase of the yield with the power input is weak and reaches saturation, and attribute this effect to the coalescence of the bubbles, which would increase their size, leading to lower pressure pulses at the end of the collapse. This saturation has been observed in several systems [87,88,89]. Other authors have pointed out that lower intensities are more effective than higher intensities at the same total power, not only in experiments on water sonolysis [90] but also in degradation process of chlorinated organocompounds [45].
The influence of ultrasound power has been related with both mechanisms: pyrolysis or radical attack. It is normally shown during the experimental work that the rate of thermolytic cleavage of bonds in the substrate molecules is accelerated at higher ultrasound intensities. On the other hand, the formation of a certain minimum number of free radicals depends on the intensity of the collapse and the number of cavitating events, which in turn depends on the operating intensity of irradiation. These enhancements are commonly interpreted by considering the bubble implosion and the resulting conditions within the bubble. Acoustic intensity is directly related to acoustic pressure, as described by equation (1):
which in turn determines the final temperatures and pressures within the bubble
where PA is the acoustic pressure amplitude, ρ is the density of the fluid, c is the speed of sound in the fluid, Tmax and Pmax are the maximum temperature and pressure at implosion, T0 is the ambient temperature of the fluid, P0 is the pressure in the bubble at its maximum size (which some authors assume to be the vapor pressure of the fluid), Pm is the peak pressure of the bubble, which is the sum of the hydrostatic and acoustic pressure amplitude (PA from equation (1)) and γ is the polytropic index of the saturating gas previously presented [38]. Thus, as the ultrasound intensity increases, higher temperatures and pressures are reached within the bubble interior, which further enhance the overall decomposition rate of the target molecule and/or the formation of organic free-radicals. These observations are consistent with previous reports of enhanced decomposition kinetics at higher acoustic intensities [19]. It should be stressed that not only the transient temperature and the internal pressure in the cavitation bubble during collapse, but also the cavitation bubble size and the bubble collapse time are all dependent on the power intensity. According to Ref. [91], the maximum bubble size is dependent on the density of the liquid, the applied frequency, the hydrostatic pressure and the acoustic pressure, as follows [91]:
where ωa is the applied acoustic frequency and Ph is the external (hydrostatic) pressure, which is 1 atm under experimental conditions. In addition, the bubble collapse time, τ, is proportional to the maximum bubble size, Rmax [91]:
where Pm is the pressure in the liquid (i.e., Pm = Ph + PA) and Pvg is the vapor pressure in the bubble. Therefore, at high acoustic intensity (i.e., large PA values) the cavitation bubbles are able to grow larger in size during a rarefaction cycle such that insufficient time is available for complete collapse during a single compression cycle. As the above equations predict and the experimental results show, there is an optimum power density which can be applied during sonochemical irradiation in order to obtain the maximum reaction rates. More modern theories, based on bubble radial dynamics simulations allowing water transport through the bubble wall, attribute the saturation of the yields with power to the fact that bubbles excited more strongly expand more in the rarefaction phase, and therefore trap more water during collapse, owing to a diffusional barrier [78,85,92]. The predictions are in reasonable agreement with the experimental results of Mark et al. [93]. Other interpretations are linked to the self-attenuation of the acoustic field near the sonotrode tip: larger excitations produce more numerous and violently oscillating bubbles, which may act as a screen for the acoustic waves [94], and dissipate a larger energy [95].
Returning to the results reported specifically on chlorinated compounds, the degradation of CCl4 has shown a linear enhanced degradation with the power [8] or some authors have even obtained a maximum [12], where product ratios were to some extent intensity dependent, and degradation rates were likewise dependent on intensity [33]. Kruus et al. [45] suggested that, at the same total power, a lower intensity is more effective than higher intensity in the sonication of chlorobenzene in water. Yiang et al. [49] reported that the rate constant of the degradation of 1,4-dichlorobenzene and 1-chloronaphthalene at 500 kHz linearly increases with increasing ultrasonic intensity, advising that the ultrasound power used was too low to find the saturation effect reported previously. Similar behavior was reported for nonvolatile compounds. For example, Zhang and Hua [54] also reported that the rate of degradation of PCBs at 20 kHz was accelerated at higher sound intensities. Gondrexon et al. [59] reported, for a continuous flow ultrasonic reactor, an increase of the rate conversion of pentachlorophenol with increasing power input, reaching values of rate conversion close to 80%.
3.4. Influence of the dissolved gas
In addition to the physicochemical properties of the target molecule, chemical variables such as dissolved gases [7,8,9,12,33,96] should be taken into account. The nature of the radicals formed in the sonolysis of water depends on the dissolved gas in the solvent: if Ar is present in the aqueous solution, OH· and H· radicals are detected [97,98] and H2O2 and H2 are formed from the radical recombinations. After the initial cleavage of water, 80% of the OH· and H· radicals formed have been reported to recombine in the cavity [99]. However, the net quantity of OH· radicals produced in the gas-filled cavity may diffuse to the gas-liquid interfacial region, where they combine to form hydrogen peroxide. The concentration of OH· radicals at the bubble interface in water has been estimated to be at mM level [100]. The formation of H2O2, a measure for OH· production, during the sonolysis of water at 514 kHz was only about 20% lower in the case of argon-saturated water than in the case of oxygen-saturated water [71]. If the aqueous solution is saturated in oxygen, OH· and HO2· can be observed [101,102] and if the aqueous solution is saturated in nitrogen [103], nitrite, nitrate and ammonium are detected. Nam et al. have reported values of the rate constants for the recombinations of the radicals produced from the sonolysis of water [53]. Not only the radicals coming from the sonolysis of water and/or dissolved gas should be taken into account, but radicals coming from the substrate may also participate [104]. Petrier et al. [56] have pointed out an important phenomenon that occurs when sonication is run in a closed system. CO2 has long been recognized as an inhibitor for sonochemical reactions [105], and incomplete removal of this gas in the medium due to the absence of bubbling can slow down sonochemical processes. Temperature, as another example, changes the cavitation intensity and the diffusion of solutes [9] and thus the rate determining step.
Again, there is some controversy over whether or not the rate of degradation of CCl4 is affected by the gaseous atmospheres employed [33], for example ozone [31], but in some cases, the speciation and even the yield were found to be dependent on the dissolved gas [8]. The static pressure not only affects the degradation efficiency (with maxima at 6 and 13 atm) but also the products ratio [29]. Drijvers et al. [46] reported that saturation with argon accelerated the degradation of chlorobenzene in comparison with air. In a later work in the presence of argon or air [46], the same authors provided different speciation in the degradation of chlorobenzene, detecting additional chlorophenol as a product when the experiment was run in the presence of oxygen. In the case of nonvolatile compounds such as 4-chlorophenol [55], the presence of ozone enhanced the rate of degradation at 20 kHz in comparison to 500 kHz (inverse situation in the absence of ozone). Pentachlorophenate degradation at 530 kHz was analyzed in the presence of air, oxygen and argon by Petrier et al. [56], yielding mineralization of the target molecule to CO2 when the solution was saturated with air or oxygen, CO production when saturated with argon, and nitrite and nitrate formation when the reaction was conducted with air-saturated solutions. Wayment and Casadonte [62] have reported that argon-saturated solutions display an enhancement in the destruction of alachlor compared with either oxygen or air. The products of the degradation result primarily from bond scission in the case of an argon-saturated solution, and both bond scission and oxygen addition when the ambient gas is oxygen. Kojima et al. [63] studied the effect of dissolved gas species on the sonochemical degradation of (4-chloro-2-methylphenoxy) acetic acid at 500 kHz. In N2, air (O2/N2), O2 and Ar atmospheres, the rate enhancement of the degradation was found to be more effective in an O2-enriched atmosphere compared to an Ar atmosphere. It was considered that a higher amount of oxidants was formed under a higher partial pressure of O2, which accelerated the (4-chloro-2-methylphenoxy) acetic acid decomposition in a radical reaction system. On the other hand, the dechlorination removal rate was higher in an Ar atmosphere, compared to those in O2/N2 atmosphere. It was found that the herbicide was most effectively decomposed by sonication in an Ar/O2 (60/40% v/v) atmosphere.
The effect of the dissolved gases may be partially explained by considering the adiabatic compression in the bubble. A monoatomic gas heats more in compression than a diatomic gas, and the higher temperatures reached may therefore enhance bond scission. The analysis can even be refined by considering the thermal conductivity of the noble gas, which decreases from He toward Xe, so that a Xenon bubble would be the hottest one. This is confirmed by various experiments [72], in which a larger OH· yield was found with Krypton than with Argon, which, in turn, showed a larger yield than with Helium. The presence of a diatomic gas decreases the final temperature of the collapse, but the presence of O2 can participate directly in the production of OH·, while the latter only originate from water vapor in the presence of a monoatomic gas. From this competition between the two effects, one may therefore expect that, when saturating the solution with an Ar-O2 mixture, a maximal OH· yield should be obtained for some composition between pure Argon and pure oxygen. This is indeed the case [102], and single-bubble simulations correctly predict this phenomenon [92]. In contrast to pyrolysis effects, nor requiring oxidating species are expected to be mainly influenced by the final collapse temperature. Finally, the inhibiting effect of CO2 can be explained by its large thermal conductivity and low specific heat ratio γ.
3.5. Interrelation between the operational variables
Finally, the interrelation between the operational variables should be stressed. Increasing the pressure results in fewer cavitation sites due to the increase in the threshold for cavitation [29,96]. At the same time, the cavitation is more intense due to the stronger forces acting during implosion of the bubbles. The temperature changes the physical properties of the liquid. Higher temperatures decrease the cavitation threshold by reducing surface tension and viscosity, so a higher number of cavitation sites will be formed. However, the increase in vapor pressure of the liquid now allows vapor to enter the forming bubbles more easily, thus cushioning their implosion. The cushioning reduces the intensity of the cavitation. A larger presence of vapor in the bubble may also cool the collapse, since vapor decreases the global polytropic index of the gas/vapor mixture. It is not entirely clear how pH affects the sonication process [33,66,65,106]. At high alkalinity levels (>11), the hydroxyl radical could decompose to the oxide ion [107]. This provides part of the explanation of the decrease in the destruction rates as the pH is increased. As the pH drops below 4, the concentration of the hydrated electron species falls rapidly. However, it is not clear if the presence of the hydrated electron is important for sonochemical oxidation. Some discrepancies have also been found among frequencies and dissolved gas influences. Wayment et al. [62] found that the reactivity under argon compared to oxygen at higher frequencies was opposite to the results of previous investigators for the sonochemical oxidation of potassium iodide [71] where an oxygen-saturated solution had higher rates of reactivity. The rates, however, were faster under argon-saturated solutions at 20 kHz. In these earlier studies, the primary mechanism of reactivity was through hydroxyl radicals produced upon aqueous sonolysis. It was conjectured that while more OH· was produced at both high and low frequencies in an oxygen atmosphere, at higher frequencies the faster bubble collapse time caused more OH· to be ejected into the bulk to react. Longer collapse times at lower frequencies allowed for greater radical recombination inside the bubble, and, consequently, lower rates of reactivity in an oxygen atmosphere. The apparent discrepancy in this case is due to the presence of at least two different mechanisms being operant, one thermal and the other radical oxidation. A systematic study aiming at optimizing ultrasonic irradiation as an advanced oxidation technology has been proposed by Hua and Hoffmann [72]. The influence of the frequency and saturating gas during sonolysis were correlated to the hydrogen peroxide and OH· radical production. The highest production of hydrogen peroxide and OH· radicals were observed for a Kr-saturated solution at 500 kHz, and the lowest at 20 KHz in He-saturated solution.
Few multivariable analyses of chlorinated compounds can be found in the literature. Gaddam and Cheung [41] studied the influence of the pressure, temperature and pH on 1,1,1-trichloroethane degradation and, by statistical analysis, they proposed optimum conditions and, especially, they suggested that the initial concentration does not influence the process. Drijvers et al. [46], in the degradation of chlorobenzene, reported no influence of pH, in contrast to that found by the same authors for the degradation of TCE [38]. Its influence depends on the possible formation of CO2 and its acid-base character. The effect of pH on the ionization of the CO2 formed was thought to be the reason for the higher degradation rate in basic solutions. As mentioned above, the specific heat ratio γ for CO2 is lower than for air, which decreases the efficiency of the adiabatic bubble collapse and consequently the degradation rate of TCE in acidic or neutral solutions. So, the fact that there is no pH effect on the rate of sonolysis of chlorobenzene indicates that no CO2 is formed or that the CO2 formation is too small to influence the average specific heat ratio γ of the gas in the collapsing cavitations. Table 1, at the end of the text, shows an extensive summary of the main results found in literature for the sonochemical degradation of chlorinated compounds.
4. Mechanisms and Kinetics
4.1. General statements
Looking to the older literature [8,9], where both thermal and electric discharge theories were discussed, a first approach of the classical sonochemical reaction scenario can be obtained and enriched with recent developments. In the previous section we pointed out that the mechanism and kinetics of the process strongly depends on the place where the reaction takes place. Despite the fact that the reaction location is determined by several interrelated aspects, the properties of pollutant and solvent have more influence. The lower the log Kow value of the organic compound, the less hydrophobic it is. Another property is the vapor pressure. Compounds with a relatively high vapor pressure tend to enter the cavitation bubble gas phase more rapidly, and hence undergo degradation more effectively. The solvent being water, hydrophobic and/or volatile chemicals have a strong tendency to diffuse into the gaseous bubble interior [67,108]. The most effective site for their destruction is the bubble-liquid interface [109] and/or the bubble interior itself [110,111], where they suffer thermal and oxidation effects [112], please see Figure 3. Chemical partition of the pollutant between the gas phase and the gas-liquid interface is also dependent on the concentration of the chemical [113]. In contrast, hydrophilic and/or non-volatile compounds tend to remain in the bulk liquid during irradiation so that the major reaction site for such chemicals is, therefore, the liquid medium at low concentration [64] and/or the bubble-liquid interface at higher concentration [66], where they may be effectively destroyed by oxidative degradation, provided that sufficient quantities of radicals are ejected into the solution during the cavity collapse.
This previous classification is not strictly closed and pyrolysis of nonvolatile solutes at the interface is also possible, its efficiency depending on the hydrophobicity of the nonvolatile compounds, that is, their ability to accumulate at the interface [114]. Riesz has proposed a hydrophobic nature [115] for the interface, and Henglein has proposed that the concentration of OH· radicals is greatest in the interface [116]. This interface presents T < 2000K, a thickness of 0.2 μm, and pressure gradients [76]. Helping the interrelated classification, pyrolysis also occurs when polar compounds are sonicated. Currell et al. [117] found that the sonication of phenol yields acetylene as well. Secondary implications are brought from the physicochemical nature of the pollutant. Volatile compounds can scavenge the radicals formed inside the cavitating bubble [118], avoiding their release into the solution or their recombination, a situation which could not occur with non-volatile compounds [119]. Heinglein et al. have determined from radiolysis and ultrasound experiments that the ability of a wide range of known scavengers to decrease H2O2 yields depends only upon their hydrophobic character [120]. No correlation between the effectiveness of the scavengers and their volatility, or their rate constants for hydroxyl scavenging has been observed. Furthermore, the yield of thermal products from the sonolysis of a solute increases as the solute's ability to accumulate at the gas interface region increases [121].
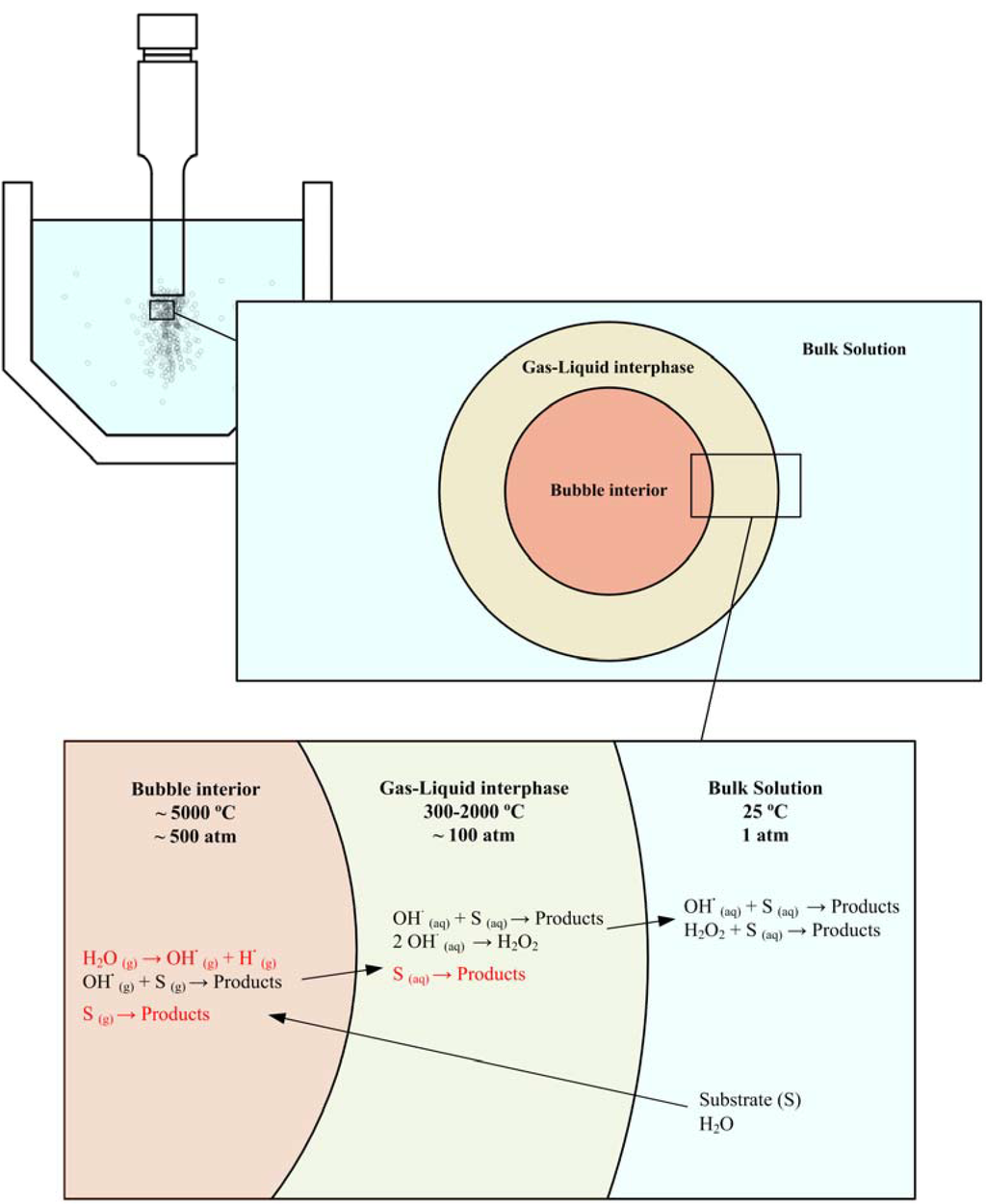
Figure 3.
General mechanism for sonochemical degradation.
Figure 3.
General mechanism for sonochemical degradation.
4.2. Contributions from the studies with chlorinated compounds
Apart from their specific interest, the studies on the degradation of chlorinated compounds deserve specific interest because they contributed greatly to building models of the chemical activity in a cavitation field. Hydrophobic and volatile chlorinated organocompounds such as CCl4 [13,30,31], chloroform [13], trihalomethanes [43] chlorobenzene [45,46,47,49,50,51] and trichloroethylene [38,50,51] have been subject to kinetic analysis and their degradation has been used to develop simulation models of kinetics for compounds which are degraded in the interior of cavitation bubbles by thermolysis (pyrolysis) and, in presence of oxygen, by oxidation reactions. Pyrolysis reactions are expected to follow simple first-order kinetics [44]. If the number of cavitation bubbles is constant throughout the experiment, the reaction rate observed is proportional to the reaction rate in the cavitations during collapse. Second, the concentration of organics in the bulk liquid phase is assumed to be proportional to the concentration in the bubble phase. As a result of these assumptions, the reactions are normally described with respect to the liquid phase with first order kinetics, and many reports have provided first (or pseudo-first) order kinetics, as shown in Table 1. However, sometimes this is not the case. Reaction rates are higher at low concentrations. The adiabatic temperature rise upon compression of a cavitation is much lower if a volatile vapor is present (due to the presence of starting material and/or reaction products). Excessive amounts of volatile compounds lower the temperature of a collapsing cavitation bubble, which lowers the rate of degradation. These facts will be commented on further in this section.
Shemer et al. [43] analyzed the sonochemical degradation of different trihalomethanes, which are compounds presenting high vapor pressures. As a conclusion, the authors reported that vapor pressure was found to be the most important parameter affecting the sonodegradation kinetics and efficiency, while the bond dissociation energy and the hydrophilic/hydrophobic characteristics of the trihalomethanes compounds were found to be of secondary importance, providing a specific difference between volatile and hydrophobic features. Other chlorinated organocompounds, now non-volatile such as p-chlorobenzoic acid [53], or even highly hydrophobic and non-volatile such as PCBs, have also been studied [54]. Chlorophenols have also been extensively studied [56,57]. Petrier et al. [56] have proposed that chlorophenol is degraded with a scheme of radical reactions occurring most probably in the solution, because chloride appears very soon after sonication has been started and CO2 evolution appears after a lag time. If the reaction was taking place inside the bubble, these compounds should be produced together at the beginning of the reaction. An interesting comparison of both behaviors was reported by Petrier et al. [52], who compared the sonochemical degradation of chlorobenzenes and chlorophenols as examples of hydrophobic and hydrophilic compounds. They suggested, studying mixtures of both compounds, that chlorobenzene is degraded inside of the bubble before chlorophenols, which are degraded in the solution. They explained this finding by the differences between the volatilities and because in the bubble, the chlorobenzene degradation replaces the OH· formation. Besides, the degradation of chlorobenzene forms CO2 and HCl, whereas chlorophenol degrades to formic acid, oxalic acid and CO2.
However, the mechanism can present several interactions with other operational variables, such as the dissolved gas, which draws a more complex picture of the sonochemical activity. Drijvers et al. [46] analyzed the mechanism of the degradation of chlorobenzene and concluded that the process takes place in the interior of the bubble, but that the degradation of organic compounds also occurs in the cavitation bubbles by reaction with oxidative species such as OH· and O. The absence of chlorophenol was observed in argon-saturated solutions in contrast to air-saturated solutions, because chlorophenol is not formed directly by the addition of OH· radicals to chlorobenzene at high temperatures during the collapse. The authors present a mechanism in order to explain the different intermediates obtained. In the absence of oxygen there are two possible initiation reactions for the thermal degradation of chlorobenzene (homolytic cleavage of the C-Cl bond as the most favorable, or of the C-H bond). The main final product was acetylene C2H2, with a concentration higher in argon than in air. At the same time, the concentration profile of chloride in argon-saturated solutions is more than twice as high as in air-saturated solutions, while the initial degradation rate of chlorobenzene increases only by about 50% by replacing air with argon as the saturating gas. This means that more phenyl radicals are formed in argon-saturated solutions, and that relatively more chlorophenyl radicals are formed in air-saturated solutions. No explanation could be found for the relatively higher number of chlorophenyl radicals in air-saturated solutions. The authors could only suggest an initiation reaction, involving oxygen directly or indirectly, which effectively competes with the initiation reactions of cleavage of C-Cl and C-H. To explain the accelerated pyrolysis of chlorobenzene in the presence of oxygen several such initiation reactions were proposed:
It is also very important to stress that all intermediate products are formed from the beginning of the sonolysis of chlorobenzene. This indicates that several consecutive reactions occur during a single cavitation event. Several organic intermediates present steady concentrations during the sonolysis of chlorobenzene; this should mean that either they are quickly degraded as fast as they are formed, or they are only formed at the start of the sonication and that they degrade very slowly. Finally, it should be noted that no phenol was detected during the sonication of the air-saturated solution of chlorobenzene. A possible explanation for the fact that only chlorophenol and no phenol was detected may be that H-abstraction by OH· radicals, as well as conversion of chlorophenyl radicals into chlorophenol by addition of oxygen, mainly takes place near the interface, where the concentration of OH· radicals is the largest [116].
An additional parameter essentially affecting the sonodegradation is the concentration of organic compound. The concentration of volatile compounds in the aqueous solution influences their concentration in the cavitation bubble, and, therefore, affects the sonolysis rate and efficiency. Depending on the time the gas bubble exists in the solution before it becomes a “hot spot”, there are two possibilities. The gas bubble lasts long enough in the solution to achieve equilibrium partitioning of the volatile compound between the gas bubble and the solution. This equilibrium is given by the Henry’s law coefficient of the volatile compound. If, on the other hand, the gas bubble is not present long enough in solution to reach equilibrium, the concentration of the compound in the collapsing bubble will be determined by its rate of diffusion inside the gas bubble. In this case, the diffusion rate is determined by the aqueous diffusion coefficient of the volatile compound [47]. In the case of chlorobenzene, the proportionality of the volatile compound concentration between the cavitation bubble and the aqueous solution is controlled by the diffusion coefficient in water, rather than by Henry's law constant [47]. This statement is strengthened by the observed linear relation between the compounds half-lives and their log Kow values. Shemer and Narkis [43] also suggested that the trihalomethanes transfer depends on the diffusion in water and consequently is limited by it.
Finally, in spite of the fact that OH· radicals are not expected to influence the breakdown of apolar compounds considerably (for example CCl4 is efficiently broken down by ultrasonic waves [37] but the breakdown of CCl4 by OH· radicals is negligible, compared to other chlorinated hydrocarbons [122]), some authors have, however, considered this path of degradation. Weavers et al. [55] analyzed the sonochemical degradation of nitrobenzene, 4-nitrophenol and 4-chlorophenol at two different frequencies. Nitrobenzene has the highest Henry's Law constant and reacts considerably faster at 20 kHz than 4-chlorophenol or 4-nitrophenol. However, as the Henry's Law constant decreases, other pathways such as OH· attack become increasingly prevalent, as observed not only with 4-nitrophenol but also with 4-chlorophenol. In addition, at 500 kHz, 4-chlorophenol reacts most rapidly due to its fast reaction rate with OH· and its high Kow, which suggests a greater partitioning to the bubble interface due to its higher hydrophobicity. In the same line, Wayment et al. [62], working with alachlor, have found that the oxidative reaction with hydroxyl radicals appears to occur in the bubble and in the interfacial region rather than in the bulk, and, as such, a higher rate of ejection of OH· from the bubble interior may lead to the decrease in the observed rate.
However, the possibility that OH· radicals can attack apolar or volatile compounds is not only supported by the observed influence of frequency but also by some observed effects of the pollutant concentration. Indeed, Drijvers et al. analyzed the mechanism of a mixture of chlorinated compounds in the mM concentration range [50] and Dewulf et al. in the μM concentration range [51]. In the mM range, a change from the first-order reaction rate to zero-order was found, when a second chlorinated compound was added to a single compound solution. From the results, it was stated that the influence on the degradation rate cannot be explained considering only the effect of the average specific heat ratio γ, but the interaction between the different intermediates was proposed as an additional factor in the mechanism degradation. Further implications were found in the μM range, where the effect of initial concentrations on the specific heat ratio was not sufficient to interpret the experimental results, which showed that the degradation rate was faster than predicted from pyrolysis kinetics [51], therefore suggesting additional removal mechanisms. The latter may originate from an OH· radical induced degradation pathway. This mechanism may be important provided that both chlorinated compound and OH· radicals are present in substantial concentrations. In this sense, the number of chlorinated molecules would be lower at micromolar levels, when compared to millimolar levels. On the other hand, however, the number of OH· radicals does not decrease with decreasing concentrations of chlorinated compound. It might even increase, since the temperature, and hence the pyrolysis of water, can be enhanced due to the lower chlorinated concentrations, affecting the specific heat ratio. This means that the ratio of OH· radical/chlorinated compound in the cavitation would increase. Whereas pyrolysis is not affected by this ratio, the reaction of chlorinated compound with OH· radicals becomes more likely at lower chlorinated compound concentrations, resulting in an increase in the significance of radical degradation by OH·. Secondly, lower initial chlorinated compound concentrations result in higher specific heat ratios, enhancing the reaction temperature in the cavitation, so that pyrolysis of both chlorinated compound and water would be faster. The possible initial pyrolysis reactions are:
The dissociation energies will determine the order of the degradation rate between chlorinated compound and water. For reaction (8), the dissociation energy is 498 kJ mol-1 and this value is dependent on the temperature. If the temperature of the collapse increases due to an increase in the specific heat ratio (for example by lower initial concentrations of the chlorinated compound) the ratio of pyrolysis rate of the chlorinated compound to that of water decreases, resulting in a higher OH· radical/chlorinated compound concentration ratio in the cavitation. In conclusion, this indirect temperature effect of decreased substrate concentrations would favor the OH· radical induced degradation over the pyrolytic degradation of the chlorinated compound.
An approach to support the thesis of OH· radical induced degradation at micromolar concentration of chlorinated compound, was to develop a new kinetic model, incorporating both pyrolytic degradation and OH· radical induced breakdown. The OH· radical induced breakdown pathway requires that the pyrolysis of water and its subsequent reactions are included. Zang et al. [54], analyzing the degradation of PCBs, which are hydrophobic and non-volatile compounds, proposed that the mechanisms of degradation were based on thermolysis and also free radical attack. They concluded that OH· attack contributes substantially to the enhanced sonochemical rates at 358 kHz, but the chloride recovery was optimal at 1071 kHz. The differential role of aqueous OH· attack was confirmed over a range of ultrasonic frequencies.
Finally, theoretical explanations have been provided to explain the observed degradation trends of phenol and chlorophenols on the basis of concentration of the pollutant at the cavitation bubble/solution interface. The chemical stability of these compounds towards radical attack in the liquid phase was also discussed. It has been observed that, due to their higher hydrophobicity, chloro-derivates were degraded much faster than the parent compounds [123].
4.3. Specific mechanisms
Jennings et al. [13] proposed a free radical mechanism for the degradation of CCl4 and CHCl3. Energy requirements suggest that if H2O can undergo homolytic dissociation, CCl4 and CHCl3 molecules, which must also be present in the cavitation bubbles, should likewise experience primary cleavage. The authors thus considered the following mechanisms:
Because of the high resonance energy of CCl3·, reaction (11) is expected to occur to some extent, in spite of the fact that C-H bonds are generally stronger than C-Cl bonds. Once the radicals are freed, their subsequent fate will depend on their concentrations and on energy considerations. The authors took into account the displacement reactions of CCl4 by H·, Cl· and OH· radicals giving HCl, Cl2 and HOCl, respectively, and CCl3·. In the case of CHCl3, H·, Cl· and OH· radicals give H2, HCl and H2O and CCl3·, CCl2·. The concentration of CCl4 and CHCl3 at the reaction site is probably greater than the concentration of primary free radicals, leading to the expectation that radical coupling will compete successfully only when high activation energies are associated with the displacement reactions available to a given radical. These arguments supported the final products obtained by the authors: CO2, O2, Cl2, HCl, C2Cl6 and C2Cl4 as final products from CCl4, and HCl, C2Cl6 and C2Cl4 from CHCl3.
Hoffmann [30,31] described a simple kinetic model where the rate-limiting step in the sonolysis of CCl4 was CCl4 pyrolysis into CCl3· and Cl·, and allowed a description of the rate of production and subsequent degradation of the intermediates for the sonolysis of CCl4. The thermal dissociation of CCl4 in an Ar gas phase is known to produce two chlorine atoms for every molecule of CCl4; fission of the second chlorine is thought to occur at ≈10% of the rate of the primary fission [124]. Since the degradation of CCl4 was observed to be a pseudo-first-order reaction, the reverse reaction CCl3· and Cl· radicals can be ignored. The disproportionation rate of this trichloromethyl radical related to its combination depends on the radical species, but is usually less than 1 [125]. The recombination of the trichloromethyl radical in the gas phase has been studied, and at 298K and 1 atm (in Ar), the rate constant of formation of C2Cl6 is ≈ 1 order of magnitude less than the re-formation of CCl4 [126]. At the same time, since the final stable Cl-containing product of CCl4 sonolysis is Cl-, they simplified the mechanism of the sonolysis degradation, which allowed them to obtain expressions for CCl4, the main intermediate, and Cl- concentration vs. time. It is very interesting to remark that the total measured Cl- accounted for only 75% of the total chlorine initially present in the CCl4. However, sonication reactions at higher initial concentrations of carbon tetrachloride, [CCl4]0, were clearly more complicated. For example, at higher initial concentrations of CCl4, the initial pyrolysis product, •CCl3 self-reacts to give the major intermediate, C2Cl6, instead of further decomposing via an additional breaking of the C-Cl bond to produce CCl2.
Gaddam [41] analyzed the degradation of 1,1,1-trichloroethane (TCA) and detected trace amounts of 1,2 dichloroethene as a byproduct, detection of which is also reported by other authors. One possible route of production of ethenes from ethanes is the reaction between chloride radicals and unsaturated derivatives of TCA, which in turn were formed by hydrogen and chloride abstraction from TCA.
The sonolysis of TCE also proceeds mainly by pyrolysis reaction [38]. The following equations represent the lowest-energy dissociation reactions of TCE [50,127]:
TCE is not degraded by OH· radicals in the bulk solution but by indirect decomposition by radicals formed thermally from other TCE molecules, for instance the chlorine atom, and takes place as follows [38]:
A wide range of volatile aliphatic compounds were identified as volatile organic intermediates during the sonolysis of TCE, and most of them were formed by oligomerisation of chloroactylene and dichloroacetylene, the chloroacetylene being formed by two stepwise Cl· atom eliminations from TCE [38].
Clark et al. [139] analyzed the perchloroethylene degradation in aqueous solution and with ethanol as co-solvent. They found that for every concentration of PCE evaluated, a higher ethanol percentage in solution produced a lower rate constant value. Moreover, at constant ethanol content, the rate constant values decreased with increasing PCE concentration. The mass balance for the experimental results also suggested that there might have been competition between PCE and ethanol molecules for access to the degradation mechanisms (pyrolysis and free-radical reactions), although there is much more ethanol than PCE. This implication was made because at constant initial PCE concentrations, as the amount of ethanol in solution increased, the amount of degraded PCE decreased. In addition, the amount of ethanol in solution decreased in lower amounts as the initial amount of PCE in solution increased. These opposing effects could be due to a limited amount of free radicals and/or surface area at the air-liquid interface during pyrolysis. Because ethanol has a higher vapor pressure and a lower boiling point than PCE, a larger fraction of ethanol would have been present in the gas phase, therefore undergoing degradation by pyrolysis in the cavitation bubble. Moreover, PCE has a higher Kow value, and thus partitions more effectively to the hydrophobic bubble interface. This suggests that the degradation of PCE might have been dominated by pyrolysis in and on the cavitation bubbles due to the suspected competition between the ethanol and PCE molecules. Besides, the high concentration of ethanol far exceeded the relatively small amount of PCE in solution. This allowed easier partitioning of the ethanol to the cavitation bubble surfaces, and, to a lesser extent, allowed ethanol to scavenge the free radicals in the bulk solution more easily than did the PCE molecules, and this may explain therefore the larger decrease of the ethanol mass compared to that of PCE, as the sonication time increased. They concluded that pyrolysis was the more dominant degradation mechanism in this experiment.
Jiang et al. [49] analyzed the reaction mechanisms of the decomposition process of chlorobenzene on sonolysis at 500 kHz in air-equilibrated aqueous solutions. The first step in the degradation would be expected to be the removal of a chloride atom from the aromatic ring and reduction to chloride ion as a result of high temperature combustion
In addition, the carbon-hydrogen bonds of chlorobenzene could be cleaved by pyrolysis
Dissociation by reaction (17) is energetically more favorable than reaction (18) due to the lower bond dissociation energy of C-Cl (400 kJ/mol) compared to that of C-H (463 kJ/mol) [128]. Brown carbonaceous particles were observed visually to appear after 50 min of sonication. It was deduced that phenyl radicals can further decompose into C4H3, C2H2 and C according to the previous and following reactions [128,129]:
These observations are consistent with soot formation under pyrolytic conditions as observed by Hart et al. [108], who showed that soot can be formed through phenyl radical combination or C2H2 reaction at high temperature, which can be further oxidized into CH4 and CO2.
Jiang also detected hydroxylated compounds, resulting from the attack of OH· radicals, but the yield detected by HPLC was quite low. In addition, the finding that the time-increasing generation of H2O2 is not significantly affected by the presence of aqueous chlorobenzene demonstrates that oxidation of chlorobenzene by free radical OH· outside the bubbles (even at the interphase of liquid-gas bubbles) is of minor importance. Acetylene, methane, CO and CO2 were the major gaseous products formed. Finally the authors suggested that the absence of nitrate and nitrite ions also indicates that chlorobenzene decomposition occurs predominantly inside the cavitation bubbles. It has been reported [130,131,132] that nitrites are the primary products of sonolysis of air-saturated water, with the generation of nitrate resulting from the oxidation of nitrite by hydrogen peroxide. Hart and Henglein indicated [130] that in the first instance, nitrogen is oxidized to nitrogen oxide and nitrous oxide by reaction with OH· and O inside the cavitation bubble. Further NO and N2O oxidation with OH· and molecular oxygen lead to nitrous acid. The reaction rate is dependent on solution pH, which decreases rapidly with sonication time [132]. It is therefore expected that nitrate and nitrite ions would be generated as soon as an air-equilibrated solution is exposed to ultrasonic irradiation. However, the generation of nitrite and nitrate is observed to be inhibited in the initial stages of sonication if volatile compounds are present. These volatile compounds diffuse to the gas-bubble interfaces and not only inhibit the interaction between free radicals and nitrogen but also decrease γ and, therefore, the collapse temperature.
5. Mass balance and speciation. Technical and Environmental viability
From the previous section, we can realize that a vast number of academic studies have been focused on kinetic and mechanistic aspects of the sonochemical degradation of chlorinated compounds, but there are few authors who comment on the mass balance, due to the difficulty to close it [45,52,54]. Looking at the literature, the sonochemical degradation of other compounds, even with a volatile behavior, can yield a totally closed mass balance [65] but, as we have noticed in the previous section analyzing the mechanisms, the complexity of the process involves a wide range of intermediates, and therefore a wide speciation. Focusing on chlorinated compounds, two different atoms can be monitored, C and Cl, the latter being easier from an analytical point of view. A total carbon mass balance is not usually obtained, for example because some brown carbonaceous particles can be separated by filtration from the reaction mixture [49]. This is consistent with soot formation under pyrolytic conditions, as mentioned previously. Even choosing Cl as the target element, some authors have admitted that the mass balance (even defining the mass balance related to chlorinated compound disappeared [31], and not related to the initial amount of starting material) after sonolysis is found to be as low as 50% [41] and frequently in the range [70-100%] [30,31,54]. The Cl· radical presents such a high reactivity that it is difficult to detect it directly by ESR [133], which could be a reasonable explanation.
Since the 1950s, when Jennings and Townsend [13] found HCl, C2Cl6 and C2Cl4 as final products of the ultrasonic degradation of CHCl3, and CO2, O2, Cl2, HCl, C2Cl6 and C2Cl4 as speciation for CCl4 sonochemical degradation, CCl4 has become a target molecule for mass balance and speciation studies. Chendke and Fogler [29] reported only HCl and HOCl as final products and Spurlock and Reifsneider [33] found CO, CO2, HClO, HCl and C2Cl6, but no information about mass balance was given. Petrier et al. have developed specific work in order to analyze the mass balance in the degradation of CCl4 [34]. Their experimental system was a cylindrical jacketed glass equipped with a Teflon holder, which accepts transducers at different frequencies, 20 and 500 kHz. In aqueous solution and during the first hour C2Cl4 and C2Cl6 were the main products, along with small amounts of dichloromethane, chloroform, trichloroethylene, 1,1,1,2-tetrachloroethane or 1,1,2,2-tetrachloroethane, pentachloroethane, hexachloropropene and hexachlorobutadiene. The same compounds were identified in the gaseous phase with, in addition, tetracholocyclopropene and pentachlorocyclopropane. The authors pointed out that the total amount of all these reaction products was very low and that only C2Cl4 could be quantified with HPLC. The maximum amount of intermediates was less than 5% of the initial aqueous CCl4 concentration and all these chlorinated products disappeared as the reaction progressed. Compounds with high molecular weight (C > 4) were no longer detected, so that they supposed that chlorinated organic species were degraded into smaller molecules rather than polymerized into longer chains. The chloride anion was the main product of CCl4 sonolysis. The nearly 100% yield obtained after five hours irradiation showed that CCl4 is really degraded and is not just volatilized during the course of the irradiation. This result has been reported elsewhere [35]. Yields of CO (5%) and CO2 (70% at 500 kHz, and 54% at 20 kHz) were obtained from the gaseous phase of the reactor headspace. They corrected these values with the CO2 dissolved in aqueous solution (by means of Henry’s Law), finally giving yields of CO2 of 95% at 500 kHz and 75% at 20 kHz. The same compound was also degraded by Hua and Hoffmann at 20 and 135 kHz [30,31], who reported chlorine mass balance after sonolysis >70% with chloride ion and hypochlorous acid as final products. Traces of hexachloroethane and tetrachloroethene were also reported. A wide speciation has also been reported for trichloroethylene degradation. Studies at 520 kHz have produced chloroacetylene, dichloroacetylene, dichlorodiacetylene, tetrachloroethylene, isomers of trichlorobutenyne, tetrachlorobutenyne, pentchlorobutadiene and hexachlorobutadiene, without any information about the mass balance [38]. Gaddam et al. [41] followed the atom balance on chlorine in the degradation of 1,1,1,-trichloroethane, accounting for an average of 50% of the chlorine. At the end of the run, the chlorine was presented as unreacted TCA, Cl-, OCl-, HOCl. Finally, trihalomethanes have provided a wide range of removal efficiency, from 60% for CHI3 to 100% for CHCl3 [43] without any comment about the speciation. Bhatnagar and Cheung [37] reported percent destruction ranging from 72-99% for chlorinated C1 and C2 volatile organic compounds and mixtures of them. In a later work [39] the same authors found total degradation for several aliphatic compounds and no chlorinated compounds were detected as final products.
Chlorinated aromatic compounds follow the same controversial scenario. Drijvers et al. [46], while degrading chlorobenzene in air- or argon-saturated aqueous solutions, have detected different intermediates depending whether air or argon was the dissolved gas. For argon-saturated solutions, methane, acetylene, butenyne, butadiyne, benzene and phenylacetylene and other chlorinated and non-chlorinated monocyclic and dicyclic hydrocarbons were detected. For air-saturated solutions, chlorophenols were detected in addition. No comments were reported related to the mass balance. Kruss et al. [45] analyzed the sonochemical degradation of chlorobenzene in aqueous solution and, in addition to chloride ions, found reaction products absorbing in the range 250-290 nm, the concentration of which increased with time, and which made the solution cloudy. Chlorophenols, chloronaphthtalene, byphenyl, mono and dichlorobiphenyls, naphtho or dibenzofuran at ppm levels were typical byproducts. The same authors claimed the impossibility to conduct a material balance of the chlorine over time and degradation efficiency (defined as produced chloride ion) higher than 85% could not be obtained. A similar degradation efficiency was obtained by Tiehm et al. [48]. Other authors have, however, reported cleaner reactions. Petrier et al. [52] expeditiously degraded chlorobenzene to chloride ions and 44% of the carbon atoms were recovered as CO and CO2, with degradation efficiency higher than 90%. In the same study, chlorophenols were also degraded by OH· radicals outside the cavitation bubble. A total carbon mass balance could not be obtained, but some brown carbonaceous particles could be separated by filtration from the reaction mixture. In a posterior work, Yiang et al. [49] reported more than 90% of the chlorine recovered as chloride ion with CO, C2H2, CH4 and CO2 as the major gaseous products of chlorobenzene degradation. Intermediates resulting from attack of OH· radicals were detected with quite a low yield and disappeared with extended ultrasonic irradiation.
Pétrier et al. [56] degraded pentachlorophenate and 90% of the chlorine was recovered in the solution as chloride ions. CO2 was also found as a product of the pentachlorophenate degradation when the solution was saturated with air or oxygen, and CO when saturated with argon. In addition, acetylene, methane and ethane should be formed along with carbon dioxide. Similar conclusions were reported for atrazine at 20 and 500 kHz [58] and by Serpone et al. [57] using 20 kHz ultrasound field, where chlorophenols were nearly quantitatively dechlorinated, with CO2 as a detected product. At 1.7 MHz, the degradation efficiency decreased to values lower than 20% at lower concentrations, or even 3% at higher concentrations [60]. Trichlorophenols presented 42% fractional conversion, and a 25% degradation efficiency at 360 kHz, which was found to be the optimal frequency [61]. A specific study of toxicity revealed that an increase of the toxicity appeared in the middle of the sonochemical treatment, and decreased for longer times.
Finally, Zhang and Hua [54] degraded polychlorinated byphenyls (PCBs) at 20 kHz with a chloride ion recovery lower than 80%, and a high speciation of ethyl benzene, diethylbiphenyl, dibutylbiphenyl, phenol, propylphenol, and id-tert-butyl phenol. Atrazine presented poor results even at 500 kHz, with a dechlorination efficiency as low as 46% [58]. (4-chloro-2-methylphenoxy) acetic acid (MCPA) was degraded at 500 kHz in different atmospheres, the dechlorination ratio being higher in O2 and air (O2/N2) atmospheres [63], although many studies report that ultrasonic degradation of organic compounds in Ar was more effective than O2, N2, and air atmospheres [46,62]. MCPA produced acetic and formic acids through the formation of intermediates such as 4-chloro-2-methyl phenol, methylhydroquinone, 4-chlorocatecol and cresol.
From this review, it is clear that the further sonochemical breakdown of intermediates and mineralization depends on intermediate features, such as volatility, polarity and dissociation in aqueous solution. The impossibility of closing the mass balance and the high speciation has provoked some authors to question the environmental viability of the process. Kruus et al. [134] reported a wide range of speciation for single and mixtures of chlorinated compounds in water, and claimed the need for a careful evaluation of sonication as a method of eliminating chlorinated organic compounds from water. Despite the fact that these conclusions are relevant enough to consider a further and careful development of sonochemical technology, it is also true that all these studies have been carried out at laboratory scale, where in-depth analysis of the experiment was not always considered. In addition, some difficulties have been reported in the scale-up of the process in the past [59] but, nowadays, the technology has improved enough to provide competitive equipment [135] and references therein]. Supporting this expectation, recent applied studies treating polluted natural ground water [136,137] or drinking water polluted with halomethanes [138] can be found in the literature.
6. Conclusions and perspectives. Economical viability
After reviewing the extensive literature in the sonochemical degradation of chlorinated compounds, it remains only to emphasize that in the case of pyrolysis, faster reaction rates are achieved than with radical reactions. This review clearly establishes that a strict protocol should be followed in order to obtain comparable results in our experimental trials. In this section, we will summarize some key aspects to take into account.
First of all, some preliminary tests and assays should be carried out to configure the experiment series:
- (i)
- The temperature monitoring system should be carefully designed to ensure strict control. This allows us to fix values for the solubility, octanol-water partition coefficient, Henry constant, vapor pressure, density and other properties in our chlorinated organocompound + water system. A summary of the theoretical values of these properties is advised to design properly not only the experimental set-up but also the analytical procedure.
- (ii)
- Previous trials should be carried in the preparation of the solutions to check the chemical inertness of the materials used, i.e., glass, teflon and septum taps, although these materials have usually been used in chlorinated compound sonolysis [34,38]. Solutions of the chlorinated compound should be prepared by stirring, and maintained at 25 ºC in volumetric flasks with (a) Teflon or glass covered magnetic bars and/or (b) glass or septum taps. Liquid and gas samples have to be sequentially withdrawn and analyzed for the possible combinations, and no more than 3% of the initial material should be lost during the chosen experiment time. Keeping in mind that headspaces should be as small as possible in order to avoid complications in the experiments [52], the real headspace above the aqueous solution in the closed sonoreactor should also be studied. To do that, the sonoreactor must be airtight in order to prevent any compound loss by air stripping, and separate control experiments should be carried out in the absence of ultrasonic irradiation, in order to measure a % of volatilization, to be compared with the estimation made from data given in the literature [37,139]. Schwarzenbach et al. [140] reported the following equation to estimate the partitioning of organic chemicals due to Henry's law:where Vw represents the volume of water phase, Vg represents the volume of gas phase; and K'H represents the dimensionless Henry's constant. The acceptance of high values of volatilization percentage is not unusual [38]. This is because it is stated [31] that in a closed system volatile solutes re-enter the treated solution and the observed losses are due to chemical reaction and not to volatilization.
- (iii)
- Due to the fact that many buffers are effective radical scavengers [141], we have to analyze very carefully the options for adjusting the pH of the solutions.
Next, the possible high volatility and low solubility of our substrate can complicate the work-up of solutions with exactly the same concentration [31] and thus, it is not surprising to find in the literature trials carried out in order to check the reproducibility [13,46]. The causes of the deviations in the results could not be attributed to the analytical procedures themselves but to some aspects of the ultrasonic procedure. At least three sources of error can be suggested [13]: (a) unless the coupling of the transducer to the reaction vessel is identical from run to run, there are variations in the intensity of ultrasound reaching the reacting medium, with consequent variations in the yield. (b) A high frequency generator does not maintain absolutely constant electrical output; the acoustical output varies accordingly. Ideally the reacting system should be monitored constantly by using some suitable energy detector and automatic control should be made for fluctuations in sound intensity. (c) The amount of gas dissolved in the reacting liquids at the start of the reaction is apparently significant. However, some available commercial systems present a fixed set-up, and an advanced electronic searching device design in order to address points (a) and (b).
Due to the fact that the reaction scenario can be strongly different at low and at high frequencies, once this operational variable is fixed, the next variable to be varied should be the initial concentration. Indeed, it has been shown that this parameter determines the main site for the sonochemical reaction and therefore the kinetics and speciation. Later, the influence of pH should be studied in the case of pollutants presenting acid-base behavior. Finally, ultrasound power should be the next variable to be studied, looking for the cavitation threshold. Calorimetric characterization of the systems is required and the two possibilities for obtaining this parameter (ultrasound intensity and/or power density) should be used. These aspects are very important for the comparison of results.
Before the beginning of the experiment, and especially in the case of volatile compounds, a stabilization time is necessary to allow the previously mentioned distribution of the compound between the gaseous and the aqueous phase in the airtight closed sonoreactor, according to Henry’s law. Sonolysis of the aqueous solution should begin only when the compound concentration in the gaseous phase has reached a steady value. At this early stage, the temperature control has to be fast and efficient. The withdrawn samples at the determined times must be transferred to vials without headspace to minimize volatilization losses and analyzed immediately after their collection.
As noted in the previous section, in most cases, the ultrasound treatment yields a great speciation of products. In the treatment of an aerated aqueous solution of one pollutant, which is the most common case, the formation of products of different natures should be monitored. On one hand, products formed from pyrolysis reactions should be analyzed, and due to the radical nature of these reactions, both products with smaller molecular weight than the target compound, and products with high weight molecular from coupling reactions, can be formed. On the other hand, hydroxylated compounds formed by the reaction of the target compound and the hydroxyl radical should also be monitored. Several anions, such as chloride along with the chlorooxoanions (ClO-, ClO3-), should also be quantified to determine the degradation grade. The production of chlorine gas can be stopped by adjusting the pH of the solution above 3. The possible formation of other anions such as NO3- and NO2- (in the case of air-equilibrated solutions) should be checked and can give information about whether the reaction takes place in the liquid phase or in the bubble. Volatile and semi-volatile products, along with CO/CO2, originated from the complete mineralization of the substrate, should be monitored in the gaseous phase. Finally, the formation of soot should also be taken into account. Once the reaction has finished, it is strongly suggested that an in-depth analysis of the experimental results should be done, with special attention paid to the mass balance. Once the mass balance has reached a reasonable satisfactory level, further sonochemical trials can be carried out.
As a final remark, economical viability is, without any doubt, the main goal to achieve to be able to consider sonochemical treatment as a competitive technology. Poor attention to this subject can be found in the literature and only few applied works have provided economical results. Sonochemical degradation of trihalomethanes [43] has provided energetic consumption of 1.4 kWh/L to 0.3 kWh/L for a degradation of 90% of a solution 10 mg/L. Degradation of volatile pollutants in natural ground water with concentrations in the μg/L range has used consumptions of 0.5 kWh/L [137], and 0.64 kWh/L with concentrations in the mg/L range [136]. Trying to improve this figure of merit, batch and circulating systems have been used in the analysis of sonochemical degradation of chlorinated compounds [42] with an energetic load of 4.6 kW L-1 in the batch reactor, and 0.64 kW L-1 in the circulating reactor. Comparative studies using different experimental set-ups should be carefully analyzed [45]. Despite this fact, some authors [37] have proposed the ultrasound treatment as comparable to the advanced oxidation technologies, (based on the relative efficiency in terms of the total power delivered per liter of water, 0.1 kW L-1), others have claimed the limitations of sonolysis for the control of trace contaminants in water, due to fact that it is relatively inefficient with respect to the input energy [65]. Some previously available commercial configurations have not been optimized for an efficient generation of transient cavitation. The high-intensity sound waves generated at the tip of the horn usually result in intense local cavitation, but the large number of bubbles formed cause a “screening effect” that reduces the sonication efficiency. Some further developments, such as the parallel-plate near-field acoustical processor [19], have been used to show that a more dispersal energy, due to a larger emitting area, can provide a higher energetic efficiency. Fortunately, nowadays there are strong efforts for improving not only the high power ultrasound generators [135], but also sonoreactor designs [142,143,144] trying to overcome the energetic cost penalty usually assigned to sonochemical technology. Other improvements of the technology should develop the combination with other advanced oxidation technologies, which will provide higher rates. These items must be considered as future research lines.

Table 1.
Summary of the main results for the sonochemical degradation of chlorinated compounds.
| k min-1 | F kHz | C μM | T K | Π W cm-3 | pH | gas | Intermediates and by products | Mechanism model | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|
| CCl4 | 0.025 | 20 | 200 | 286 | 0.65 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.070 | 500 | 200 | 286 | 0.1 | nat.to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.044 | 205 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.049 | 358 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.055 | 618 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.039 | 1078 | 200 | 286 | 0.06 | nat. to 3.5 | --- | C2Cl6, C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CCl4 | 0.043 ± 0.002 | 20 | 396 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CCl4 | 0.198 | 20 | 195 | 298 | 1.23 | 11.8 | Ar | C2Cl6, C2Cl4, Cl-, HOCl | pyrolysis | [31] |
| CCl4 | 0.234 | 20 | 19.5 | 298 | 1.23 | 11.8 | Ar | C2Cl6, C2Cl4, Cl-, HOCl | pyrolysis | [31] |
| CHCl3 | 0.028 | 205 | 200 | 286 | 0.08 | nat. to 3.5 | --- | C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CHCl3 | 0.043 ± 0.005 | 20 | 1590 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CHCl3 | 0.028 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| CHCl3 | 0.0248 ± 0.0008 | 20 | 84 | 298 | 0.1843.75 | 5.4-5.8unbf | air | not studied | pyrolysis | [43] |
| CH2Cl2 | 0.016 | 205 | 200 | 286 | 0.08 | nat. to 3.5 | --- | C2Cl4, Cl-, HOCl HCl, CO2 | pyrolysis | [30] |
| CH2Cl2 | 0.033 ± 0.002 | 20 | 2471 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| CH2Cl2 | 0.0393 ± 0.0027 | --- | 1176-11764 | 288-293 | 0.23 | nat. to 3 | --- | not studied, only HCl | not commented | [39] |
| CH2Cl2 | 0.016 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,2-DCA | 0.021 ± 0.003 | 20 | 1364 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.021 ± 0.001 | 20 | 1255 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.0455 ± 0.0011 | 520 | 5 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0364 ± 0.0012 | 520 | 50 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0345 ± 0.0013 | 520 | 250 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0232 ± 0.0008 | 520 | 500 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0230 ± 0.0009 | 520 | 1000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0208 ± 0.0009 | 520 | 2000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| CH2Cl2 | 0.016 | 205 | 150 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,2-DCA | 0.021 ± 0.003 | 20 | 1364 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.021 ± 0.001 | 20 | 1255 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis | [37] |
| TCE | 0.0455 ± 0.0011 | 520 | 5 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0364 ± 0.0012 | 520 | 50 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0345 ± 0.0013 | 520 | 250 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0232 ± 0.0008 | 520 | 500 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0230 ± 0.0009 | 520 | 1000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0208 ± 0.0009 | 520 | 2000 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0127 ± 0.0008 | 520 | 3440 | 302 | 0.06 | 7 | air | not studied | pyrolysis + radical attack | [50] |
| TCE | 0.0617 ± 0.0012 | 520 | 1670 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.0389 ± 0.0011 | 520 | 3440 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.0194 ± 0.0014 | 520 | 6680 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| TCE | 0.026 ± 0.004 | 20 | 3340 | 305 | 0.43 | 7-2.9 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.033 ± 0.005 | 520 | 3340 | 302 | 0.095 | 7-2.4 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.031 ± 0.003 | 520 | 3340 | 302 | 0.095 | 4.7 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.039 ± 0.003 | 520 | 3340 | 302 | 0.095 | 7 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.043 ± 0.004 | 520 | 3340 | 302 | 0.095 | 10 | air | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| TCE | 0.062 ± 0.0034 | 520 | 3340 | 302 | 0.095 | --- | Ar | C2Cl2, C2Cl4 C2HCl, C4Cl2, C4HCl3, C4Cl4, C4HCl5, C4Cl6 | pyrolysis | [38] |
| 1,1,1-TCA | 0.046 ± 0.003 | 20 | 824 | 293-298 | 0.23 | nat. | --- | not detected | pyrolysis + radical attack | [37] |
| 1,1,1-TCA | 0.0324 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0528 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0582 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,1-TCA | 0.0378 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.0126 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.192 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.192 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 1,1,2-TCA | 0.0114 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0246 | 205 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0378 | 358 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.0438 | 618 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| C2Cl6 | 0.027 | 1078 | 1 | 283 | 0.08 | nat. | Ar | not studied | pyrolysis | [36] |
| 2Cl-PhOH | 0.0048 ± 0.0004 | 20 | 83 | 306 | 0.5 | nat. to 5.7 | air | chlorohydroquinone, catechol, 3-chlorocatechol | solution bulk at <75 μM gas bubble/liquid interface at >75 μM | [57] |
| 3Cl-PhOH | 0.0044 ± 0.0005 | 20 | 78 | 306 | 0.5 | nat. to 5.4 | air | chlorohydroquinone, 4-chlorocatechol, 3-chlorocatechol | solution bulk at low C gas bubble/liquid interface at high C | [57] |
| 4Cl-PhOH | 0.0033 ± 0.0002 | 20 | 75 | 306 | 0.5 | nat. to 5.1 | air | hydroquinone, 4-chlororesorcinol, 4-chlorocatechol, Cl- | solution bulk at low C gas bubble/liquid interface at high C | [57] |
| 4Cl-PhOH | 0.00156 | 20 | 0.5 | 293 | 0.12 | nat. | air | 4-chlorocatechol, hydroquinone, Cl- formic, oxalic acid, CO2 | radical attack | [52] |
| 4Cl-PhOH | 0.0291 | 500 | 0.5 | 293 | 0.12 | nat. | air | 4-chlorocatechol, hydroquinone, Cl- formic, oxalic acid, CO2 | radical attack | [52] |
| 4Cl-PhOH | 0.0039 | 1.7 MHz | 27 | 298 | 0.18 | --- | --- | C6H5O3Cl | pyrolysis | [59] |
| 4Cl-PhOH | 0.0017 ± 0.00008 | 20 | 100 | 298±5 | 0.24 | --- | O2 | 4-chlorocatechol | pyrolysis + radical attack | [55] |
| 4Cl-PhOH | 0.021 ± 0.0005 | 515 | 100 | 298±5 | 0.24 | --- | O2 | 4-chlorocatechol | pyrolysis + radical attack | [55] |
| 2,4- Cl-PhOH | 0.0189 | 360 | 800-950 | 293±5 | 0.2 | 4 | air | ---- | ---- | [48] |
| 2,3,5- Cl-PhOH | 0.0111 | 360 | 800-950 | 293±5 | 0.2 | 4 | air | ------ | ------ | [61] |
| ClC6H5 | 0.0072 | 20 | 0.5 | 293 | 0.12 | nat. | air | HCl, CO, CO2, C2H2, soot | pyrolysis | [52] |
| ClC6H5 | 0.0486 | 500 | 0.5 | 293 | 0.12 | nat. | air | HCl, CO, CO2, C2H2, soot | pyrolysis | [52] |
| ClC6H5 | 0.0177 ± 0.0008 | 520 | 1720 | 302 | 0.095 | 7 | air | main by-products: methane, acetlyene, butenyne, butadiyne, benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0184 ± 0.0007 | 520 | 1720 | 302 | 0.095 | 4.7 | air | main by-products: methane, acetlyene, butenyne, butadiyne, benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0181 ± 0.0004 | 520 | 1720 | 302 | 0.095 | 10 | air | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0190 ± 0.0007 | 520 | 1720 | 302 | 0.095 | unbf | air | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0285 ± 0.00108 | 520 | 1720 | 302 | 0.095 | 7 | Ar | main by-products: methane, acetlyene, butenyne, butadiyne and benzene, phenylacetylene, styrene, C7H7Cl, chlorophenol | pyrolysis | [46] |
| ClC6H5 | 0.0243 ± 0.0007 | 520 | 860 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0176 ± 0.0006 | 520 | 1720 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0057 ± 0.0005 | 520 | 3440 | 302 | 0.095 | 7 | --- | not studied | pyrolysis | [50] |
| ClC6H5 | 0.0160 ± 0.0004 | 520 | 500 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0127 ± 0.0004 | 520 | 1000 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0065 ± 0.0003 | 520 | 2000 | 302 | 0.06 | --- | --- | benzene, phenylacetylene, styrene, biphenyl, byphenylene, dihalogenated benzene, monoahlogenated phenol, 1-halo-4-ethynyl-benzene and monohalogenated bypehnyl | pyrolysis | [50] |
| ClC6H5 | 0.0365 ± 0.0027 | 520 | 1 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0335 ± 0.0009 | 520 | 5 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0329 ± 0.0011 | 520 | 25 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0302 ± 0.0010 | 520 | 50 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0190 ± 0.0012 | 520 | 100 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0154 ± 0.0006 | 520 | 250 | 302 | 0.06 | 7 | air | ----- | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.0025 ± 0.0001 | 520 | 3440 | 302 | 0.06 | 7 | air | ------ | pyrolysis + radical attack | [50] |
| ClC6H5 | 0.020 ± 0.001 | 500 | 500 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| ClC6H5 | 0.035 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| ClC6H5 | 0.026 ± 0.001 | 500 | 200 | 293 | 0.1 | --- | air | CO, C2H2, CH4, CO2, soot | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.054 ± 0.001 | 500 | 200 | 293 | 0.2 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.009 ± 0.001 | 500 | 200 | 293 | 0.04 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.022 ± 0.001 | 500 | 500 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.036 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1,4-Cl2C6H4 | 0.028 ± 0.001 | 500 | 300 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| pCBA | ----- | 20 | 5, 10, 20 | 298±2 | 0.095 | --- | air | ----- | radical attack | [53] |
| 1Cl-naphthalene | 0.036 ± 0.001 | 500 | 40 | 293 | 0.1 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1Cl-naphthalene | 0.038 ± 0.001 | 500 | 200 | 293 | 0.2 | --- | air | not studied | pyrolysis + radical attack | [49] |
| 1Cl-naphthalene | 0.008 ± 0.001 | 500 | 200 | 293 | 0.04 | --- | air | not studied | pyrolysis + radical attack | [49] |
| ClC6H5CH3 | 0.02637 ±0.00138 | 520 | 680 | 302 | 0.097 | --- | --- | ------ | pyrolysis | [44] |
| ClC6H5CH3 | 0.02923 ±0.00225 | 520 | 340 | 302 | 0.097 | --- | --- | ------- | pyrolysis | [44] |
| ClC6H5CH3 | 0.04145 ±0.00223 | 520 | 170 | 302 | 0.097 | --- | --- | --------- | pyrolysis | [44] |
| 2-PCB | 0.123 ± 0.002 | 20 | 4.6 | 288 | 0.24340.1/1.3240.1/165 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 3.6 10-3 ± 7.8 10-5 | 205 | 4.6 | 288 | 0.243160/29160/400 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 8.5 10-3 ± 4.2 10-5 | 358 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 6.9 10-3 ± 1.4 10-5 | 618 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2-PCB | 4.1 10-3 ± 1.2 10-5 | 1071 | 4.6 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 4-PCB | 0.096 ± 0.002 | 20 | 5.4 | 288 | 0.243 | --- | Ar | biphenyl, toluene, ethylbenzene, diethylbiphenyl, dibutenylbiphenyl phenol, propylphenol, di-tert-butylphenol, cyclohexenyl diphenol | pyrolysis + radical attack | [54] |
| 2,4,5-PCB | 0.156 ± 0.002 | 20 | 0.076 | 288 | 0.243 | --- | Ar | ethyl benzene, diethylbiphenyl, trichlorophenol | pyrolysis + radical attack | [54] |
| Alachlor | 0.0016 | 20 | 370 | 291 | 0.25 | --- | Air | MW=149, MW=225, MW=241, MW=253, MW=285 | pyrolysis + radical attack | [62] |
| Alachlor | 0.0375 | 300 | 370 | 291 | 0.25 | --- | Ar | MW=149, MW=225, MW=241, MW=253, MW=285 | pyrolysis + radical attack | [62] |
| MCPA | 0.0066 | 500 | 500 | 298 | 0.214 | --- | N2 | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.047 | 500 | 500 | 298 | 0.214 | --- | O2 | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.029 | 500 | 500 | 298 | 0.214 | --- | Ar | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
| MCPA | 0.042 | 500 | 500 | 298 | 0.214 | --- | Air | 4-chloro-2-methylphenol | pyrolysis + radical attack | [63] |
Alachlor: 2-chloro-2'6'-diethyl-N-(mehoxymethyl)acetanilideMCPA:(4-chloro-2-methylphenoxy)acetic acid;pCBA: p-chlorobenzoic acidnat.: naturalunbf : unbuffered
Acknowledgements
J.G.-G. and M.D.E. thank Caja de Ahorros del Mediterráneo. J.G.-G. thanks the Ministry of Industry, Tourism and Trade for its financial support under contract DEX-560620-2008-135. J.G.-G., M.D.E. and I.T. thank Generalidad Valenciana for its financial support under projects AORG09/051 and ACOMP09/128. J.G.-G. and I.T. also thank Generalidad Valenciana for its financial support under grant FPA/2009/024. O.L. thanks Generalidad Valenciana (project AINV07/044) and Universidad de Alicante (project INV08-31) for funding scientific exchange missions in Alicante. Authors thank Mary Thompson for her help with the English edition.
References and Notes
- Vogel, T.M.; Criddle, C.S.; McCarty, P.L. Transformations of halogenated aliphatic compounds. Environ. Sci. Technol. 1987, 21, 722–737. [Google Scholar] [CrossRef]
- Hitchman, M.L.; Spackman, R.A.; Ross, N.C.; Agra, C. Disposal methods for chlorinated aromatic waste. Chem. Soc. Rev. 1995, 24, 423–430. [Google Scholar]
- Entezari, M.H.; Mostafai, M.; Sarafraz-yazdi, A. A combination of ultrasound and a bio-catalyst: removal of 2-chlorophenol from aqueous solution. Ultrason. Sonochem. 2005, 12, 137–141. [Google Scholar] [CrossRef]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 1. Combinative and hybrid sonophotochemical oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 3409–3420. [Google Scholar]
- Shannon, M.A.; Bohn, P.W.; Elimelech, M.; Georgiadis, J.G.; Mariñas, B.J.; Mayes, A.M. Science and technology for water purification in the coming decades. Nature 2008, 452, 301–310. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Griffing, V. The Chemical effects of ultrasonics. J. Chem. Phys. 1952, 20, 939–942. [Google Scholar]
- Fitzgerald, M.E.; Griffing, V.; Sullivan, J. Chemical effects of ultrasonics-“Hot Spot” Chemistry. J. Chem. Phys. 1956, 25, 926–933. [Google Scholar] [CrossRef]
- Sehgal, C.M.; Wang, S.Y. Threshold intensities and kinetics of sonoreaction of thymine in aqueous solutions at low ultrasonic intensities. J. Am. Chem. Soc. 1981, 103, 6606–6611. [Google Scholar]
- Levy, S.V.; Low, C.R.R. Ultrasound in Synthesis; Springer-Verlag: London, UK, 1989. [Google Scholar]
- Kimura, T.; Sakamoto, T.; Leveque, J.M.; Sohmiya, H.; Fujita, M.; Ikeda, S.; Ando, T. Standarization of ultrasonic power for sonochemical reaction. Ultrason. Sonochem. 1996, 3, S157–S161. [Google Scholar] [CrossRef]
- Weissler, A.; Cooper, H.W.; Snyder, S. Chemical effect of ultrasonic waves: oxidation of potassium iodide solution by carbon tetrachloride. J. Am. Chem. Soc. 1950, 72, 1769–1775. [Google Scholar] [CrossRef]
- Jennings, B.H.; Townsend, S.N. The sonochemical reactions of carbon tetrachloride and chloroform in aqueous suspension in an inert atmosphere. J. Phys. Chem. 1961, 65, 1574–1579. [Google Scholar] [CrossRef]
- Paniwnyk, L.; Cai, H.; Albu, S.; Mason, T.J.; Cole, R. The enhancement and scale up of the extraction of anti-oxidants from Rosmarinus officinalis using ultrasound. Ultrason. Sonochem. 2009, 16, 287–292. [Google Scholar] [CrossRef]
- Mason, T.J.; Cordemans, E. Practical considerations for process optimization in Synthetic Organic Sonochemistry. In Synthetic Organic Sonochemistry; Luche, J.L., Ed.; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Grönroos, A.; Kyllönen, H.; Korpijärvi, K.; Pirkonen, P.; Paavola, T.; Jokela, J.; Rintala, J. Ultrasound assisted method to increase soluble chemical oxygen demand (SCOD) of sewage sludge for digestion. Ultrason. Sonochem. 2005, 12, 115–120. [Google Scholar] [CrossRef]
- Kyllönen, H.; Pirkonen, P.; Hintikka, V.; Parvinen, P.; Grönroos, A.; Sekki, H. Ultrasonically aided mineral processing technique for remediation of soil contaminated by heavy metals. Ultrason. Sonochem. 2004, 11, 211–216. [Google Scholar] [CrossRef]
- Gallego-Juárez, J.A.; Rodríguez Corral, G.; Riera, E.; Vázquez-Martinez, F.; Acosta-Aparicio, V.M.; Campos-Pozuelo, C. Development of industrial models of high power stepped-plate sonic and ultrasonic transducer for use in fluids. IEEE Ultrasonic Sympos. Proc. 2001, 571–578. [Google Scholar]
- Hua, I.; Höchemer, R.; Hoffmann, M.R. Sonochemical degradation of p-Nitrophenol in a Parallel-Plate near-field acoustical processor. Environ. Sci. Technol. 1995, 29, 2790–2795. [Google Scholar] [CrossRef]
- Xie, R.; Xing, Y.; Ghani, Y.A.; Ooi, K.-E.; Ng, S.-W. Full-scale demostration of an ultrasonic disintegration technology in enhancing anaerobic digestion of mixed primary and thickened secondary sewage sludge. J. Environ. Eng. Sci. 2007, 6, 533–541. [Google Scholar]
- Nickel, K.; Neis, U. Ultrasonic disintegration of biosolids for improved biodegradation. Ultrason. Sonochem. 2007, 14, 450–455. [Google Scholar] [CrossRef]
- Khanal, S.K.; Grewell, D.; Sung, S.; van Leeuwen, J. Ultrasound applications in wastewater sludge pretreatment: A review. Crit. Rev. Env. Sci. Technol. 2007, 37, 277–313. [Google Scholar] [CrossRef]
- Appels, L.; Bayens, J.; Degrève, J.; Dewil, R. Principles and potential of the anaerobic digestion of waste-activated sludge. Prog. Energy Combust. Sci. 2008, 34, 755–781. [Google Scholar] [CrossRef]
- Laborde, J.L.; Bouyer, C.; Caltagirone, J.P.; Gérard, A. Acoustic cavitation field prediction at low and high frequency ultrasound. Ultrasonics 1998, 36, 581–587. [Google Scholar] [CrossRef]
- Servant, G.; Caltagirone, J.P.; Gérard, A.; Laborde, J.L.; Hita, A. Numerical simulation of cavitation bubble dynamics induced by ultrasound waves in a high frequency reactor. Ultrason. Sonochem. 2000, 7, 217–227. [Google Scholar] [CrossRef]
- Sáez, V.; Frías-Ferrer, A.; Iniesta, J.; González-García, J.; Aldaz, A.; Riera, E. Characterization of a 20 kHz sonoreactor. Part I: Analysis of mechanical effects by classical and numerical methods. Ultrason. Sonochem. 2005, 12, 59–65. [Google Scholar]
- Mandroyan, A.; Doche, M.L.; Hihn, J.Y.; Viennet, R.; Bailly, Y.; Simonin, L. Modification of the ultrasound induced activity by the presence of an electrode in a sono-reactor working at two low frequencies (20 and 40 kHz). Part II: Mapping flow velocities by particle image velocimetry (PIV). Ultrason. Sonochem. 2009, 16, 97–104. [Google Scholar]
- Sáez, V.; Frías-Ferrer, A.; Iniesta, J.; González-García, J.; Aldaz, A.; Riera, E. Characterization of a 20 kHz sonoreactor. Part II: Analysis of chemical effects by classical and electrochemical methods. Ultrason. Sonochem. 2005, 12, 67–72. [Google Scholar]
- Chendke, P.K.; Fogler, H.S. Sonoluminescence and Sonochemical reactions of aqueous carbon tetrachloride solutions. J. Phys. Chem. 1983, 87, 1362–1369. [Google Scholar] [CrossRef]
- Hung, H.M.; Hoffmann, M.R. Kinetics and Mechanism of the sonolytic degradation of chlorinated hydrocarbons: frequency effects. J. Phys. Chem. A 1999, 103, 2734–2739. [Google Scholar] [CrossRef]
- Hua, I.; Hoffmann, M.R. Kinetics and Mechanism of the sonolytic degradation of CCl4 intermediates and byproducts. Environ. Sci. Technol. 1996, 30, 864–871. [Google Scholar] [CrossRef]
- Rajan, R; Kumar, R.; Gandhi, K.S. Modelling of sonochemical decomposition of CCl4 in aqueous solution. Environ. Sci. Technol. 1998, 32, 1128–1133. [Google Scholar]
- Spurlock, L.A.; Reifsneider, S.B. Chemistry of ultrasound. I. A reconsideration of first principles and the aplications to a Dialkyl sulfide. J. Am. Chem. Soc. 1970, 92, 6112–6117. [Google Scholar]
- Francony, A.; Pétrier, C. Sonochemical degradation of carbon tetrachloride in aqueous solution at two frequencies: 20 kHz and 500 kHz. Ultrason. Sonochem. 1996, 3, S77–S82. [Google Scholar] [CrossRef]
- Petrier, C.; Francony, A. Incidence of wave-frequency on the reaction rates during ultrasonic wastewater treatment. Wat. Sci. Tech. 1997, 35, 175–180. [Google Scholar] [CrossRef]
- Colussi, A.J.; Hung, H.-M.; Hoffmann, M.R. Sonochemical degradation rates of volatiles solutes. J. Phys. Chem. A. 1999, 103, 2696–2699. [Google Scholar]
- Bhatnagar, A.; Cheung, H.M. Sonochemical destruction of chlorinated C1 and C2 volatile organic compounds in dilute aqueous solution. Environ. Sci. Technol. 1994, 28, 1481–1486. [Google Scholar] [CrossRef]
- Drijvers, D.; De Baets, R.; De Visscher, A.; van Langenhove, H. Sonolysis of trichloroethylene in aqueous solution: volatile organic intermediates. Ultrason. Sonochem. 1996, 3, S83–S90. [Google Scholar] [CrossRef]
- Cheung, H.M.; Bhatnagar, A.; Jansen, G. Sonochemical destruction of Chlorinated hydrocarbons in dilute aqueous solution. Environ. Sci. Technol. 1991, 25, 1510–1512. [Google Scholar] [CrossRef]
- Inazu, K.; Nagata, Y.; Maeda, Y. Decomposition of Chlorinated hydrocarbons in aqueous solutions by ultrasonic irradiation. Chem. Lett. 1993, 22, 57–60. [Google Scholar] [CrossRef]
- Gaddam, K.; Cheung, H.-M. Effects of pressure, temperature, and pH on the sonochemical destruction of 1,1,1-trichloroethane in dilute aqueous solution. Ultrason. Sonochem. 2001, 8, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.M.; Kurup, S. Sonochemical destruction of CFC 11 and CFC 13 in dilute aqueous solution. Environ. Sci. Technol. 1994, 28, 1619–1622. [Google Scholar] [CrossRef]
- Shemer, H.; Narkis, N. Sonochemical removal of trihalomethanes from aqueous solutions. Ultrason. Sonochem. 2005, 12, 495–499. [Google Scholar] [CrossRef]
- De Visscher, A.; Van Eenoo, P.; Drijvers, D.; Van Langenhove, H. Kinetics Model for the sonochemical degradation of monocyclic aromatic compounds in aqueous solution. J. Phys. Chem. 1996, 100, 11636–11642. [Google Scholar]
- Kruus, P.; Burk, R.C.; Entezari, M.H.; Otson, R. Sonication of aqueous solutions of chlorobenzene. Ultrason. Sonochem. 1997, 4, 229–233. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Vervaet, K. Sonolysis of chlorobenzene in aqueous solution: organic intermediates. Ultrason. Sonochem. 1998, 5, 13–19. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Herrygers, V. Sonolysis of fluoro-, chloro-, bromo- and iodobenzene: a comparative study. Ultrason. Sonochem. 2000, 7, 87–95. [Google Scholar]
- Tiehm, A.; Kohnagel, I.; Neis, U. Removal of chlorinated pollutants by a combination of ultrasound and biodegradation. Water. Sci. Technol. 2001, 43, 297–303. [Google Scholar]
- Jiang, Y.; Pétrier, C.; David Waite, T. Kinetics and mechanisms of ultrasonic degradation of volatile chlorinated in aqueous solutions. Ultrason. Sonochem. 2002, 9, 317–323. [Google Scholar] [CrossRef]
- Drijvers, D.; van Langenhove, H.; Nguyen Thi Kim, L.; Bray, L. Sonolysis of an aqueous mixture of trichloroethylene and chlorobenzene. Ultrason. Sonochem. 1999, 6, 115–121. [Google Scholar] [CrossRef]
- Dewulf, J.; van Langenhove, H.; De Visscher, A.; Sabbe, S. Ultrasonic degradation of trichloroethylene and chlorobenzene at micromolar concentrations: kinetics and modelling. Ultrason. Sonochem. 2001, 8, 143–150. [Google Scholar] [CrossRef]
- Petrier, C.; Jiang, Y.; Lamy, M.-F. Ultrasound and Environment: sonochemical destruction of chloroaromatic derivatives. Environ. Sci. Technol. 1998, 32, 1316–1318. [Google Scholar] [CrossRef]
- Nam, S.-N.; Han, S.-K.; Kang, J.-W.; Choi, H. Kinetics and mechanisms of the sonolytic destruction of non-volatile organic compounds: investigation of the sonochemical reaction zone using several OH· monitoring techniques. Ultrason. Sonochem. 2003, 10, 139–147. [Google Scholar]
- Zhang, G.; Hua, I. Cavitation chemistry of polychlorinated biphenyls: decomposition mechanisms and rates. Environ. Sci. Technol. 2000, 34, 1529–1534. [Google Scholar] [CrossRef]
- Weavers, L.K.; Ling, F.H.; Hoffmann, M.R. Aromatic compound degradation in water using a combination of sonolysis and ozonolysis. Environ. Sci. Technol. 1998, 32, 2727–2733. [Google Scholar] [CrossRef]
- Petrier, C.; Micolle, M.; Merlin, G.; Luche, J.-L.; Reverdy, G. Characteristics of pentachlorophenate degradation in aqueous solution by means of ultrasound. Environ. Sci. Technol. 1992, 26, 1639–1642. [Google Scholar]
- Serpone, N.; Terzian, R.; Hidaka, H.; Pelizzetti, E. Ultrasonic induced dehalogenation and oxidation of 2-, 3-,and 4-chlorophenol in air-equilibrated aqueous media. Similarities with irradiated semiconductor particulates. J. Phys. Chem. 1994, 98, 2634–2640. [Google Scholar]
- Petrier, C.; David, B.; Laguian, S. Ultrasonic degradation at 20 kHz and 500 kHz of atrazine and pentachlorophenol in aqueous solution: preliminary results. Chemosphere 1996, 32, 1709–1718. [Google Scholar] [CrossRef]
- Gondrexon, N.; Renaudin, V.; Petrier, C.; Boldo, P.; Bernis, A.; Gonthier, Y. Degradation of pentachlorophenol aqueous solutions using a continuous flow ultrasonic reactor: experimental performance and modelling. Ultrason. Sonochem. 1999, 5, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Chen, Y.; Wu, M.; Wang, H.; Yingwu, Y.; Lü, Z. Sonochemistry of degrading p-chlorophenol in water by high frequency ultrasound. Ultrason. Sonochem. 2004, 11, 43–46. [Google Scholar] [CrossRef]
- Tiehm, A.; Neis, U. Ultrasonic dehalogenation and toxicity reduction of trichlorophenol. Ultrason. Sonochem. 2005, 12, 121–125. [Google Scholar] [CrossRef]
- Wayment, D.G.; Casadonte, D.J., Jr. Frequency effect on the sonochemical remediation of alachlor. Ultrason. Sonochem. 2002, 9, 251–257. [Google Scholar] [CrossRef]
- Kojima, Y.; Fujita, T.; Ona, E.P.; Matsuda, H.; Koda, S.; Tanahashi, N.; Asakura, Y. Effects of disolved gas species on ultrasonic degradation of (4-chloro-2-methylphenoxy) acetic acid (MCPA) in aqueous solution. Ultrason. Sonochem. 2005, 12, 359–365. [Google Scholar] [CrossRef]
- Pétrier, C.; Lamy, M.-F.; Francony, A.; Renaudin, V.; Gondrexon, N.; David, B.; Benahcene, A. Sonochemical degradation of phenol in dilute aqueous solutions: comparison of the reaction rates at 20 and 487 kHz. J. Phys. Chem. 1994, 98, 10514–10520. [Google Scholar] [CrossRef]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Oxidation of hydrogen sulfide in aqueous solution by ultrasonic irradiation. Environ. Sci. Technol. 1992, 26, 2420–2428. [Google Scholar] [CrossRef]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Ultrasonic irradiation of p-nitrophenol in aqueous solution. J. Phys. Chem. 1991, 95, 3630–3638. [Google Scholar] [CrossRef]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Sonolysis of hydrocarbons in aqueous solution. Radiat. Phys. Chem. 1990, 36, 511–516. [Google Scholar]
- Kotronarou, A.; Mills, G.; Hoffmann, M.R. Decomposition of parathion in aqueous solution by ultrasonic irradiation. Environ. Sci. Technol. 1992, 26, 1460–1462. [Google Scholar] [CrossRef]
- Kondo, T.; Krishna, C.M.; Riesz, P. Sonolysis of concentrated aqueous solutions of nonvolatile solutes: spin-trapping evidence for free radicals formed by pyrolysis. Radiat. Res. 1998, 118, 211–229. [Google Scholar]
- Suslick, K.S.; Didenko, Y.; Fang, M.M.; Hyeon, T.; Kolbeck, K.J.; McNamara III, W.B.; Mdeleleni, M.M.; Wong, M. Acoustic cavitation and its chemcal consequences. Phil. Trans. R. Soc. Lond. A. 1999, 357, 335–353. [Google Scholar] [CrossRef]
- Petrier, C.; Jeunet, A.; Luche, J.-L.; Reverdy, G. Unexpected frequency effects on the rate of oxidative processes induced by ultrasound. J. Am. Chem. Soc. 1992, 114, 3148–3150. [Google Scholar] [CrossRef]
- Hua, I.; Hoffmann, M.R. Optimization of ultrasonic irradiation as an advanced oxidation technology. Environ. Sci. Technol. 1997, 31, 2237–2243. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. Engineering design method for cavitational reactors: I.Sonochemical reactors. Aiche J. 2000, 46, 372–379. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Hua, I.; Höchemer, R. Application of ultrasonic irradiation for the degradation of chemical contaminants in water. Ultrason. Sonochem. 1996, 3, S163–S172. [Google Scholar] [CrossRef]
- Zheng, W.; Maurin, M.; Tarr, M.A. Enhancement of sonochemical degradation of phenol using hydrogen atom scavengers. Ultrason. Sonochem. 2005, 12, 313–317. [Google Scholar] [CrossRef]
- Suslick, K.S.; Hammerton, D.A.; Cline, R.E. The sonochemical hot spot. J. Am. Chem. Soc. 1986, 108, 5641–5642. [Google Scholar] [CrossRef]
- Lepoint, T.; Mullie, F. What exactly is cavitation chemistry? Sonochem. 1994, 1, S13–S22. [Google Scholar] [CrossRef]
- Toegel, R.; Gompf, R.; Pecha, R.; Lohse, D. Does water vapor prevent upscaling sonoluminescence? Phys. Rev. Lett. 2000, 85, 3165–3168. [Google Scholar]
- Petrier, C.; Francony, A. Ultrasonic waste-water treatment: incidence of ultrasonic frequency on the rate of phenol and carbon tetrachloride degradation. Ultrason. Sonochem. 1997, 4, 295–300. [Google Scholar] [CrossRef]
- Mettin, R. Bubble structures in acoustic cavitation. In Bubble and Particle Dynamics in Acoustic Fields: Modern Trends and Applications; Doinikov, A.A., Ed.; Research Signpost: Kerala, India, 2005; pp. 1–36. [Google Scholar]
- Mettin, R. From a single bubble to bubble structures in acoustic cavitation. In Oscillations, Waves and Interactions; Kurz, T., Parlitz, U., Kaatze, U., Eds.; Universitätsverlag Göttingen: Göttingen, Germany, 2007; pp. 171–198. [Google Scholar]
- Yasui, K.; Tuziuti, T.; Kozuka, T.; Towata, A.; Iida, Y. Influence of ambient bubble radius on sonoluminescence and sonochemical reactions. In Revista de Acústica (Special Issue) Volume 38, 19th International Congress on Acoustics, Madrid, Spain, 2-7 September 2007; NLA-01-013. pp. 1–6.
- Mettin, R. Bubble motion and instability. In Proceedings of 11th Meeting of the European Society of Sonochemistry, La Grande-Motte, France, 1-5 June 2008; p. 95.
- Calvisi, M.L.; Lindua, O.; Blake, J.R.; Szeri, A.J. Shape stability and violent collapse of microbubbles in acoustic traveling waves. Phys. Fluids 2007, 19, 047101. [Google Scholar] [CrossRef]
- Storey, B.D.; Szeri, A.J. Water vapour, sonoluminescence and sonochemistry. Proc. R. Soc. London, Ser. A 2000, 456, 1685–1709. [Google Scholar]
- Kang, J.-W.; Hung, H.-M.; Lin, A.; Hoffmann, M.R. Sonolytic destruction of methyl tert-butyl ether by ultrasonic irradiation: the role of O3, H2O2, frequency and power density. Environ. Sci. Technol. 1999, 33, 3199–3205. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A. Chemical action of pulsed ultrasound: observation of an unprecedented intensity effect. J. Phys. Chem. 1990, 94, 3625–3628. [Google Scholar] [CrossRef]
- Gogate, P.R.; Shirgaonkar, I.Z.; Sivakurmar, M.; Senthilkumar, P.; Vichare, N.P.; Pandit, A.B. Cavitation reactors: efficiency assessment using a model reaction. Aiche J. 2001, 47, 2526–2538. [Google Scholar] [CrossRef]
- Ondruska, B.; Lifka, J.; Hoffmann, J. Aquasonolysis of Ether - Effect of frequency and acoustic power of ultrasound. Chem. Eng. Tech. 2000, 23, 588–592. [Google Scholar] [CrossRef]
- Entezari, M.H.; Kruus, P. Effect of frequency on sonochemical reactions II. Temperature and intensity effects. Ultrason. Sonochem. 1996, 3, 19–24. [Google Scholar] [CrossRef]
- Apfel, R.E. Acoustic cavitation prediction. J. Acoust. Soc. Am. 1981, 69, 1624–1633. [Google Scholar]See also the chapter 4 The forced bubble in The Acoustic Bubble by T. G. Leighton; Academic Press: London, UK, 1994.
- Storey, B.D.; Szeri, A.J. A reduced model of cavitation physics for use in sonochemistry. Proc. R. Soc. London, Ser. A 2001, 457, 1685–1700. [Google Scholar]
- Mark, G.; Tauber, A.; Laupert, R.; Schuchmann, H.; Schulz, D.; Mues, A.; von Sonntag, C. OH - radical formation by ultrasound in aqueous solution - Part II: Terephthalate and Fricke dosimetry and the influence of various conditions on the sonolytic yield. Ultrason. Sonochem. 1998, 5, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Vanhille, C.; Campos-Pozuelo, C. Nonlinear ultrasonic waves in bubbly liquids with unhomogeneous bubble distribution: numerical experiments. Ultrason. Sonochem. 2009, 19, 669–685. [Google Scholar] [CrossRef]
- Louisnard, O. Nonlinear attenuation of sound waves by inertial cavitation bubbles. In To appear in Physics Procedia, International Congress on Ultrasonics, Santiago de Chile, Chile, 11-17 January 2009.
- Schmitt, F.O.; Johnson, C.H.; Olson, A.R. Oxidations promoted by ultrasonic radiation. J. Am. Chem. Soc. 1929, 51, 370–375. [Google Scholar] [CrossRef]
- Makino, K.; Mossoba, M.; Riesz, P. Chemical effects of ultrasound on aqueous solution. Formation of hydroxyl radicals and hydrogen atoms. J. Phys. Chem. 1983, 87, 1369–1377. [Google Scholar]
- Riesz, P.; Berdhal, D.; Christman, C.L. Free radical generation by ultrasound in aqueous and nonaqueous solutions. Environ. Health Perspect. 1985, 64, 233–252. [Google Scholar] [CrossRef]
- Fischer, C.H.; Hart, E.J.; Henglein, A. H/D Isotope exchange in the HD-H2O system under the influence of ultrasound. J. Phys. Chem. 1986, 90, 3059–3060. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A.; Ibañez, F. Radical Scavenging in the sonolysis of aqueous solutions of I-, Br- and N3-. J. Phys. Chem. 1991, 95, 6044–6047. [Google Scholar] [CrossRef]
- Fischer, Ch.-H.; Hart, E.J.; Henglein, A. Ultrasonic irradiation of water in the presence of 18,18O2: Isotope exchange and isotopic distribution of H2O2. J. Phys. Chem. 1986, 90, 1954–1956. [Google Scholar] [CrossRef]
- Hart, E.J.; Henglein, A. Free radical and free atom reactions in the sonolysis of aqueous iodide and formate solutions. J. Phys. Chem. 1985, 89, 4342–4347. [Google Scholar] [CrossRef]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Isotopic exchange in the sonolysis of aqueous solutions containing 14,14N2 and 15,15N2. J. Phys. Chem. 1986, 90, 5989–5991. [Google Scholar] [CrossRef]
- Misik, V.; Riesz, P. Recent application of EPR and spin trapping to sonochemical studies of organic liquids and aqueous solutions. Ultrason. Sonochem. 1996, 3, S173–S186. [Google Scholar]
- Kondo, T.; Krishna, C.M.; Riesz, P. Pyrolysis radicals formed by ultrasound in aqueous solutions of nucleotides: a spin trapping study. Int. J. Radiat. Biol. 1990, 57, 23–33. [Google Scholar] [CrossRef]
- Guo, A.; Zheng, Z.; Zheng, S.; Hu, W.; Feng, R. Effects of various sono-oxidation parameters on the removal of aqueous 2,4-dinitrophenol. Ultrason. Sonochem. 2005, 12, 461–465. [Google Scholar] [CrossRef]
- Spinks, J.W.T.; Woods, R.J. An introduction to radiation chemistry, 3rd ed.; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Hart, E.J.; Fischer, C.-H.; Henglein, A. Pyrolysis of acetylene in sonolytic cavitation bubbles in aqueous solution. J. Phys. Chem. 1990, 94, 284–290. [Google Scholar] [CrossRef]
- Alegria, A.E.; Lion, Y.; Kondo, T.; Riesz, P. Sonolysis of aqueous surfactant solutions. Probing the interfacial region of cavitation bubbles by spin trapping. J. Phys. Chem. 1989, 93, 4908–4913. [Google Scholar]
- Krishna, C.M.; Lion, Y.; Kondo, T.; Riesz, P. Thermal decomposition of methanol in the sonolysis of methanol-water mixtures. Spin-trapping evidence for isotope exchange reactions. J. Phys. Chem. 1987, 91, 5847–5850. [Google Scholar]
- Hart, E.J.; Fischer, Ch.-H.; Henglein, A. Isotopic exchange in the sonolysis of aqueous solutions containing D2 and CH4. J. Phys. Chem. 1987, 91, 4166–4169. [Google Scholar] [CrossRef]
- Gutierrez, M.; Henglein, A.; Fischer, Ch.-H. Hot spot kinetics of the sonolysis of aqueous acetate solutions. Int. J. Radiat. Biol. 1986, 50, 313–321. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T.; Krishna, C.M. Free radical formation by ultrasound in aqueous solutions. A spin trapping study. Free Radical Res. Commun. 1990, 10, 27–35. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T. Free radical formation induced by ultrasound and its biological implications. Free Radical Biol. Med. 1992, 13, 247–270. [Google Scholar] [CrossRef]
- Riesz, P.; Kondo, T.; Krishna, C.M. Sonochemistry of volatile and non-volatile solutes in aqueous solutions: e.p.r. and spin trapping studies. Ultrasonics 1990, 28, 295–303. [Google Scholar]
- Henglein, A. Sonochemistry: historical developments and modern aspects. Ultrasonics 1987, 25, 6–16. [Google Scholar] [CrossRef]
- Currell, D.L.; Wilheim, G.; Nagy, S. The effect of certain variables on the ultrasonic cleavage of phenol and of pyridine. J. Am. Chem. Soc. 1963, 85, 127–130. [Google Scholar] [CrossRef]
- Weissler, A. Formation of hydrogen peroxide by ultrasonic waves: free radicals. J. Am. Chem. Soc. 1959, 81, 1077–1081. [Google Scholar] [CrossRef]
- Anbar, M.; Pecht, I. On the sonochemical formation of hydrogen peroxide in water. J. Phys. Chem. 1964, 68, 352–355. [Google Scholar] [CrossRef]
- Henglein, A.; Kormann, C. Scavenging of OH radicals produced in the sonolysis of water. Int. J. Radiat. Biol. 1985, 48, 251–258. [Google Scholar] [CrossRef]
- Krishna, C.M.; Kondo, T.; Riesz, P. Sonochemistry of alcohol-water mixtures: spin-trapping evidence for thermal decomposition and isotope-exchange reactions. J. Phys. Chem. 1989, 93, 5166–5172. [Google Scholar] [CrossRef]
- Atkinson, R. Kinetics and Mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds under atmospheric conditions. Chem. Rev. 1985, 85, 69–201. [Google Scholar]
- Bapat, P.S.; Gogate, P.R.; Pandit, A.B. Theoretical analysis of sonochemical degradation of phenol and its chloro-derivatives. Ultrason. Sonochem. 2008, 15, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Lim, K.P.; Kumaran, S.S.; Kiefer, J.H. Thermal decomposition of carbon tetrachloride. J. Phys. Chem. 1993, 97, 1914–1919. [Google Scholar] [CrossRef]
- Nohhebel, D.C.; Walton, J.C. Free-Radical Chemistry; Cambridge University Press: Cambridge, UK, 1974; p. 572. [Google Scholar]
- Ellermann, T. Fine structure of the CCl3 UV absorption spectrum and CCl3 kinetics. Chem. Phys. Lett. 1992, 189, 175–181. [Google Scholar]
- Yokoyama, K.; Fujisawa, G.; Yokoyama, A. The mechanism of the unimolecular dissociation of trichloroethylene CHCl=CCl2 in the ground electronic state. J. Chem. Phys. 1995, 102, 7902–7909. [Google Scholar] [CrossRef]
- Ritter, E.R.; Bozelli, J.W.; Dean, A.M. Kinetic study on thermal decomposition of chlorobenzene diluted in H2. J. Phys. Chem. 1990, 94, 2493–2504. [Google Scholar] [CrossRef]
- Kern, R.D.; Xie, K.; Chen, H. A Shock tube study of chlorobenzene pyrolysis. Combust. Sci. Technol. 1992, 85, 77–86. [Google Scholar] [CrossRef]
- Mead, E.L.; Sutherland, R.G.; Verral, R.E. The effect on water in the presence of dissolved gases. Can. J. Chem. 1976, 54, 1114–1120. [Google Scholar] [CrossRef]
- Hart, E.J.; Henglein, A. Sonolytic decomposition of nitrous oxide in aqueous solution. J. Phys. Chem. 1986, 90, 5992–5995. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, Y.-N. Solubility and decomposition kinetics of nitrous acid in aqueous solution. J. Phys. Chem. 1988, 92, 6294–6302. [Google Scholar] [CrossRef]
- Janzen, E.G. Electron Spin Resonance. Anal. Chem. 1974, 46, 478R–490R. [Google Scholar] [CrossRef]
- Kruus, P.; Beutel, L.; Aranda, R.; Penchuk, J.; Otson, R. Formation of complex organochlorine species in water due to cavitation. Chemosphere 1998, 36, 1811–1824. [Google Scholar] [CrossRef]
- Gallego-Juárez, J.A.; Rodriguez, G.; Acosta, V.; Riera, E. Power ultrasonic transducers with extensive radiators for industrial processing. Ultrason. Sonochem. [CrossRef]
- Krüger, O.; Schulze, Th.-L.; Peters, D. Sonochemical treatment of natural ground water at different high frequencies: preliminary results. Ultrason. Sonochem. 1999, 6, 123–128. [Google Scholar] [CrossRef]
- Peters, D. Sonolytic degradation of volatile pollutants in natural ground water: conclusions from a model study. Ultrason. Sonochem. 2001, 8, 221–226. [Google Scholar] [CrossRef]
- Guo, Z.; Gu, C.; Zheng, Z.; Feng, R.; Jiang, F.; Gao, G.; Zheng, Y. Sonodegradation of halomethane mixtures in chlorinated drinking water. Ultrason. Sonochem. 2006, 13, 487–492. [Google Scholar] [CrossRef]
- Clark II, C.J.; Annable, M.D.; Rao, P.S. Evaluation of sonochemical destruction of PCE in situ flushing waste fluids. J. Environ. Eng. 2000, 126, 1033–1038. [Google Scholar] [CrossRef]
- Schwarzenbach, R.P.; Gschwend, P.M.; Imboden, D.M. Environmental organic chemistry; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH·/O·-) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar]
- Louisnard, O.; González-García, J.; Tudela, I.; Klíma, J.; Sáez, V.; Vargas-Hernández, Y. FEM simulation of a sono-reactor accounting for vibrations of the boundaries. Ultrason. Sonochem. 2009, 16, 250–259. [Google Scholar] [CrossRef]
- Ashokkumar, M.; Lee, J.; Iida, Y.; Yasui, K.; Kozuka, T.; Tuziuti, T.; Towata, A. The detection and control of stable and transient acoustic cavitation bubbles. Phys. Chem. Chem. Phys. 2009, 11, 10118–10121. [Google Scholar] [CrossRef]
- Moholkar, V.S. Mechanistic optimization of a dual frequency sonochemical reactor. Chem. Eng. Sci. 2009, 64, 5255–5267. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).