

Genomic Analysis of the Uncultured AKYH767 Lineage from a Wastewater Treatment Plant Predicts a Facultatively Anaerobic Heterotrophic Lifestyle and the Ability to Degrade Aromatic Compounds



Abstract
1. Introduction
2. Materials and Methods
2.1. Characteristics of WWTPs, Sampling Sources, and Metagenome Sequencing
2.2. Assembly and Taxonomic Identification of MAGs
2.3. Metagenome Sequencing Using the Oxford Nanopore Technique and Assembly of the Complete Genome of the AKYH767 Bacterium
2.4. Annotation of MAGs, Phylogenetic Analysis, and Metabolic Reconstruction
3. Results
3.1. Genomic Features of the AKYH767 Lineage
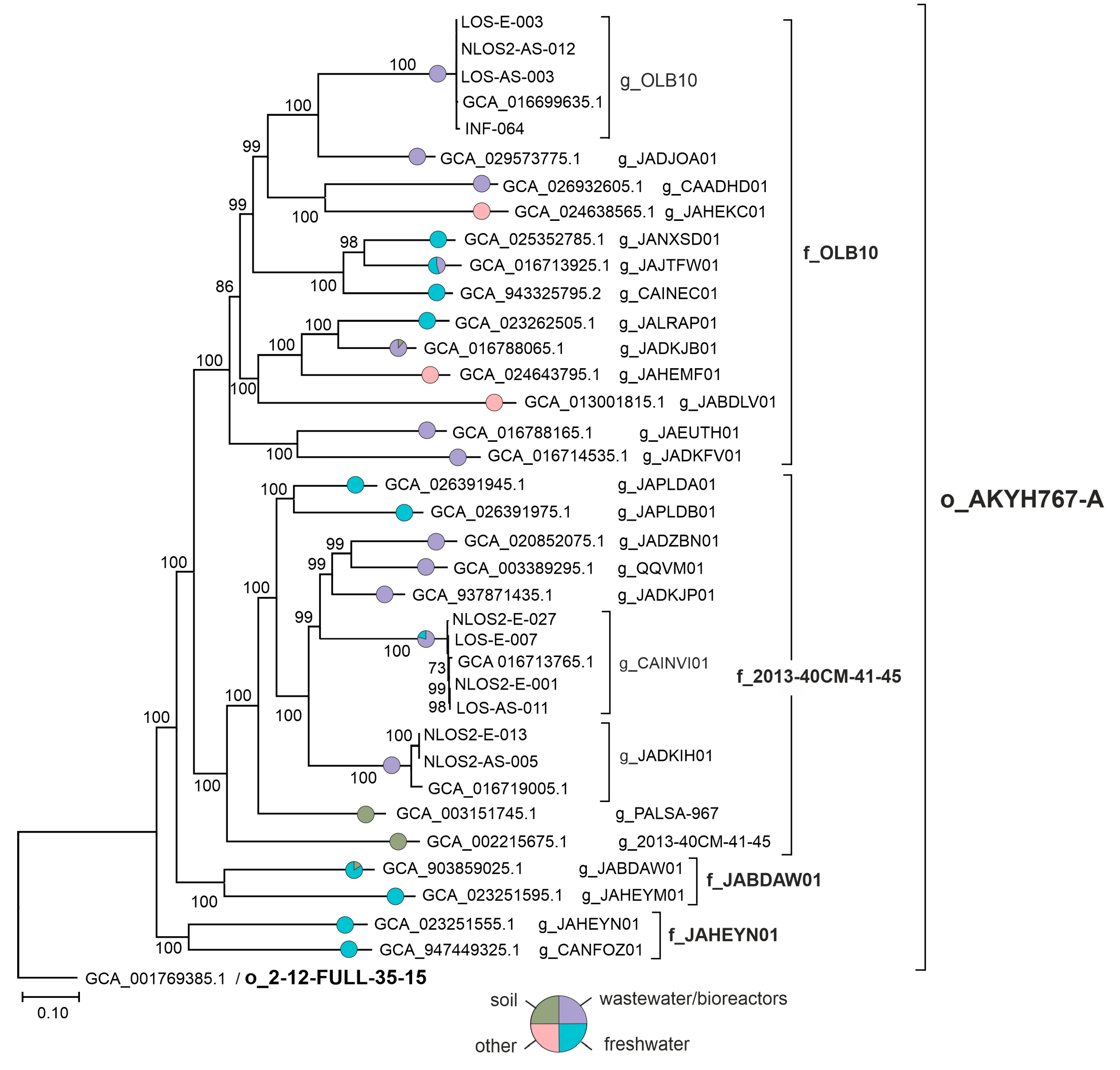
3.2. Phylogenetic Placement of AKYH767 Genomes
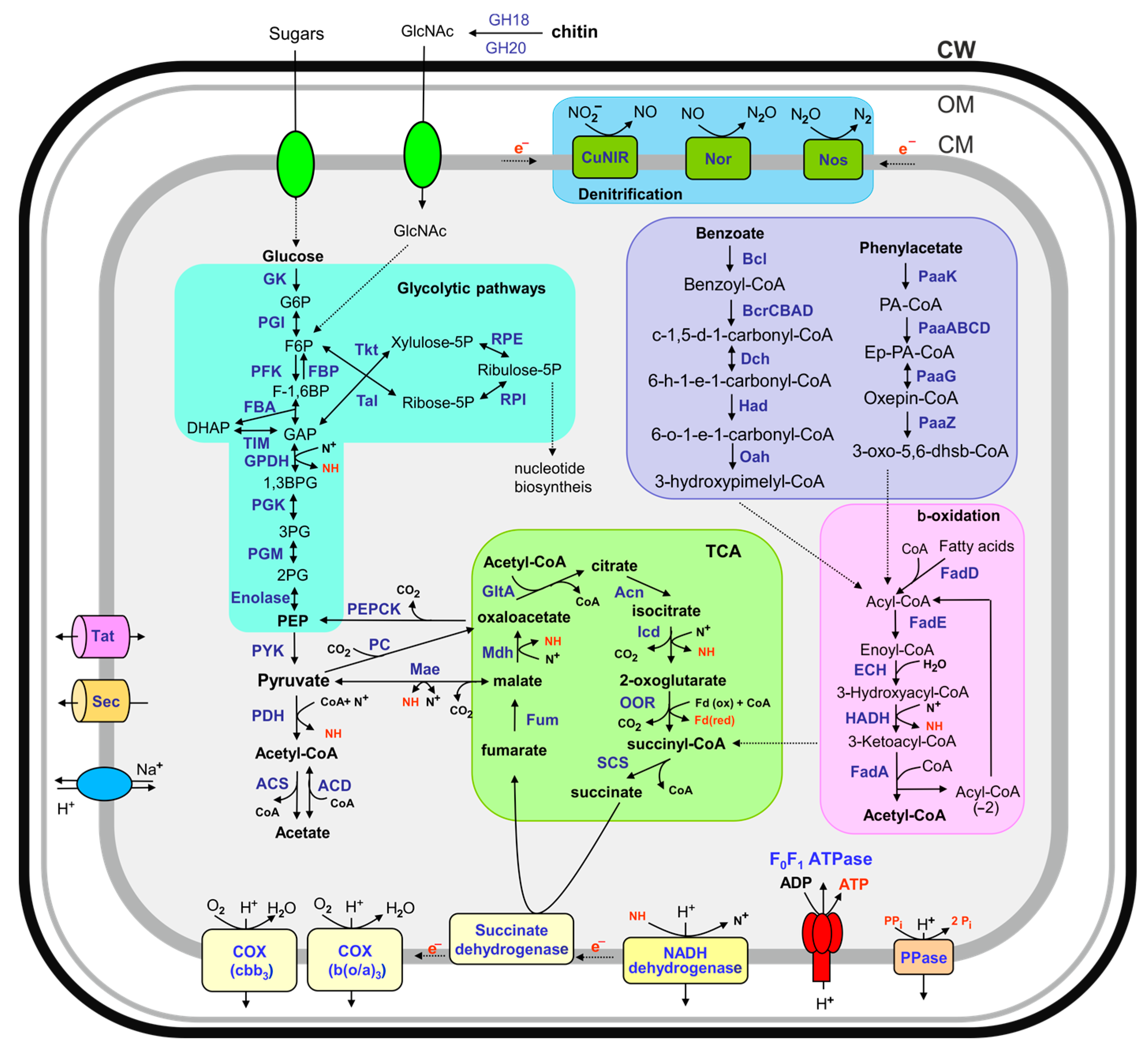
3.3. Central Metabolic Pathways
3.4. Possible Growth Substrates
3.5. Description of New Taxa
- Candidatus Pollutiaquabacterales ord. nov.
- Candidatus Pollutiaquabacteraceae fam. nov.
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sharma, M.; Agarwal, S.; Agarwal Malik, R.; Kumar, G.; Pal, D.B.; Mandal, M.; Sarkar, A.; Bantun, F.; Haque, S.; Singh, P.; et al. Recent advances in microbial engineering approaches for wastewater treatment: A review. Bioengineered 2023, 14, 2184518. [Google Scholar] [CrossRef] [PubMed]
- Seviour, R.J.; Nielsen, P.H. (Eds.) Microbial Ecology of Activated Sludge; IWA Publishing: London, UK, 2010; p. 688. [Google Scholar]
- Nielsen, P.H.; Daims, H.; Lemmer, H. (Eds.) FISH Handbook for Biological Wastewater Treatment; IWA Publishing: London, UK, 2009; p. 200. [Google Scholar]
- Chen, G.; Bai, R.; Zhang, Y.; Zhao, B.; Xiao, Y. Application of metagenomics to biological wastewater treatment. Sci. Total Environ. 2022, 807, 150737. [Google Scholar] [CrossRef] [PubMed]
- Bovio-Winkler, P.; Guerrero, L.D.; Erijman, L.; Oyarzúa, P.; Suárez-Ojeda, M.E.; Cabezas, A.; Etchebehere, C. Genome-centric metagenomic insights into the role of Chloroflexi in anammox, activated sludge and methanogenic reactors. BMC Microbiol. 2023, 23, 45. [Google Scholar] [CrossRef] [PubMed]
- Bovio-Winkler, P.; Cabezas, A.; Etchebehere, C. Unveiling the hidden diversity and functional role of Chloroflexota in full-scale wastewater treatment plants through genome-centric analyses. ISME Commun. 2024, 4, ycae050. [Google Scholar] [CrossRef] [PubMed]
- Mardanov, A.V.; Beletsky, A.V.; Ravin, N.V.; Botchkova, E.A.; Litti, Y.V.; Nozhevnikova, A.N. Genome of a novel bacterium “Candidatus Jettenia ecosi” reconstructed from the metagenome of an anammox bioreactor. Front. Microbiol. 2019, 10, 2442. [Google Scholar] [CrossRef] [PubMed]
- Begmatov, S.; Dorofeev, A.G.; Kadnikov, V.V.; Beletsky, A.V.; Pimenov, N.V.; Ravin, N.V.; Mardanov, A.V. The structure of microbial communities of activated sludge of large-scale wastewater treatment plants in the city of Moscow. Sci. Rep. 2022, 12, 3458. [Google Scholar] [CrossRef]
- Begmatov, S.; Beletsky, A.V.; Dorofeev, A.G.; Pimenov, N.V.; Mardanov, A.V.; Ravin, N.V. Metagenomic insights into the wastewater resistome before and after purification at large-scale wastewater treatment plants in the Moscow city. Sci. Rep. 2024, 14, 6349. [Google Scholar] [CrossRef]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.; Zhang, Q.; Brown, M.R.; Li, Z.; Van Nostrand, J.D.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef]
- Derwis, D.; Al-Hazmi, H.E.; Majtacz, J.; Ciesielski, S.; Mąkinia, J. Enhancing nitrogen removal in the partial denitrification/anammox processes for SO4-rich wastewater treatment: Insights into autotrophic and mixotrophic strategies. J. Environ. Manag. 2024, 358, 120908. [Google Scholar] [CrossRef]
- Guo, M.; Wang, J.; You, J.; Zong, Y.; Fu, C. The Influence of DO on the Microbiological Community of the A2O Treatment of Municipal Wastewater in Alpine Regions. Water Air Soil Pollut. 2022, 233, 470. [Google Scholar] [CrossRef]
- Cai, C.H.; Then, C.K.; Lin, Y.L.; Shih, C.C.; Li, C.C.; Yang, T.S. Associative analysis of sludge microbiota and wastewater degradation efficacy within swine farm sludge systems. Heliyon 2024, 10, e39997. [Google Scholar] [CrossRef]
- Bond, P.L.; Hugenholtz, P.; Keller, J.; Blackall, L.L. Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl. Environ. Microbiol. 1995, 61, 1910–1916. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 2020, 16, e1007981. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhao, X.M.; Coelho, L.P. SemiBin2: Self-supervised contrastive learning leads to better MAGs for short- and long-read sequencing. Bioinformatics 2023, 39, i21–i29. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Rodriguez, R.L.M.; Konstantinidis, K.T. The Enveomics Collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2019, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Mauch, E.; Serra, M.L.; Andrei, A.S. Complete genome sequence of an uncultivated freshwater Bacteroidota lineage. Microbiol. Resour. Announc. 2022, 11, e0076622. [Google Scholar] [CrossRef] [PubMed]
- Bendezú, F.O.; de Boer, P.A. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 2008, 190, 1792–1811. [Google Scholar] [CrossRef] [PubMed]
- Singleton, C.M.; Petriglieri, F.; Kristensen, J.M.; Kirkegaard, R.H.; Michaelsen, T.Y.; Andersen, M.H.; Kondrotaite, Z.; Karst, S.M.; Dueholm, M.S.; Nielsen, P.H.; et al. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 2021, 12, 2009. [Google Scholar] [CrossRef]
- Pitcher, R.S.; Brittain, T.; Watmough, N.J. Cytochrome cbb(3) oxidase and bacterial microaerobic metabolism. Biochem. Soc. Trans. 2002, 30, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Godocíková, J.; Zámocký, M.; Bucková, M.; Obinger, C.; Polek, B. Molecular diversity of katG genes in the soil bacteria Comamonas. Arch. Microbiol. 2010, 192, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Huxley, L.; Quirk, P.G.; Cotton, N.P.; White, S.A.; Jackson, B. The specificity of proton-translocating transhydrogenase for nicotinamide nucleotides. Biochim. Biophys. Acta 2011, 1807, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Stenlid, J. Evolution of Family 18 Glycoside Hydrolases: Diversity, Domain Structures and Phylogenetic Relationships. J. Mol. Microbiol. Biotechnol. 2009, 16, 208–223. [Google Scholar] [CrossRef]
- Wang, X.; Isbrandt, T.; Strube, M.L.; Paulsen, S.S.; Nielsen, M.W.; Buijs, Y.; Schoof, E.M.; Larsen, T.O.; Gram, L.; Zhang, S.D. Chitin degradation machinery and secondary metabolite profiles in the marine bacterium Pseudoalteromonas rubra S4059. Mar. Drugs 2021, 19, 108. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Gumerov, V.M.; Rakitin, A.L.; Beletsky, A.V.; Damsté, J.S.; Muyzer, G.; Mardanov, A.V.; Ravin, N.V. Genome analysis of Chitinivibrio alkaliphilus gen. nov., sp. nov., a novel extremely haloalkaliphilic anaerobic chitinolytic bacterium from the candidate phylum Termite Group 3. Environ. Microbiol. 2014, 16, 1549–1565. [Google Scholar] [CrossRef]
- Jeckelmann, J.M.; Erni, B. Transporters of Glucose and other carbohydrates in Bacteria. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1129–1153. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 Update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Price, M.N.; Deutschbauer, A.M.; Arkin, A.P. GapMind: Automated annotation of amino acid biosynthesis. Msystems 2020, 53, e00291-20. [Google Scholar] [CrossRef]
- Dosselaere, F.; Vanderleyden, J. A metabolic node in action: Chorismate-utilizing enzymes in microorganisms. Crit. Rev. Microbiol. 2001, 27, 75–131. [Google Scholar] [CrossRef]
- Fuchs, G.; Boll, M.; Heider, J. Microbial degradation of aromatic compounds—From one strategy to four. Nat. Rev. Microbiol. 2011, 9, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Luengo, J.M.; García, J.L.; Olivera, E.R. The phenylacetyl-CoA catabolon: A complex catabolic unit with broad biotechnological applications. Mol. Microbiol. 2001, 39, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Mascaraque, V.; Ismail, W.; Voss, M.; Perera, J.; Eisenreich, W.; Haehnel, W.; Fuchs, G. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. USA 2010, 107, 14390–14395. [Google Scholar] [CrossRef] [PubMed]
- Ismail, W.; Gescher, J. Epoxy Coenzyme A Thioester pathways for degradation of aromatic compounds. Appl. Environ. Microbiol. 2012, 78, 5043–5051. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Rosselló-Móra, R.; Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017, 11, 2399–2406. [Google Scholar] [CrossRef]
- Fatone, F.; Di Fabio, S.; Bolzonella, D.; Cecchi, F. Fate of aromatic hydrocarbons in Italian municipal wastewater systems: An overview of wastewater treatment using conventional activated-sludge processes (CASP) and membrane bioreactors (MBRs). Water Res. 2011, 45, 93–104. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, T.; Li, X.; Yao, J.; Liu, W.; Chang, S.; Chen, Y. The fate and enhanced removal of polycyclic aromatic hydrocarbons in wastewater and sludge treatment system: A review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 1425–1475. [Google Scholar] [CrossRef]
- Syafiuddin, A.; Boopathy, R. A review of polycyclic aromatic hydrocarbons and their substitutions in full-scale wastewater treatment plants. Environ. Qual. Manag. 2021, 31, 21–37. [Google Scholar] [CrossRef]
- Vogelsang, C.; Grung, M.; Jantsch, T.G.; Tollefsen, K.E.; Liltved, H. Occurrence and removal of selected organic micropollutants at mechanical, chemical and advanced wastewater treatment plants in Norway. Water Res. 2006, 40, 3559–3570. [Google Scholar] [CrossRef] [PubMed]
- Mezzanotte, V.; Anzano, M.; Collina, E.; Marazzi, F.A.; Lasagni, M. Distribution and removal of polycyclic aromatic hydrocarbons in two Italian municipal wastewater treatment plants in 2011–2013. Polycycl. Aromat. Compd. 2015, 36, 213–228. [Google Scholar] [CrossRef]
- Cao, W.; Qiao, M.; Liu, B.; Zhao, X. Occurrence of parent and substituted polycyclic aromatic hydrocarbons in typical wastewater treatment plants and effluent receiving rivers of Beijing, and risk assessment. J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 2018, 53, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Haddad, L.; Ali, M.; Pronk, M.; van Loosdrecht, M.C.M.; Saikaly, P.E. Demystifying polyphosphate-accumulating organisms relevant to wastewater treatment: A review of their phylogeny, metabolism, and detection. Environ. Sci. Ecotechnol. 2024, 21, 100387. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| MAG ID | Source | Completeness, % | Contamination, % | Coding Density, % | Contigs | Contig N50 (nt) | MAG Length, Mb | GC Content, % | Protein-Coding Genes | tRNA Genes | rRNA Operons * |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NLOS2-E-001 | Effluent, NLOS2 | 100 | 0.11 | 0.907 | 1 | 3,719,830 | 3.72 | 54 | 3005 | 42 | 3 |
| LOS-E-007 | Effluent, LOS | 90.24 | 0.71 | 0.928 | 256 | 17,413 | 2.82 | 54 | 2436 | 42 | 2 |
| LOS-AS-011 | AS, LOS | 96.81 | 1.91 | 0.926 | 387 | 15,555 | 3.44 | 54 | 2927 | 45 | 2 |
| NLOS2-E-027 | Effluent, NLOS2 | 93.13 | 1.96 | 0.924 | 407 | 15,220 | 3.55 | 54 | 3040 | 49 | 3 |
| LOS-E-003 | Effluent, LOS | 99.94 | 0.48 | 0.927 | 70 | 88,674 | 3.08 | 37 | 2585 | 64 | 1 |
| LOS-AS-003 | AS, LOS | 99.92 | 0.46 | 0.925 | 60 | 122,921 | 3.12 | 37 | 2664 | 69 | 2 |
| NLOS2-AS-012 | AS, NLOS2 | 100 | 0.4 | 0.925 | 133 | 37,341 | 3.11 | 37 | 2681 | 60 | 1 |
| INF-064 | Influent, NLOS2 | 97.29 | 1.46 | 0.928 | 318 | 11,795 | 2.76 | 37 | 2567 | 44 | 1 |
| NLOS2-E-013 | Effluent, NLOS2 | 96.37 | 1.02 | 0.902 | 321 | 17,020 | 3.53 | 39 | 3023 | 33 | 2 |
| NLOS2-AS-005 | AS, NLOS2 | 97.36 | 0.22 | 0.902 | 211 | 30,160 | 3.82 | 39 | 3166 | 38 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begmatov, S.; Beletsky, A.V.; Mardanov, A.V.; Ravin, N.V. Genomic Analysis of the Uncultured AKYH767 Lineage from a Wastewater Treatment Plant Predicts a Facultatively Anaerobic Heterotrophic Lifestyle and the Ability to Degrade Aromatic Compounds. Water 2025, 17, 1061. https://doi.org/10.3390/w17071061
Begmatov S, Beletsky AV, Mardanov AV, Ravin NV. Genomic Analysis of the Uncultured AKYH767 Lineage from a Wastewater Treatment Plant Predicts a Facultatively Anaerobic Heterotrophic Lifestyle and the Ability to Degrade Aromatic Compounds. Water. 2025; 17(7):1061. https://doi.org/10.3390/w17071061
Chicago/Turabian StyleBegmatov, Shahjahon, Alexey V. Beletsky, Andrey V. Mardanov, and Nikolai V. Ravin. 2025. "Genomic Analysis of the Uncultured AKYH767 Lineage from a Wastewater Treatment Plant Predicts a Facultatively Anaerobic Heterotrophic Lifestyle and the Ability to Degrade Aromatic Compounds" Water 17, no. 7: 1061. https://doi.org/10.3390/w17071061
APA StyleBegmatov, S., Beletsky, A. V., Mardanov, A. V., & Ravin, N. V. (2025). Genomic Analysis of the Uncultured AKYH767 Lineage from a Wastewater Treatment Plant Predicts a Facultatively Anaerobic Heterotrophic Lifestyle and the Ability to Degrade Aromatic Compounds. Water, 17(7), 1061. https://doi.org/10.3390/w17071061